
Flame Retardants-Mediated Interferon Signaling in the Pathogenesis of Nonalcoholic Fatty Liver Disease




Abstract
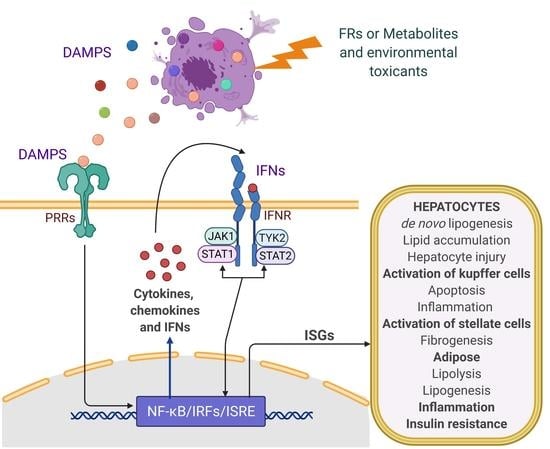
:
1. Introduction
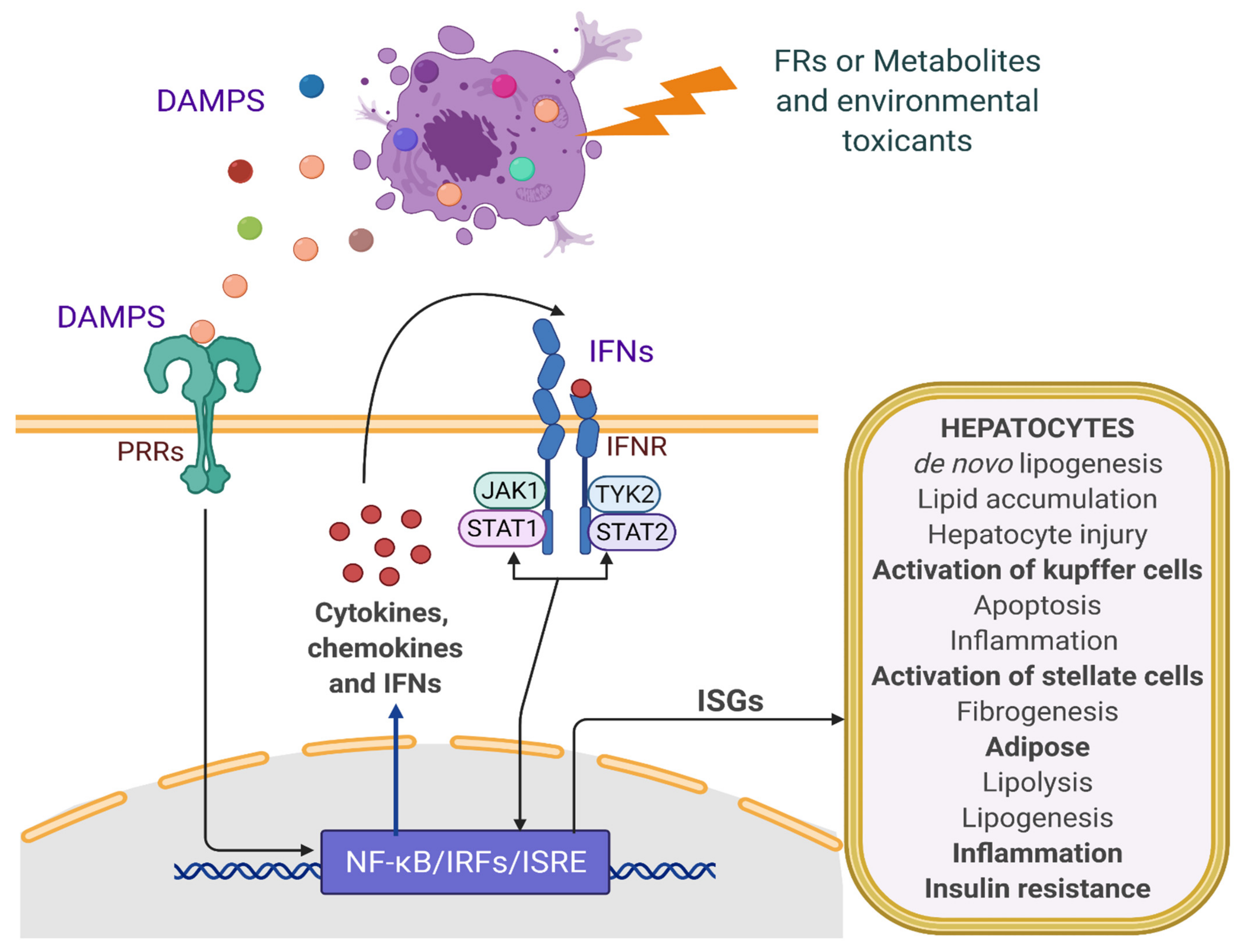
2. Pathobiology of NAFLD: Role of IFN and Inflammatory Signaling
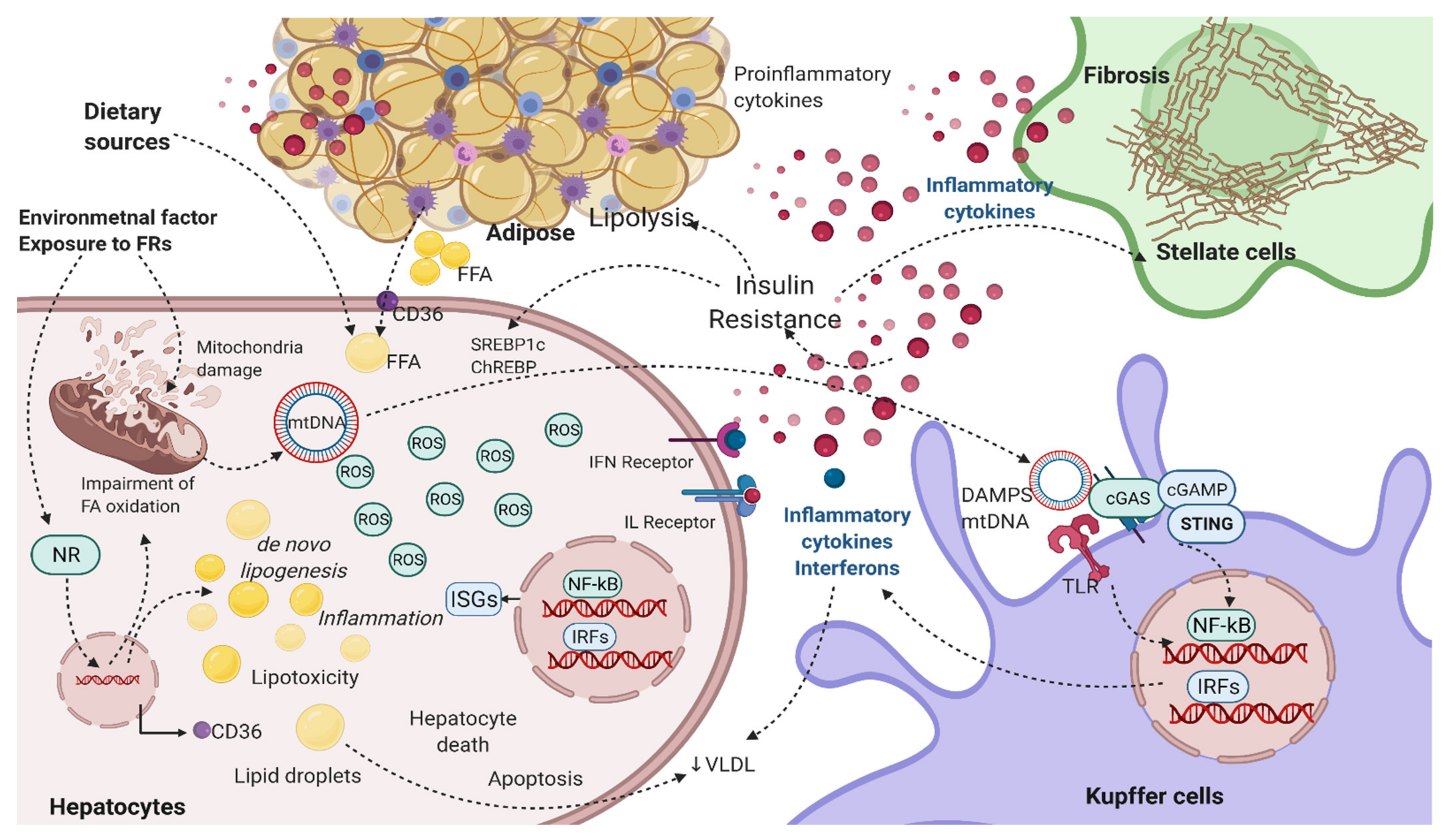
3. Biological Actions of FRs: Role in Inflammatory and Cytokine Signaling
4. Modulation of IFN Signaling by FRs and Role in NAFLD
5. Role of FRs in Modulating Other Signaling Linked to NAFLD Biology
6. Emerging Pharmacotherapeutics for NAFLD Targeting Immune or Inflammatory Signaling
7. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ACC | acetyl-CoA carboxylase |
| AhR | aryl hydrocarbon receptor |
| ALT | alanine aminotransferase |
| AOP | adverse outcome pathways |
| AST | aspartate Aminotransferase |
| ATP | adenosine triphosphate |
| Bcl2 | b-cell lymphoma 2 |
| BFR | brominated flame retardants |
| cAMP | cyclic adenosine monophosphate |
| CAR | chimeric antigen receptor |
| CCR5 | c-c chemokine receptor type 5 |
| CD36 | cluster of differentiation 36 |
| cGAMP | cyclic guanosine monophosphate-adenosine monophosphate |
| cGAS | cyclic guanosine monophosphate-adenosine monophosphate synthase |
| ChREBP | carbohydrate-responsive element-binding protein |
| CYP450 | cytochrome P450 |
| DAMPs | damage-associated molecular patterns |
| DGAT | diacylglycerol acyltransferase |
| ERK | extracellular-signal-regulated kinase |
| FAS | fatty acid synthase |
| FR | flame retardants |
| FXR | farnesoid X receptor |
| GLUT4 | glucose transporter type 4 |
| HCC | hepatocellular carcinoma |
| HMGB1 | high mobility group box protein 1 |
| HFD | high-fat diet |
| IFN | interferon |
| IFNGR | interferon-gamma receptor |
| IL | interleukin |
| IRF | interferon regulatory factor |
| IRS | insulin receptor substrate |
| ISGs | interferon-stimulated genes |
| ISRE | interferon-stimulated response elements |
| JAK1 | Janus kinase 1 |
| JNK | c-jun N-terminal protein kinase |
| LXR | liver X receptor |
| MAPK | mitogen-activated protein kinase |
| MCP | monocyte chemoattractant protein |
| MDC | metabolic disrupting chemicals |
| MIE | molecular initiating events |
| mtDNA | mitochondrial DNA |
| mTOR | mammalian target of rapamycin |
| NAFLD | nonalcoholic fatty liver disease |
| NASH | nonalcoholic steatohepatitis |
| NLRs | nucleotide-binding oligomerization domain-like receptors |
| NR | nuclear receptor |
| NXR | nuclear xenobiotic receptors |
| OPFRs | organophosphorus flame retardants |
| PAMP | pathogen-associated molecular patterns |
| PBDEs | polybrominated diphenyl ethers |
| PBMC | peripheral blood mononuclear cells |
| PI3K | phosphoinositide 3-kinase |
| PPAR | peroxisome proliferator-activated receptor |
| PRR | pattern recognition receptors |
| PXR | pregnane X receptor |
| RAGE | receptor for advanced glycation end products |
| ROS | reactive oxygen species |
| RXR | retinoid X receptor |
| SCD-1 | stearoyl-CoA desaturase-1 |
| SOCS | suppressor of cytokine signaling |
| SREBP | sterol regulatory element-binding protein |
| STAT | signal transducer and activator of transcription |
| STING | stimulator of interferon genes |
| T2D | type 2 diabetes mellitus |
| TAFLD | toxicant-associated fatty liver disease |
| TASH | toxicant-associated steatohepatitis |
| TGFβ | transforming growth factor-beta |
| TH | thyroid hormone |
| TLR | Toll-like receptor |
| TNF | tumor necrosis factor |
| TYK2 | tyrosine kinase 2 |
| VLDL | very-low-density lipoprotein |
References
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease—Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [Green Version]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef]
- Farrell, G.C.; Larter, C.Z. Nonalcoholic fatty liver disease: From steatosis to cirrhosis. Hepatology 2006, 43, S99–S112. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Blissett, D.; Blissett, R.; Henry, L.; Stepanova, M.; Younossi, Y.; Racila, A.; Hunt, S.; Beckerman, R. The economic and clinical burden of nonalcoholic fatty liver disease in the United States and Europe. Hepatology 2016, 64, 1577–1586. [Google Scholar] [CrossRef]
- Sanyal, A.J. Past, present and future perspectives in nonalcoholic fatty liver disease. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Alves-Bezerra, M.; Cohen, D.E. Triglyceride metabolism in the liver. Compr. Physiol. 2018, 8, 1–22. [Google Scholar] [CrossRef]
- Martin, S.; Parton, R.G. Lipid droplets: A unified view of a dynamic organelle. Nat. Rev. Mol. Cell Biol. 2006, 7, 373–378. [Google Scholar] [CrossRef]
- Byrne, C.D.; Targher, G. What’s new in NAFLD pathogenesis, biomarkers and treatment? Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 70–71. [Google Scholar] [CrossRef]
- Nassir, F.; Rector, R.S.; Hammoud, G.M.; Ibdah, J.A. Pathogenesis and prevention of hepatic steatosis. Gastroenterol. Hepatol. 2015, 11, 167–175. [Google Scholar]
- Fielding, C.M.; Angulo, P. Hepatic steatosis and steatohepatitis: Are they really two distinct entities? Curr. Hepatol. Rep. 2014, 13, 151–158. [Google Scholar] [CrossRef] [Green Version]
- Fang, Y.L.; Chen, H.; Wang, C.L.; Liang, L. Pathogenesis of non-alcoholic fatty liver disease in children and adolescence: From “two hit theory” to “multiple hit model”. World J. Gastroenterol. 2018, 24, 2974–2983. [Google Scholar] [CrossRef]
- Aron-Wisnewsky, J.; Vigliotti, C.; Witjes, J.; Le, P.; Holleboom, A.G.; Verheij, J.; Nieuwdorp, M.; Clément, K. Gut microbiota and human NAFLD: Disentangling microbial signatures from metabolic disorders. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 279–297. [Google Scholar] [CrossRef] [PubMed]
- Polyzos, S.A.; Kountouras, J.; Mantzoros, C.S. Obesity and nonalcoholic fatty liver disease: From pathophysiology to therapeutics. Metabolism 2019, 92, 82–97. [Google Scholar] [CrossRef]
- Foulds, C.E.; Treviño, L.S.; York, B.; Walker, C.L. Endocrine-disrupting chemicals and fatty liver disease. Nat. Rev. Endocrinol. 2017, 13, 445–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heindel, J.J.; Blumberg, B.; Cave, M.; Machtinger, R.; Mantovani, A.; Mendez, M.A.; Nadal, A.; Palanza, P.; Panzica, G.; Sargis, R.; et al. Metabolism disrupting chemicals and metabolic disorders. Reprod. Toxicol. 2017, 68, 3–33. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, T.; Hidaka, T.; Kumagai, Y.; Yamamoto, M. Environmental pollutants and the immune response. Nat. Immunol. 2020, 21, 1486–1495. [Google Scholar] [CrossRef] [PubMed]
- Wahlang, B.; Jin, J.; Beier, J.I.; Hardesty, J.E.; Daly, E.F.; Schnegelberger, R.D.; Falkner, K.C.; Prough, R.A.; Kirpich, I.A.; Cave, M.C. Mechanisms of Environmental Contributions to Fatty Liver Disease. Curr. Environ. Health Rep. 2019, 6, 80–94. [Google Scholar] [CrossRef] [PubMed]
- Wahlang, B.; Beier, J.I.; Clair, H.B.; Bellis-Jones, H.J.; Falkner, K.C.; McClain, C.J.; Cave, M.C. Toxicant-associated steatohepatitis. Toxicol. Pathol. 2013, 41, 343–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwingel, P.A.; Cotrim, H.P.; Salles, B.R.; Almeida, C.E.; Dos Santos, C.R.; Nachef, B.; Andrade, A.R.; Zoppi, C.C. Anabolic-androgenic steroids: A possible new risk factor of toxicant-associated fatty liver disease. Liver Int. 2011, 31, 348–353. [Google Scholar] [CrossRef] [PubMed]
- Cave, M.; Falkner, K.C.; Ray, M.; Joshi-Barve, S.; Brock, G.; Khan, R.; Bon Homme, M.; McClain, C.J. Toxicant-associated steatohepatitis in vinyl chloride workers. Hepatology 2010, 51, 474–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibson, E.A.; Stapleton, H.M.; Calero, L.; Holmes, D.; Burke, K.; Martinez, R.; Cortes, B.; Nematollahi, A.; Evans, D.; Herbstman, J.B. Flame retardant exposure assessment: Findings from a behavioral intervention study. J. Expo. Sci. Environ. Epidemiol. 2019, 29, 33–48. [Google Scholar] [CrossRef]
- Kemmlein, S.; Hahn, O.; Jann, O. Emissions of organophosphate and brominated flame retardants from selected consumer products and building materials. Atmos. Environ. 2003, 37, 5485–5493. [Google Scholar] [CrossRef]
- González-Rubio, S.; Ballesteros-Gómez, A.; Asimakopoulos, A.G.; Jaspers, V.L.B. A review on contaminants of emerging concern in European raptors (2002−2020). Sci. Total Environ. 2020, 760, 143337. [Google Scholar] [CrossRef]
- Yang, J.; Zhao, Y.; Li, M.; Du, M.; Li, X.; Li, Y. A review of a class of emerging contaminants: The classification, distribution, intensity of consumption, synthesis routes, environmental effects and expectation of pollution abatement to organophosphate flame retardants (opfrs). Int. J. Mol. Sci. 2019, 20, 2874. [Google Scholar] [CrossRef] [Green Version]
- Maddela, N.R.; Venkateswarlu, K.; Kakarla, D.; Megharaj, M. Inevitable human exposure to emissions of polybrominated diphenyl ethers: A perspective on potential health risks. Environ. Pollut. 2020, 266, 115240. [Google Scholar] [CrossRef]
- Wang, R.; Tang, J.; Xie, Z.; Mi, W.; Chen, Y.; Wolschke, H.; Tian, C.; Pan, X.; Luo, Y.; Ebinghaus, R. Occurrence and spatial distribution of organophosphate ester flame retardants and plasticizers in 40 rivers draining into the Bohai Sea, north China. Environ. Pollut. 2015, 198, 172–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blum, A.; Behl, M.; Birnbaum, L.S.; Diamond, M.L.; Phillips, A.; Singla, V.; Sipes, N.S.; Stapleton, H.M.; Venier, M. Organophosphate Ester Flame Retardants: Are They a Regrettable Substitution for Polybrominated Diphenyl Ethers? Environ. Sci. Technol. Lett. 2019, 6, 638–649. [Google Scholar] [CrossRef] [PubMed]
- Van der Veen, I.; de Boer, J. Phosphorus flame retardants: Properties, production, environmental occurrence, toxicity and analysis. Chemosphere 2012, 88, 1119–1153. [Google Scholar] [CrossRef] [PubMed]
- Mitro, S.D.; Dodson, R.E.; Singla, V.; Adamkiewicz, G.; Elmi, A.F.; Tilly, M.K.; Zota, A.R. Consumer Product Chemicals in Indoor Dust: A Quantitative Meta-analysis of U.S. Studies. Environ. Sci. Technol. 2016, 50, 10661–10672. [Google Scholar] [CrossRef] [PubMed]
- Rantakokko, P.; Kumar, E.; Braber, J.; Huang, T.; Kiviranta, H.; Cequier, E.; Thomsen, C. Concentrations of brominated and phosphorous fl ame retardants in Finnish house dust and insights into children’s exposure. Chemosphere 2019, 223, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Poma, G.; Glynn, A.; Malarvannan, G.; Covaci, A.; Darnerud, P.O. Dietary intake of phosphorus flame retardants (PFRs) using Swedish food market basket estimations. Food Chem. Toxicol. 2017, 100, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Zhao, F.; Chen, M.; Gao, F.; Shen, H.; Hu, J. Organophosphorus Flame Retardants in Pregnant Women and Their Transfer to Chorionic Villi. Environ. Sci. Technol. 2017, 51, 6489–6497. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Isobe, T.; Muto, M.; Tue, N.M.; Katsura, K.; Malarvannan, G.; Sudaryanto, A.; Chang, K.H.; Prudente, M.; Viet, P.H.; et al. Organophosphorus flame retardants (PFRs) in human breast milk from several Asian countries. Chemosphere 2014, 116, 91–97. [Google Scholar] [CrossRef]
- Ding, J.; Xu, Z.; Huang, W.; Feng, L.; Yang, F. Organophosphate ester flame retardants and plasticizers in human placenta in Eastern China. Sci. Total Environ. 2016, 554–555, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Qiao, L.; Zheng, X.B.; Zheng, J.; Lei, W.X.; Li, H.F.; Wang, M.H.; He, C.T.; Chen, S.J.; Yuan, J.G.; Luo, X.J.; et al. Analysis of human hair to assess exposure to organophosphate flame retardants: Influence of hair segments and gender differences. Environ. Res. 2016, 148, 177–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castorina, R.; Butt, C.; Stapleton, H.M.; Avery, D.; Harley, K.G.; Holland, N.; Eskenazi, B.; Bradman, A. Flame retardants and their metabolites in the homes and urine of pregnant women residing in California (the CHAMACOS cohort). Chemosphere 2017, 179, 159–166. [Google Scholar] [CrossRef] [Green Version]
- Saillenfait, A.M.; Ndaw, S.; Robert, A.; Sabaté, J.P. Recent biomonitoring reports on phosphate ester flame retardants: A short review. Arch. Toxicol. 2018, 92, 2749–2778. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Jin, J.; Wang, Y.; Hu, J.; Xu, M.; Sun, Y.; Ma, Y. Concentrations of organophosphorus, polybromobenzene, and polybrominated diphenyl ether flame retardants in human serum, and relationships between concentrations and donor ages. Chemosphere 2017, 171, 654–660. [Google Scholar] [CrossRef] [PubMed]
- Lunder, S.; Hovander, L.; Athanassiadis, I.; Bergman, Å. Significantly higher polybrominated diphenyl ether levels in young U.S. children than in their mothers. Environ. Sci. Technol. 2010, 44, 5256–5262. [Google Scholar] [CrossRef] [PubMed]
- Harrad, S.; de Wit, C.A.; Abdallah, M.A.-E.; Bergh, C.; Björklund, J.A.; Covaci, A.; Darnerud, P.O.; de Boer, J.; Diamond, M.; Huber, S.; et al. Indoor Contamination with Hexabromocyclododecanes, Polybrominated Diphenyl Ethers, and Perfluoroalkyl Compounds: An Important Exposure Pathway for People? Environ. Sci. Technol. 2010, 44, 3221–3231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schreder, E.D.; Uding, N.; La Guardia, M.J. Inhalation a significant exposure route for chlorinated organophosphate flame retardants. Chemosphere 2016, 150, 499–504. [Google Scholar] [CrossRef]
- Makinen, M.S.E.; Makinen, M.R.A.; Koistinen, J.T.B.; Pasanen, A.-L.; Pasanen, P.O.; Kalliokoski, P.J.; Korpi, A.M. Respiratory and dermal exposure to organophosphorus flame retardants and tetrabromobisphenol A at five work environments. Environ. Sci. Technol. 2009, 43, 941–947. [Google Scholar] [CrossRef]
- Gravel, S.; Aubin, S.; Labrèche, F. Assessment of Occupational Exposure to Organic Flame Retardants: A Systematic Review. Ann. Work Expo. Health 2019, 63, 386–406. [Google Scholar] [CrossRef] [PubMed]
- Estill, C.F.; Slone, J.; Mayer, A.; Chen, I.C.; La Guardia, M.J. Worker exposure to flame retardants in manufacturing, construction and service industries. Environ. Int. 2020, 135, 105349. [Google Scholar] [CrossRef] [PubMed]
- HBM4EU-Science and Policy for a Healthy Future. Available online: https://www.hbm4eu.eu/ (accessed on 19 April 2021).
- Costa, L.G.; de Laat, R.; Tagliaferri, S.; Pellacani, C. A mechanistic view of polybrominated diphenyl ether (PBDE) developmental neurotoxicity. Toxicol. Lett. 2014, 230, 282–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makey, C.M.; McClean, M.D.; Braverman, L.E.; Pearce, E.N.; He, X.-M.; Sjödin, A.; Weinberg, J.M.; Webster, T.F. Polybrominated Diphenyl Ether Exposure and Thyroid Function Tests in North American Adults. Environ. Health Perspect. 2016, 124, 420–425. [Google Scholar] [CrossRef] [Green Version]
- Johnson, P.I.; Stapleton, H.M.; Mukherjee, B.; Hauser, R.; Meeker, J.D. Associations between brominated flame retardants in house dust and hormone levels in men. Sci. Total Environ. 2013, 445–446, 177–184. [Google Scholar] [CrossRef] [Green Version]
- Schreiber, T.; Gassmann, K.; Götz, C.; Hübenthal, U.; Moors, M.; Krause, G.; Merk, H.F.; Nguyen, N.H.; Scanlan, T.S.; Abel, J.; et al. Polybrominated diphenyl ethers induce developmental neurotoxicity in a human in vitro model: Evidence for endocrine disruption. Environ. Health Perspect. 2010, 118, 572–578. [Google Scholar] [CrossRef] [Green Version]
- Martin, O.V.; Evans, R.M.; Faust, M.; Kortenkamp, A. A Human Mixture Risk Assessment for Neurodevelopmental Toxicity Associated with Polybrominated Diphenyl Ethers Used as Flame Retardants. Environ. Health Perspect. 2017, 125, 087016. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Peng, L.; Zhang, W.; Liu, C.; Yang, Q.; Zheng, S.; Bao, M.; Huang, Y.; Wu, K. Adipose tissue levels of polybrominated diphenyl ethers and breast cancer risk in Chinese women: A case–control study. Environ. Res. 2018, 167, 160–168. [Google Scholar] [CrossRef]
- Bajard, L.; Melymuk, L.; Blaha, L. Prioritization of hazards of novel flame retardants using the mechanistic toxicology information from ToxCast and Adverse Outcome Pathways. Environ. Sci. Eur. 2019, 31, 14. [Google Scholar] [CrossRef] [Green Version]
- Hood, E. Endocrine Disruption and Flame-Retardant Chemicals: PBDE-99 Effects on Rat Sexual Development. Environ. Health Perspect. 2006, 114, A112. [Google Scholar] [CrossRef] [Green Version]
- Dishaw, L.V.; Macaulay, L.J.; Roberts, S.C.; Stapleton, H.M. Exposures, mechanisms, and impacts of endocrine-active flame retardants. Curr. Opin. Pharmacol. 2014, 19, 125–133. [Google Scholar] [CrossRef] [Green Version]
- Kojima, H.; Takeuchi, S.; Van den Eede, N.; Covaci, A. Effects of primary metabolites of organophosphate flame retardants on transcriptional activity via human nuclear receptors. Toxicol. Lett. 2016, 245, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Diamanti-Kandarakis, E.; Bourguignon, J.P.; Giudice, L.C.; Hauser, R.; Prins, G.S.; Soto, A.M.; Zoeller, R.T.; Gore, A.C. Endocrine-disrupting chemicals: An Endocrine Society scientific statement. Endocr. Rev. 2009, 30, 293–342. [Google Scholar] [CrossRef] [PubMed]
- Gore, A.C.; Chappell, V.A.; Fenton, S.E.; Flaws, J.A.; Nadal, A.; Prins, G.S.; Toppari, J.; Zoeller, R.T. EDC-2: The Endocrine Society’s Second Scientific Statement on Endocrine-Disrupting Chemicals. Endocr. Rev. 2015, 36, 1–150. [Google Scholar] [CrossRef] [PubMed]
- Llm, J.S.; Lee, D.H.; Jacobs, D.R. Association of brominated flame retardants with diabetes and metabolic syndrome in the U.S. population, 2003–2004. Diabetes Care 2008, 31, 1802–1807. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Li, S.; Liu, L.; Wang, L.; Xiao, X.; Sun, Z.; Wang, X.; Wang, C.; Wang, M.; Li, L.; et al. Environmental exposure to BDE47 is associated with increased diabetes prevalence: Evidence from community-based case-control studies and an animal experiment. Sci. Rep. 2016, 6, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Zhao, F.; Li, Y.; Zhang, S.; Ding, M.; Hu, J. Association of Aryl Organophosphate Flame Retardants Triphenyl Phosphate and 2-Ethylhexyl Diphenyl Phosphate with Human Blood Triglyceride and Total Cholesterol Levels. Environ. Sci. Technol. Lett. 2019, 6, 532–537. [Google Scholar] [CrossRef]
- Ongono, J.S.; Dow, C.; Gambaretti, J.; Severi, G.; Boutron-Ruault, M.C.; Bonnet, F.; Fagherazzi, G.; Mancini, F.R. Dietary exposure to brominated flame retardants and risk of type 2 diabetes in the French E3N cohort. Environ. Int. 2019, 123, 54–60. [Google Scholar] [CrossRef]
- Eslam, M.; Sanyal, A.J.; George, J.; Sanyal, A.; Neuschwander-Tetri, B.; Tiribelli, C.; Kleiner, D.E.; Brunt, E.; Bugianesi, E.; Yki-Järvinen, H.; et al. MAFLD: A Consensus-Driven Proposed Nomenclature for Metabolic Associated Fatty Liver Disease. Gastroenterology 2020, 158, 1999–2014.e1. [Google Scholar] [CrossRef] [PubMed]
- Day, C.P.; James, O.F.W. Steatohepatitis: A tale of two “Hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef]
- Guturu, P.; Duchini, A. Etiopathogenesis of Nonalcoholic Steatohepatitis: Role of Obesity, Insulin Resistance and Mechanisms of Hepatotoxicity. Int. J. Hepatol. 2012, 2012, 212865. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Moschen, A.R. Evolution of inflammation in nonalcoholic fatty liver disease: The multiple parallel hits hypothesis. Hepatology 2010, 52, 1–8. [Google Scholar] [CrossRef]
- Tilg, H.; Adolph, T.E.; Moschen, A.R. Multiple Parallel Hits Hypothesis in NAFLD—Revisited After a Decade. Hepatology 2020, 73, 833–842. [Google Scholar] [CrossRef]
- Angrish, M.M.; Kaiser, J.P.; McQueen, C.A.; Chorley, B.N. Tipping the balance: Hepatotoxicity and the 4 apical key events of hepatic steatosis. Toxicol. Sci. 2016, 150, 261–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amacher, D.E. The mechanistic basis for the induction of hepatic steatosis by xenobiotics. Expert Opin. Drug Metab. Toxicol. 2011, 7, 949–965. [Google Scholar] [CrossRef]
- Yamaguchi, K.; Yang, L.; McCall, S.; Huang, J.; Xing, X.Y.; Pandey, S.K.; Bhanot, S.; Monia, B.P.; Li, Y.X.; Diehl, A.M. Inhibiting triglyceride synthesis improves hepatic steatosis but exacerbates liver damage and fibrosis in obese mice with nonalcoholic steatohepatitis. Hepatology 2007, 45, 1366–1374. [Google Scholar] [CrossRef] [PubMed]
- Listenberger, L.L.; Han, X.; Lewis, S.E.; Cases, S.; Farese, R.V.; Ory, D.S.; Schaffer, J.E. Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc. Natl. Acad. Sci. USA 2003, 100, 3077–3082. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.; Metlakunta, A.; Dedousis, N.; Zhang, P.; Sipula, I.; Dube, J.J.; Scott, D.K.; O’Doherty, R.M. Depletion of liver kupffer cells prevents the development of diet-induced hepatic steatosis and insulin resistance. Diabetes 2010, 59, 347–357. [Google Scholar] [CrossRef] [Green Version]
- Cai, Y.; Li, H.; Liu, M.; Pei, Y.; Zheng, J.; Zhou, J.; Luo, X.; Huang, W.; Ma, L.; Yang, Q.; et al. Disruption of adenosine 2A receptor exacerbates NAFLD through increasing inflammatory responses and SREBP1c activity. Hepatology 2018, 68, 48–61. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.B.; Liu, Y.; Liu, C.; Xiang, X.; Wang, J.; Cheng, Z.; Shah, S.V.; Zhang, S.; Zhang, L.; Zhuang, X.; et al. Immature myeloid cells induced by a high-fat diet contribute to liver inflammation. Hepatology 2009, 50, 1412–1420. [Google Scholar] [CrossRef] [Green Version]
- Cai, J.; Zhang, X.J.; Li, H. Role of Innate Immune Signaling in Non-Alcoholic Fatty Liver Disease. Trends Endocrinol. Metab. 2018, 29, 712–722. [Google Scholar] [CrossRef]
- Schuster, S.; Cabrera, D.; Arrese, M.; Feldstein, A.E. Triggering and resolution of inflammation in NASH. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 349–364. [Google Scholar] [CrossRef]
- Pestka, S.; Baron, S. Definition and Classification of the Interferons. Methods Enzymol. 1981, 78, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Handa, P.; Vemulakonda, A.; Kowdley, K.V.; Uribe, M.; Méndez-Sánchez, N. Mitochondrial DNA from hepatocytes as a ligand for TLR9: Drivers of nonalcoholic steatohepatitis? World J. Gastroenterol. 2016, 22, 6965. [Google Scholar] [CrossRef]
- Ganz, M.; Szabo, G. Immune and inflammatory pathways in NASH. Hepatol. Int. 2013, 7, S771–S781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arrese, M.; Cabrera, D.; Kalergis, A.M.; Feldstein, A.E. Innate Immunity and Inflammation in NAFLD/NASH. Dig. Dis. Sci. 2016, 61, 1294–1303. [Google Scholar] [CrossRef] [Green Version]
- Platanias, L.C. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev. Immunol. 2005, 5, 375–386. [Google Scholar] [CrossRef]
- Der, S.D.; Zhou, A.; Williams, B.R.G.; Silverman, R.H. Identification of genes differentially regulated by interferon α, β, or γ using oligonucleotide arrays. Proc. Natl. Acad. Sci. USA 1998, 95, 15623–15628. [Google Scholar] [CrossRef] [Green Version]
- Pestka, S.; Krause, C.D.; Walter, M.R. Interferons, interferon-like cytokines, and their receptors. Immunol. Rev. 2004, 202, 8–32. [Google Scholar] [CrossRef] [PubMed]
- Wack, A.; Terczyńska-Dyla, E.; Hartmann, R. Guarding the frontiers: The biology of type III interferons. Nat. Immunol. 2015, 16, 802–809. [Google Scholar] [CrossRef]
- Qiao, J.T.; Cui, C.; Qing, L.; Wang, L.S.; He, T.Y.; Yan, F.; Liu, F.Q.; Shen, Y.H.; Hou, X.G.; Chen, L. Activation of the STING-IRF3 pathway promotes hepatocyte inflammation, apoptosis and induces metabolic disorders in nonalcoholic fatty liver disease. Metabolism 2018, 81, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Li, H.; Ma, L.; Zhou, J.; Guo, X.; Woo, S.L.; Pei, Y.; Knight, L.R.; Deveau, M.; Chen, Y.; et al. Expression of STING Is Increased in Liver Tissues From Patients With NAFLD and Promotes Macrophage-Mediated Hepatic Inflammation and Fibrosis in Mice. Gastroenterology 2018, 155, 1971–1984.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.N.; Su, Y. Remdesivir attenuates high fat diet (HFD)-induced NAFLD by regulating hepatocyte dyslipidemia and inflammation via the suppression of STING. Biochem. Biophys. Res. Commun. 2020, 526, 381–388. [Google Scholar] [CrossRef]
- Ishikawa, H.; Barber, G.N. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 2008, 455, 674–678. [Google Scholar] [CrossRef]
- Gaidt, M.M.; Ebert, T.S.; Chauhan, D.; Ramshorn, K.; Pinci, F.; Zuber, S.; O’Duill, F.; Schmid-Burgk, J.L.; Hoss, F.; Buhmann, R.; et al. The DNA Inflammasome in Human Myeloid Cells Is Initiated by a STING-Cell Death Program Upstream of NLRP3. Cell 2017, 171, 1110–1124.e18. [Google Scholar] [CrossRef]
- Abe, T.; Barber, G.N. Cytosolic-DNA-Mediated, STING-Dependent Proinflammatory Gene Induction Necessitates Canonical NF- B Activation through TBK1. J. Virol. 2014, 88, 5328–5341. [Google Scholar] [CrossRef] [Green Version]
- Kumari, M.; Wang, X.; Lantier, L.; Lyubetskaya, A.; Eguchi, J.; Kang, S.; Tenen, D.; Roh, H.C.; Kong, X.; Kazak, L.; et al. IRF3 promotes adipose inflammation and insulin resistance and represses browning. J. Clin. Investig. 2016, 126, 2839–2854. [Google Scholar] [CrossRef] [Green Version]
- Honda, K.; Takaoka, A.; Taniguchi, T. Type I Inteferon Gene Induction by the Interferon Regulatory Factor Family of Transcription Factors. Immunity 2006, 25, 349–360. [Google Scholar] [CrossRef] [Green Version]
- Ghazarian, M.; Revelo, X.S.; Nøhr, M.K.; Luck, H.; Zeng, K.; Lei, H.; Tsai, S.; Schroer, S.A.; Park, Y.J.; Chng, M.H.Y.; et al. Type I interferon responses drive intrahepatic T cells to promote metabolic syndrome. Sci. Immunol. 2017, 2, eaai7616. [Google Scholar] [CrossRef] [Green Version]
- Mitsumoto, K.; Watanabe, R.; Nakao, K.; Yonenaka, H.; Hashimoto, T.; Kato, N.; Kumrungsee, T.; Yanaka, N. Time-course microarrays reveal early activation of the immune transcriptome in a choline-deficient mouse model of liver injury. Life Sci. 2017, 184, 103–111. [Google Scholar] [CrossRef]
- Wieser, V.; Adolph, T.E.; Grander, C.; Grabherr, F.; Enrich, B.; Moser, P.; Moschen, A.R.; Kaser, S.; Tilg, H. Adipose type i interferon signalling protects against metabolic dysfunction. Gut 2018, 67, 157–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.-A.; Zhang, R.; Zhang, S.; Deng, S.; Jiang, D.; Zhong, J.; Yang, L.; Wang, T.; Hong, S.; Guo, S.; et al. Interferon regulatory factor 7 deficiency prevents diet-induced obesity and insulin resistance. Am. J. Physiol. Metab. 2013, 305, E485–E495. [Google Scholar] [CrossRef] [Green Version]
- Hao, J.; Zhang, Y.; Lv, X.; Xu, N.; Liu, Q.; Zhao, S.; Feng, X.; Xing, L.; Kang, P.; Li, G.; et al. IFN-induces lipogenesis in mouse mesangial cells via the JAK2/STAT1 pathway. Am. J. Physiol. Cell Physiol. 2013, 304, 760–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grunfeld, C.; Soued, M.; Adi, S.; Moser, A.H.; Dinarello, C.A.; Feingold, K.R. Evidence for Two Classes of Cytokines That Stimulate Hepatic Lipogenesis: Relationships among Tumor Necrosis Factor, Interleukin-1 and Interferon-Alpha*. Endocrinology 1990, 127, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Tarantino, G.; Costantini, S.; Citro, V.; Conforti, P.; Capone, F.; Sorice, A.; Capone, D. Interferon-alpha 2 but not Interferon-gamma serum levels are associated with intramuscular fat in obese patients with nonalcoholic fatty liver disease 11 Medical and Health Sciences 1103 Clinical Sciences. J. Transl. Med. 2019, 17, 8. [Google Scholar] [CrossRef]
- Feingold, K.R.; Soued, M.; Serio, M.K.; Moser, A.H.; Dinarello, C.A.; Grunfeld, C. Multiple cytokines stimulate hepatic lipid synthesis in vivo. Endocrinology 1989, 125, 267–274. [Google Scholar] [CrossRef]
- Wada, T.; Hoshino, M.; Kimura, Y.; Ojima, M.; Nakano, T.; Koya, D.; Tsuneki, H.; Sasaoka, T. Both type I and II IFN induce insulin resistance by inducing different isoforms of SOCS expression in 3T3-L1 adipocytes. Am. J. Physiol. Metab. 2011, 300, E1112–E1123. [Google Scholar] [CrossRef] [Green Version]
- Ueki, K.; Kondo, T.; Kahn, C.R. Suppressor of Cytokine Signaling 1 (SOCS-1) and SOCS-3 Cause Insulin Resistance through Inhibition of Tyrosine Phosphorylation of Insulin Receptor Substrate Proteins by Discrete Mechanisms. Mol. Cell. Biol. 2004, 24, 5434–5446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ueki, K.; Kondo, T.; Tseng, Y.H.; Kahn, C.R. Central role of suppressors of cytokine signaling proteins in hepatic steatosis, insulin resistance, and the metabolic syndrome in the mouse. Proc. Natl. Acad. Sci. USA 2004, 101, 10422–10427. [Google Scholar] [CrossRef] [Green Version]
- Hardardottir, I.; Doerrler, W.; Feingold, K.R.; Grunfeld, C. Cytokines stimulate lipolysis and decrease lipoprotein lipase activity in cultured fat cells by a prostaglandin independent mechanism. Biochem. Biophys. Res. Commun. 1992, 186, 237–243. [Google Scholar] [CrossRef]
- Doerrler, W.; Feingold, K.R.; Grunfeld, C. Cytokines induce catabolic effects in cultured adipocytes by multiple mechanisms. Cytokine 1994, 6, 478–484. [Google Scholar] [CrossRef]
- Truong, N.T.T.; Lydic, T.A.; Bazil, J.N.; Suryadevara, A.; Olson, L.K. Regulation of lipid metabolism in pancreatic beta cells by interferon gamma: A link to anti-viral function. Cytokine 2020, 133. [Google Scholar] [CrossRef]
- Luo, X.-Y.; Takahara, T.; Kawai, K.; Fujino, M.; Sugiyama, T.; Tsuneyama, K.; Tsukada, K.; Nakae, S.; Zhong, L.; Li, X.-K. IFN-γ deficiency attenuates hepatic inflammation and fibrosis in a steatohepatitis model induced by a methionine- and choline-deficient high-fat diet. Am. J. Physiol. Liver Physiol. 2013, 305, G891–G899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu Shi, S.; García Martin, R.; Duncan, R.E.; Choi, D.; Lu, S.-Y.; Schroer, S.A.; Cai, E.P.; Luk, C.T.; Hopperton, K.E.; Domenichiello, A.F.; et al. Hepatocyte-specific Deletion of Janus Kinase 2 (JAK2) Protects against Diet-induced Steatohepatitis and Glucose Intolerance. J. Biol. Chem. 2012, 287, 10277–10288. [Google Scholar] [CrossRef] [Green Version]
- Sos, B.C.; Harris, C.; Nordstrom, S.M.; Tran, J.L.; Balázs, M.; Caplazi, P.; Febbraio, M.; Applegate, M.A.B.; Wagner, K.U.; Weiss, E.J. Abrogation of growth hormone secretion rescues fatty liver in mice with hepatocytespecific deletion of JAK2. J. Clin. Invest. 2011, 121, 1412–1423. [Google Scholar] [CrossRef]
- Themanns, M.; Mueller, K.M.; Kessler, S.M.; Golob-Schwarzl, N.; Mohr, T.; Kaltenecker, D.; Bourgeais, J.; Paier-Pourani, J.; Friedbichler, K.; Schneller, D.; et al. Hepatic deletion of Janus Kinase 2 counteracts oxidative stress in mice. Sci. Rep. 2016, 6, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Yu, C.Y.; Deng, W.M. The role of pro-inflammatory cytokines in lipid metabolism of metabolic diseases. Int. Rev. Immunol. 2019, 38, 249–266. [Google Scholar] [CrossRef]
- Niederreiter, L.; Tilg, H. Cytokines and fatty liver diseases. Liver Res. 2018, 2, 14–20. [Google Scholar] [CrossRef]
- Tilg, H.; Diehl, A.M. Cytokines in Alcoholic and Nonalcoholic Steatohepatitis. N. Engl. J. Med. 2000, 343, 1467–1476. [Google Scholar] [CrossRef] [PubMed]
- Negrin, K.A.; Flach, R.J.R.; DiStefano, M.T.; Matevossian, A.; Friedline, R.H.; Jung, D.; Kim, J.K.; Czech, M.P. IL-1 Signaling in obesity-induced hepatic lipogenesis and steatosis. PLoS ONE 2014, 9, e107265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stienstra, R.; Saudale, F.; Duval, C.; Keshtkar, S.; Groener, J.E.M.; Van Rooijen, N.; Staels, B.; Kersten, S.; Müller, M. Kupffer cells promote hepatic steatosis via interleukin-1β-dependent suppression of peroxisome proliferator-activated receptor α activity. Hepatology 2010, 51, 511–522. [Google Scholar] [CrossRef]
- Fève, B.; Bastard, J.P. The role of interleukins in insulin resistance and type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2009, 5, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Cobbina, E.; Akhlaghi, F. Non-alcoholic fatty liver disease (NAFLD)–pathogenesis, classification, and effect on drug metabolizing enzymes and transporters. Drug Metab. Rev. 2017, 49, 197–211. [Google Scholar] [CrossRef]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef]
- Brown, M.S.; Goldstein, J.L. Selective versus Total Insulin Resistance: A Pathogenic Paradox. Cell Metab. 2008, 7, 95–96. [Google Scholar] [CrossRef] [Green Version]
- Hectors, T.L.M.; Vanparys, C.; Van Gaal, L.F.; Jorens, P.G.; Covaci, A.; Blust, R. Insulin resistance and environmental pollutants: Experimental evidence and future perspectives. Environ. Health Perspect. 2013, 121, 1273–1281. [Google Scholar] [CrossRef] [Green Version]
- Moon, Y.A.; Liang, G.; Xie, X.; Frank-Kamenetsky, M.; Fitzgerald, K.; Koteliansky, V.; Brown, M.S.; Goldstein, J.L.; Horton, J.D. The Scap/SREBP pathway is essential for developing diabetic fatty liver and carbohydrate-induced hypertriglyceridemia in animals. Cell Metab. 2012, 15, 240–246. [Google Scholar] [CrossRef] [Green Version]
- Iizuka, K.; Bruick, R.K.; Liang, G.; Horton, J.D.; Uyeda, K. Deficiency of carbohydrate response element-binding protein (ChREBP) reduces lipogenesis as well as glycolysis. Proc. Natl. Acad. Sci. USA 2004, 101, 7281–7286. [Google Scholar] [CrossRef] [Green Version]
- Kaiser, J.P.; Lipscomb, J.C.; Wesselkamper, S.C. Putative mechanisms of environmental chemical-induced steatosis. Int. J. Toxicol. 2012, 31, 551–563. [Google Scholar] [CrossRef] [Green Version]
- Hotamisligil, G.S.; Murray, D.L.; Choy, L.N.; Spiegelman, B.M. Tumor necrosis factor alpha inhibits signaling from the insulin receptor. Proc. Natl. Acad. Sci. USA 1994, 91, 4854–4858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hotamisligil, G.S.; Peraldi, P.; Budavari, A.; Ellis, R.; White, M.F.; Spiegelman, B.M. IRS-1-mediated inhibition of insulin receptor tyrosine kinase activity in TNF-α- and obesity-induced insulin resistance. Science 1996, 271, 665–668. [Google Scholar] [CrossRef]
- Tang, Y.; Bian, Z.; Zhao, L.; Liu, Y.; Liang, S.; Wang, Q.; Han, X.; Peng, Y.; Chen, X.; Shen, L.; et al. Interleukin-17 exacerbates hepatic steatosis and inflammation in non-alcoholic fatty liver disease. Clin. Exp. Immunol. 2011, 166, 281–290. [Google Scholar] [CrossRef]
- Herbstman, J.B.; Sjödin, A.; Apelberg, B.J.; Witter, F.R.; Haiden, R.U.; Patterson, D.G.; Panny, S.R.; Needham, L.L.; Goldman, L.R. Birth delivery mode modifies the associations between prenatal polychlorinated biphenyl (PCB) and polybrominated diphenyl ether (PBDE) and neonatal thyroid hormone levels. Environ. Health Perspect. 2008, 116, 1376–1382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Julander, A.; Karlsson, M.; Hagström, K.; Ohlson, C.G.; Engwall, M.; Bryngelsson, I.L.; Westberg, H.; van Bavel, B. Polybrominated diphenyl ethers—Plasma levels and thyroid status of workers at an electronic recycling facility. Int. Arch. Occup. Environ. Health 2005, 78, 584–592. [Google Scholar] [CrossRef] [PubMed]
- Turyk, M.E.; Persky, V.W.; Imm, P.; Knobeloch, L.; Chatterton, R.; Anderson, H.A. Hormone disruption by PBDEs in adult male sport fish consumers. Environ. Health Perspect. 2008, 116, 1635–1641. [Google Scholar] [CrossRef]
- Branchi, I.; Capone, F.; Alleva, E.; Costa, L.G. Polybrominated diphenyl ethers: Neurobehavioral effects following developmental exposure. Neurotoxicology 2003, 24, 449–462. [Google Scholar] [CrossRef]
- Ping He; Aiguo Wang; Qiang Niu; Lijuan Guo; Tao Xia; Xuemin Chen Toxic effect of PBDE-47 on thyroid development, learning, and memory, and the interaction between PBDE-47 and PCB153 that enhances toxicity in rats. Toxicol. Ind. Health 2011, 27, 279–288. [CrossRef]
- Szabo, D.T.; Richardson, V.M.; Ross, D.G.; Diliberto, J.J.; Kodavanti, P.R.S.; Birnbaum, L.S. Effects of perinatal PBDE exposure on hepatic phase I, phase II, phase III, and deiodinase 1 gene expression Involved in thyroid hormone metabolism in male rat pups. Toxicol. Sci. 2009, 107, 27–39. [Google Scholar] [CrossRef] [Green Version]
- Vuong, A.M.; Braun, J.M.; Webster, G.M.; Thomas Zoeller, R.; Hoofnagle, A.N.; Sjödin, A.; Yolton, K.; Lanphear, B.P.; Chen, A. Polybrominated diphenyl ether (PBDE) exposures and thyroid hormones in children at age 3 years. Environ. Int. 2018, 117, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Sinha, R.A.; Singh, B.K.; Yen, P.M. Direct effects of thyroid hormones on hepatic lipid metabolism. Nat. Rev. Endocrinol. 2018, 14, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.-Y.; Li, Y.-X.; Zhang, T.; Cai, D.; Ruan, J.-J.; Huang, M.-Z.; Wang, L.; Zhang, J.-Q.; Qiu, R.-L. Effect of E-waste Recycling on Urinary Metabolites of Organophosphate Flame Retardants and Plasticizers and Their Association with Oxidative Stress. Environ. Sci. Technol. 2017, 51, 2427–2437. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Jin, Y.; Wu, Y.; Liu, L.; Fu, Z. Exposure of male mice to two kinds of organophosphate flame retardants (OPFRs) induced oxidative stress and endocrine disruption. Environ. Toxicol. Pharmacol. 2015, 40, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Su, F.; Hong, P.P.; Zhang, Q.; Zhao, M. 1 H NMR-based metabolomic analysis of nine organophosphate flame retardants metabolic disturbance in Hep G2 cell line. Sci. Total Environ. 2019, 665, 162–170. [Google Scholar] [CrossRef] [PubMed]
- Papalou, O.; Kandaraki, E.A.; Papadakis, G.; Diamanti-Kandarakis, E. Endocrine disrupting chemicals: An occult mediator of metabolic disease. Front. Endocrinol. 2019, 10, 112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spahis, S.; Delvin, E.; Borys, J.M.; Levy, E. Oxidative Stress as a Critical Factor in Nonalcoholic Fatty Liver Disease Pathogenesis. Antioxid. Redox Signal. 2017, 26, 519–541. [Google Scholar] [CrossRef]
- Kang, H.; Moon, H.B.; Choi, K. Toxicological responses following short-term exposure through gavage feeding or water-borne exposure to Dechlorane Plus in zebrafish (Danio rerio). Chemosphere 2016, 146, 226–232. [Google Scholar] [CrossRef] [PubMed]
- Bruchajzer, E.; Frydrych, B.; Sporny, S.; Szymańska, J.A. The effect of short-term intoxication of rats with pentabromodiphenyl ether (in mixture mimic commercial products). Hum. Exp. Toxicol. 2011, 30, 363–378. [Google Scholar] [CrossRef]
- Bondy, G.S.; Lefebvre, D.E.; Aziz, S.; Cherry, W.; Coady, L.; MacLellan, E.; Armstrong, C.; Barker, M.; Cooke, G.; Gaertner, D.; et al. Toxicologic and immunologic effects of perinatal exposure to the brominated diphenyl ether (BDE) mixture DE-71 in the Sprague-Dawley rat. Environ. Toxicol. 2013, 28, 215–228. [Google Scholar] [CrossRef] [PubMed]
- Dunnick, J.K.; Brix, A.; Cunny, H.; Vallant, M.; Shockley, K.R. Characterization of polybrominated diphenyl ether toxicity in Wistar Han rats and use of liver microarray data for predicting disease susceptibilities. Toxicol. Pathol. 2012, 40, 93–106. [Google Scholar] [CrossRef] [Green Version]
- SUN, R.B.; SHANG, S.; ZHANG, W.; LIN, B.C.; WANG, Q.; SHI, Y.; XI, Z.G. Endocrine Disruption Activity of 30-day Dietary Exposure to Decabromodiphenyl Ethane in Balb/C Mouse. Biomed. Environ. Sci. 2018, 31, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Suvorov, A.; Takser, L. Global Gene Expression Analysis in the Livers of Rat Offspring Perinatally Exposed to Low Doses of 2,2′,4,4′-Tetrabromodiphenyl Ether. Environ. Health Perspect. 2010, 118, 97–102. [Google Scholar] [CrossRef] [Green Version]
- van der Ven, L.T.M.; van de Kuil, T.; Verhoef, A.; Leonards, P.E.G.; Slob, W.; Cantón, R.F.; Germer, S.; Hamers, T.; Visser, T.J.; Litens, S.; et al. A 28-day oral dose toxicity study enhanced to detect endocrine effects of a purified technical pentabromodiphenyl ether (pentaBDE) mixture in Wistar rats. Toxicology 2008, 245, 109–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, Z.; Zhang, Z.; Lu, D.; Ding, B.; Shu, L.; Zhang, Q.; Wang, C. Organophosphorus Flame Retardants Impair Intracellular Lipid Metabolic Function in Human Hepatocellular Cells. Chem. Res. Toxicol. 2019, 32, 1250–1258. [Google Scholar] [CrossRef]
- Adams, S.; Wiersielis, K.; Yasrebi, A.; Conde, K.; Armstrong, L.; Guo, G.L.; Roepke, T.A. Sex- and age-dependent effects of maternal organophosphate flame-retardant exposure on neonatal hypothalamic and hepatic gene expression. Reprod. Toxicol. 2020, 94, 65–74. [Google Scholar] [CrossRef]
- Krivoshiev, B.V.; Beemster, G.T.S.; Sprangers, K.; Cuypers, B.; Laukens, K.; Blust, R.; Husson, S.J. Transcriptome profiling of HepG2 cells exposed to the flame retardant 9,10-dihydro-9-oxa-10-phosphaphenanthrene 10-oxide (DOPO). Toxicol. Res. 2018, 7, 492–502. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Zhu, W.; Chen, L.; Yan, J.; Teng, M.; Zhou, Z. Neonatal triphenyl phosphate and its metabolite diphenyl phosphate exposure induce sex- and dose-dependent metabolic disruptions in adult mice. Environ. Pollut. 2018, 237, 10–17. [Google Scholar] [CrossRef]
- Dunnick, J.K.; Shockley, K.R.; Morgan, D.L.; Travlos, G.S.; Gerrish, K.; Ton, T.V.T.; Wilson, R.; Brar, S.S.; Brix, A.E.; Waidyanatha, S.; et al. Hepatic Transcriptomic Patterns in the Neonatal Rat After Pentabromodiphenyl Ether Exposure. Toxicol. Pathol. 2020, 48, 338–349. [Google Scholar] [CrossRef] [PubMed]
- Patisaul, H.B.; Roberts, S.C.; Mabrey, N.; Mccaffrey, K.A.; Gear, R.B.; Braun, J.; Belcher, S.M.; Stapleton, H.M. Accumulation and Endocrine Disrupting Effects of the Flame Retardant Mixture Firemaster® 550 in Rats: An Exploratory Assessment. J. Biochem. Mol. Toxicol. 2013, 27, 124–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, C.; Zhu, L.; Kang, Q.; Lee, H.K.; Li, D.; Chung, A.C.K.; Cai, Z. Chronic exposure to tetrabromodiphenyl ether (BDE-47) aggravates hepatic steatosis and liver fibrosis in diet-induced obese mice. J. Hazard. Mater. 2019, 378. [Google Scholar] [CrossRef]
- Sun, Y.; Wang, Y.; Liang, B.; Chen, T.; Zheng, D.; Zhao, X.; Jing, L.; Zhou, X.; Sun, Z.; Shi, Z. Hepatotoxicity of decabromodiphenyl ethane (DBDPE) and decabromodiphenyl ether (BDE-209) in 28-day exposed Sprague-Dawley rats. Sci. Total Environ. 2020, 705, 135783. [Google Scholar] [CrossRef] [PubMed]
- Saquib, Q.; Siddiqui, M.A.; Ahmed, J.; Al-Salim, A.; Ansari, S.M.; Faisal, M.; Al-Khedhairy, A.A.; Musarrat, J.; AlWathnani, H.A.; Alatar, A.A.; et al. Hazards of low dose flame-retardants (BDE-47 and BDE-32): Influence on transcriptome regulation and cell death in human liver cells. J. Hazard. Mater. 2016, 308, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Gu, Y.; Fan, X. Chlorinated phosphorus flame retardants exert oxidative damage to SMMC-7721 human hepatocarcinoma cells. Sci. Total Environ. 2020, 705, 135777. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Wang, P.; Du, Z.; Wang, G.; Gao, S. Oxidative stress, cell cycle arrest, DNA damage and apoptosis in adult zebrafish (Danio rerio) induced by tris(1,3-dichloro-2-propyl) phosphate. Aquat. Toxicol. 2018, 194, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.L.; Long, C.Y.; Wang, J.L.; Yu, M.; Chen, J.X. Involvement of oxidative stress in tri-ortho-cresyl phosphate-induced liver injury in male mice. Hum. Exp. Toxicol. 2016, 35, 1093–1101. [Google Scholar] [CrossRef]
- Pereira, L.C.; Cabral Miranda, L.F.C.; Franco-Bernardes, M.F.; Tasso, M.J.; Duarte, F.V.; Inácio Varela, A.T.; Rolo, A.P.; Marques Palmeira, C.M.; Dorta, D.J. Mitochondrial damage and apoptosis: Key features in BDE-153-induced hepatotoxicity. Chem. Biol. Interact. 2018, 291, 192–201. [Google Scholar] [CrossRef]
- Bruchajzer, E.; Frydrych, B.; Kilanowicz, A.; Sapota, A.; Szymańska, J.A. Selected oxidative stress parameters after single and repeated administration of octabromodiphenyl ether to rats. Int. J. Occup. Med. Environ. Health 2014, 27, 808–820. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Wang, L.; Ji, C.; Wu, H.; Zhao, J.; Tang, J. Toxicological effects of tris(2-chloropropyl) phosphate in human hepatic cells. Chemosphere 2017, 187, 88–96. [Google Scholar] [CrossRef]
- Dunnick, J.K.; Nyska, A. Characterization of liver toxicity in F344/N rats and B6C3F1 mice after exposure to a flame retardant containing lower molecular weight polybrominated diphenyl ethers. Exp. Toxicol. Pathol. 2009, 61, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Shao, J.; White, C.C.; Dabrowski, M.J.; Kavanagh, T.J.; Eckert, M.L.; Gallagher, E.P. The role of mitochondrial and oxidative injury in BDE 47 toxicity to human fetal liver hematopoietic stem cells. Toxicol. Sci. 2008, 101, 81–90. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Li, X.; Liu, J.; Zhou, G.; Yu, Y.; Jing, L.; Shi, Z.; Zhou, X.; Sun, Z. The effects of decabromodiphenyl ether on glycolipid metabolism and related signaling pathways in mice. Chemosphere 2019, 222, 849–855. [Google Scholar] [CrossRef]
- Khalil, A.; Cevik, S.E.; Hung, S.; Kolla, S.; Roy, M.A.; Suvorov, A. Developmental exposure to 2,2′,4,4′-Tetrabromodiphenyl ether permanently alters blood-liver balance of lipids in male mice. Front. Endocrinol. 2018, 9, 548. [Google Scholar] [CrossRef] [PubMed]
- Farmahin, R.; Gannon, A.M.; Gagné, R.; Rowan-Carroll, A.; Kuo, B.; Williams, A.; Curran, I.; Yauk, C.L. Hepatic transcriptional dose-response analysis of male and female Fischer rats exposed to hexabromocyclododecane. Food Chem. Toxicol. 2019, 133, 110262. [Google Scholar] [CrossRef] [PubMed]
- Farhat, A.; Buick, J.K.; Williams, A.; Yauk, C.L.; O’Brien, J.M.; Crump, D.; Williams, K.L.; Chiu, S.; Kennedy, S.W. Tris(1,3-dichloro-2-propyl) phosphate perturbs the expression of genes involved in immune response and lipid and steroid metabolism in chicken embryos. Toxicol. Appl. Pharmacol. 2014, 275, 104–112. [Google Scholar] [CrossRef]
- Du, Z.; Zhang, Y.; Wang, G.; Peng, J.; Wang, Z.; Gao, S. TPhP exposure disturbs carbohydrate metabolism, lipid metabolism, and the DNA damage repair system in zebrafish liver. Sci. Rep. 2016, 6, 21827. [Google Scholar] [CrossRef] [Green Version]
- Yanagisawa, R.; Koike, E.; Win-Shwe, T.T.; Yamamoto, M.; Takano, H. Impaired lipid and glucose homeostasis in hexabromocyclododecane- exposed mice fed a high-fat diet. Environ. Health Perspect. 2014, 122, 277–283. [Google Scholar] [CrossRef] [Green Version]
- Bruchajzer, E.; Frydrych, B.; Sporny, S.; Szymańska, J.A. Toxicity of penta- and decabromodiphenyl ethers after repeated administration to rats: A comparative study. Arch. Toxicol. 2010, 84, 287–299. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Yan, J.; Teng, M.; Yan, S.; Zhou, Z.; Zhu, W. In utero and lactational exposure to BDE-47 promotes obesity development in mouse offspring fed a high-fat diet: Impaired lipid metabolism and intestinal dysbiosis. Arch. Toxicol. 2018, 92, 1847–1860. [Google Scholar] [CrossRef]
- Bao, J.; Liu, Y.; Li, L.; Chen, M.; Liu, J.; Niu, Y.; Liu, J.; Liang, Y. Biological effects of new-generation dialkyl phosphinate flame retardants and their hydrolysates in BALB/C mice. Environ. Toxicol. 2017, 32, 1578–1586. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Liu, S.; Guo, X.; Zhang, Y.; Zhang, X.; Li, M.; Cheng, S. Responses of Mouse Liver to Dechlorane Plus Exposure by Integrative Transcriptomic and Metabonomic Studies. Environ. Sci. Technol. 2012, 46, 10758–10764. [Google Scholar] [CrossRef] [PubMed]
- Al-Salem, A.M.; Saquib, Q.; Al-Khedhairy, A.A.; Siddiqui, M.A.; Ahmad, J. Tris(2-chloroethyl) phosphate (tcep) elicits hepatotoxicity by activating human cancer pathway genes in hepg2 cells. Toxics 2020, 8, 109. [Google Scholar] [CrossRef]
- Li, Z.; Tang, X.; Zhu, L.; Qi, X.; Cao, G.; Lu, G. Cytotoxic Screening and Transcriptomics Reveal Insights into the Molecular Mechanisms of Trihexyl Phosphate-Triggered Hepatotoxicity. Environ. Sci. Technol. 2020, 54, 11464–11475. [Google Scholar] [CrossRef] [PubMed]
- Blanco, J.; Mulero, M.; Domingo, J.L.; Sanchez, D.J. Perinatal Exposure to BDE-99 Causes Decreased Protein Levels of Cyclin D1 via GSK3β Activation and Increased ROS Production in Rat Pup Livers. Toxicol. Sci. 2014, 137, 491–498. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Huang, X.; Li, Z.; Cao, G.; Zhu, X.; She, S.; Huang, T.; Lu, G. Evaluation of hepatotoxicity induced by 2-ethylhexyldiphenyl phosphate based on transcriptomics and its potential metabolism pathway in human hepatocytes. J. Hazard. Mater. 2021, 413, 125281. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, X.; Chen, C.; An, J.; Shang, Y.; Li, H.; Xia, H.; Yu, J.; Wang, C.; Liu, Y.; et al. Regulation of TBBPA-induced oxidative stress on mitochondrial apoptosis in L02cells through the Nrf2 signaling pathway. Chemosphere 2019, 226, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Mynster Kronborg, T.; Frohnert Hansen, J.; Nielsen, C.H.; Ramhøj, L.; Frederiksen, M.; Vorkamp, K.; Feldt-Rasmussen, U. Effects of the Commercial Flame Retardant Mixture DE-71 on Cytokine Production by Human Immune Cells. PLoS ONE 2016, 11, e0154621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, H.R.; Kamau, P.W.; Loch-Caruso, R. Involvement of reactive oxygen species in brominated diphenyl ether-47-induced inflammatory cytokine release from human extravillous trophoblasts in vitro. Toxicol. Appl. Pharmacol. 2014, 274, 283–292. [Google Scholar] [CrossRef] [Green Version]
- Yasmin, S.; Whalen, M. Flame retardants, hexabromocyclododecane (HCBD) and tetrabromobisphenol a (TBBPA), alter secretion of tumor necrosis factor alpha (TNFα) from human immune cells. Arch. Toxicol. 2018, 92, 1483–1494. [Google Scholar] [CrossRef]
- Almughamsi, H.; Whalen, M.M. Hexabromocyclododecane and tetrabromobisphenol A alter secretion of interferon gamma (IFN-γ) from human immune cells. Arch. Toxicol. 2016, 90, 1695–1707. [Google Scholar] [CrossRef] [Green Version]
- Koike, E.; Yanagisawa, R.; Takano, H. Brominated flame retardants, hexabromocyclododecane and tetrabromobisphenol A, affect proinflammatory protein expression in human bronchial epithelial cells via disruption of intracellular signaling. Toxicol. Vitr. 2016, 32, 212–219. [Google Scholar] [CrossRef]
- Verstraete, S.G.; Wojcicki, J.M.; Perito, E.R.; Rosenthal, P. Bisphenol a increases risk for presumed non-alcoholic fatty liver disease in Hispanic adolescents in NHANES 2003–2010. Environ. Health 2018, 17, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Yoo, E.R.; Li, A.A.; Cholankeril, G.; Tighe, S.P.; Kim, W.; Harrison, S.A.; Ahmed, A. Elevated urinary bisphenol A levels are associated with non-alcoholic fatty liver disease among adults in the United States. Liver Int. 2019, 39, 1335–1342. [Google Scholar] [CrossRef]
- Nakagawa, Y.; Suzuki, T.; Ishii, H.; Ogata, A. Biotransformation and cytotoxicity of a brominated flame retardant, tetrabromobisphenol A, and its analogues in rat hepatocytes. Xenobiotica 2007, 37, 693–708. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, W.; Li, A.; Song, M. Tetrachlorobisphenol A induced immunosuppression and uterine injury in mice. Ecotoxicol. Environ. Saf. 2021, 207, 111527. [Google Scholar] [CrossRef]
- Dunnick, J.K.; Morgan, D.L.; Elmore, S.A.; Gerrish, K.; Pandiri, A.; Ton, T.V.; Shockley, K.R.; Merrick, B.A. Tetrabromobisphenol A activates the hepatic interferon pathway in rats. Toxicol. Lett. 2017, 266, 32–41. [Google Scholar] [CrossRef] [Green Version]
- Chappell, V.A.; Janesick, A.; Blumberg, B.; Fenton, S.E. Tetrabromobisphenol-A Promotes Early Adipogenesis and Lipogenesis in 3T3-L1 Cells. Toxicol. Sci. 2018, 166, 332–344. [Google Scholar] [CrossRef]
- Wang, X.; Wei, L.; Zhu, J.; He, B.; Kong, B.; Jin, Y.; Fu, Z. Tetrabromoethylcyclohexane (TBECH) exhibits immunotoxicity in murine macrophages. Environ. Toxicol. 2020, 35, 159–166. [Google Scholar] [CrossRef]
- Jing, L.; Sun, Y.; Wang, Y.; Liang, B.; Chen, T.; Zheng, D.; Zhao, X.; Zhou, X.; Sun, Z.; Shi, Z. Cardiovascular toxicity of decabrominated diphenyl ethers (BDE-209) and decabromodiphenyl ethane (DBDPE) in rats. Chemosphere 2019, 223, 675–685. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liu, S.; Xu, H.; Zheng, H.; Bai, C.; Pan, W.; Zhou, H.; Liao, M.; Huang, C.; Dong, Q. Maternal exposure to low dose BDE209 and Pb mixture induced neurobehavioral anomalies in C57BL/6 male offspring. Toxicology 2019, 418, 70–80. [Google Scholar] [CrossRef]
- Zhi, H.; Wu, J.P.; Lu, L.M.; Li, Y.; Chen, X.Y.; Tao, J.; Mai, B.X. Decabromodiphenyl ether (BDE-209) enhances foam cell formation in human macrophages via augmenting Toll-like receptor 4-dependent lipid uptake. Food Chem. Toxicol. 2018, 121, 367–373. [Google Scholar] [CrossRef]
- Li, X.; Li, N.; Rao, K.; Huang, Q.; Ma, M. In Vitro Immunotoxicity of Organophosphate Flame Retardants in Human THP-1-Derived Macrophages. Environ. Sci. Technol. 2020, 54, 8900–8908. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.-F.; Zhang, Y.-Q.; Fan, S.-H.; Zhuang, J.; Zheng, Y.-L.; Lu, J.; Wu, D.-M.; Shan, Q.; Hu, B. Troxerutin protects against 2,2,4,4-tetrabromodiphenyl ether (BDE-47)-induced liver inflammation by attenuating oxidative stress-mediated NAD +-depletion. J. Hazard. Mater. 2015, 283, 98–109. [Google Scholar] [CrossRef] [PubMed]
- Pereira, L.C.; Souza, A.O.; Tasso, M.J.; Oliveira, A.M.C.; Duarte, F.V.; Palmeira, C.M.; Dorta, D.J. Exposure to decabromodiphenyl ether (BDE-209) produces mitochondrial dysfunction in rat liver and cell death. J. Toxicol. Environ. Health Part A Curr. Issues 2017, 80, 1129–1144. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Su, G.; Giesy, J.P.; Letcher, R.J.; Li, G.; Agrawal, I.; Li, J.; Yu, L.; Wang, J.; Gong, Z. Acute exposure to tris(1,3-dichloro-2-propyl) phosphate (TDCIPP) causes hepatic inflammation and leads to hepatotoxicity in zebrafish. Sci. Rep. 2016, 6, 19045. [Google Scholar] [CrossRef] [Green Version]
- Rau, M.; Schilling, A.-K.; Meertens, J.; Hering, I.; Weiss, J.; Jurowich, C.; Kudlich, T.; Hermanns, H.M.; Bantel, H.; Beyersdorf, N.; et al. Progression from Nonalcoholic Fatty Liver to Nonalcoholic Steatohepatitis Is Marked by a Higher Frequency of Th17 Cells in the Liver and an Increased Th17/Resting Regulatory T Cell Ratio in Peripheral Blood and in the Liver. J. Immunol. 2016, 196, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Ferreyra Solari, N.E.; Inzaugarat, M.E.; Baz, P.; De Matteo, E.; Lezama, C.; Galoppo, M.; Galoppo, C.; Cherñavsky, A.C. The role of innate cells is coupled to a Th1-polarized immune response in pediatric nonalcoholic steatohepatitis. J. Clin. Immunol. 2012, 32, 611–621. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, W.; Shimizu, T.; Hino, A.; Kurokawa, M. Effects of decabrominated diphenyl ether (DBDE) on developmental immunotoxicity in offspring mice. Environ. Toxicol. Pharmacol. 2008, 26, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Chen, Q.; Yi, J.; Lan, X.; Lu, K.; Du, X.; Guo, Z.; Guo, Y.; Geng, M.; Li, D.; et al. IFN-γ contributes to the hepatic inflammation in HFD-induced nonalcoholic steatohepatitis by STAT1β/TLR2 signaling pathway. Mol. Immunol. 2021, 134, 118–128. [Google Scholar] [CrossRef]
- Knight, B.; Lim, R.; Yeoh, G.C.; Olynyk, J.K. Interferon-γ exacerbates liver damage, the hepatic progenitor cell response and fibrosis in a mouse model of chronic liver injury. J. Hepatol. 2007, 47, 826–833. [Google Scholar] [CrossRef]
- Bhattacharya, A.; Hegazy, A.N.; Deigendesch, N.; Kosack, L.; Cupovic, J.; Kandasamy, R.K.; Hildebrandt, A.; Merkler, D.; Kühl, A.A.; Vilagos, B.; et al. Superoxide Dismutase 1 Protects Hepatocytes from Type I Interferon-Driven Oxidative Damage. Immunity 2015, 43, 974–986. [Google Scholar] [CrossRef]
- Roh, Y.S.; Kim, J.W.; Park, S.; Shon, C.; Kim, S.; Eo, S.K.; Kwon, J.K.; Lim, C.W.; Kim, B. Toll-Like Receptor-7 Signaling Promotes Nonalcoholic Steatohepatitis by Inhibiting Regulatory T Cells in Mice. Am. J. Pathol. 2018, 188, 2574–2588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, T.; Fujii, M.; Sandel, J.; Shibazaki, Y.; Wakamatsu, K.; Mark, M.; Yoneyama, H. Linagliptin alleviates hepatic steatosis and inflammation in a mouse model of non-alcoholic steatohepatitis. Med. Mol. Morphol. 2014, 47, 137–149. [Google Scholar] [CrossRef]
- Afrin, R.; Arumugam, S.; Rahman, A.; Wahed, M.I.I.; Karuppagounder, V.; Harima, M.; Suzuki, H.; Miyashita, S.; Suzuki, K.; Yoneyama, H.; et al. Curcumin ameliorates liver damage and progression of NASH in NASH-HCC mouse model possibly by modulating HMGB1-NF-κB translocation. Int. Immunopharmacol. 2017, 44, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Sharifnia, T.; Antoun, J.; Verriere, T.G.C.; Suarez, G.; Wattacheril, J.; Wilson, K.T.; Peek, R.M.; Abumrad, N.N.; Flynn, C.R. Hepatic TLR4 signaling in obese NAFLD. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 309, G270–G278. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Wang, X.; Lan, X.; Li, Y.; Liu, L.; Yi, J.; Li, J.; Sun, Q.; Wang, Y.; Li, H.; et al. Down-regulation of miR-144 elicits proinflammatory cytokine production by targeting toll-like receptor 2 in nonalcoholic steatohepatitis of high-fat-diet-induced metabolic syndrome E3 rats. Mol. Cell. Endocrinol. 2015, 402, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Mills, E.L.; Kelly, B.; O’Neill, L.A.J. Mitochondria are the powerhouses of immunity. Nat. Immunol. 2017, 18, 488–498. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, S.E.; Sena, L.A.; Chandel, N.S. Mitochondria in the regulation of innate and adaptive immunity. Immunity 2015, 42, 406–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riley, J.S.; Tait, S.W. Mitochondrial DNA in inflammation and immunity. EMBO Rep. 2020, 21, e49799. [Google Scholar] [CrossRef]
- Walker, M.A.; Volpi, S.; Sims, K.B.; Walter, J.E.; Traggiai, E. Powering the immune system: Mitochondria in immune function and deficiency. J. Immunol. Res. 2014, 2014, 164309. [Google Scholar] [CrossRef]
- Dutta, S.; Das, N.; Mukherjee, P. Picking up a Fight: Fine Tuning Mitochondrial Innate Immune Defenses Against RNA Viruses. Front. Microbiol. 2020, 11, 1990. [Google Scholar] [CrossRef] [PubMed]
- Backer, J.M.; Weinstein, I.B. Interaction of Benzo(a)pyrene and Its Dihydrodiol-Epoxide Derivative with Nuclear and Mitochondrial DNA in C3H10T½ Cell Cultures. Cancer Res. 1982, 42, 2764–2769. [Google Scholar] [PubMed]
- Zolkipli-Cunningham, Z.; Falk, M.J. Clinical effects of chemical exposures on mitochondrial function. Toxicology 2017, 391, 90–99. [Google Scholar] [CrossRef]
- Yuan, S.; Zhu, K.; Ma, M.; Zhu, X.; Rao, K.; Wang, Z. In vitro oxidative stress, mitochondrial impairment and G1 phase cell cycle arrest induced by alkyl-phosphorus-containing flame retardants. Chemosphere 2020, 248, 126026. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Li, N.; Yuan, S.; Ji, X.; Ma, M.; Rao, K.; Wang, Z. Aryl- and alkyl-phosphorus-containing flame retardants induced mitochondrial impairment and cell death in Chinese hamster ovary (CHO-k1) cells. Environ. Pollut. 2017, 230, 775–786. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.H.; Li, X.H.; Xu, Y.; Xu, Y.; Sun, S.C. Exposure to PBDE47 affects mouse oocyte quality via mitochondria dysfunction-induced oxidative stress and apoptosis. Ecotoxicol. Environ. Saf. 2020, 198, 110662. [Google Scholar] [CrossRef]
- Le, Y.; Shen, H.; Yang, Z.; Lu, D.; Wang, C. Comprehensive analysis of organophosphorus flame retardant-induced mitochondrial abnormalities: Potential role in lipid accumulation. Environ. Pollut. 2021, 274, 116541. [Google Scholar] [CrossRef]
- Pessayre, D.; Fromenty, B. NASH: A mitochondrial disease. J. Hepatol. 2005, 42, 928–940. [Google Scholar] [CrossRef]
- Mao, K.; Ji, F.; Breen, P.; Sewell, A.; Han, M.; Sadreyev, R.; Correspondence, G.R. Mitochondrial Dysfunction in C. elegans Activates Mitochondrial Relocalization and Nuclear Hormone Receptor-Dependent Detoxification Genes. Cell Metab. 2019, 29, 1182–1191.e4. [Google Scholar] [CrossRef]
- Wei, Y.; Rector, R.S.; Thyfault, J.P.; Ibdah, J.A. Nonalcoholic fatty liver disease and mitochondrial dysfunction. World J. Gastroenterol. 2008, 14, 193–199. [Google Scholar] [CrossRef]
- Chen, Q.; Sun, L.; Chen, Z.J. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat. Immunol. 2016, 17, 1142–1149. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.; Liu, F. The cGAS-cGAMP-STING pathway: A molecular link between immunity and metabolism. Diabetes 2019, 68, 1099–1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shu, H.B.; Wang, Y.Y. Adding to the STING. Immunity 2014, 41, 871–873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Martinez, I.; Santoro, N.; Chen, Y.; Hoque, R.; Ouyang, X.; Caprio, S.; Shlomchik, M.J.; Coffman, R.L.; Candia, A.; Mehal, W.Z. Hepatocyte mitochondrial DNA drives nonalcoholic steatohepatitis by activation of TLR9. J. Clin. Investig. 2016, 126, 859–864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Y.; Liu, Y.; An, W.; Song, J.; Zhang, Y.; Zhao, X. STING-mediated inflammation in Kupffer cells contributes to progression of nonalcoholic steatohepatitis. J. Clin. Investig. 2019, 129, 546–555. [Google Scholar] [CrossRef] [PubMed]
- Henriksen, E.J.; Diamond-Stanic, M.K.; Marchionne, E.M. Oxidative stress and the etiology of insulin resistance and type 2 diabetes. Free Radic. Biol. Med. 2011, 51, 993–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dodson, R.E.; Van Den Eede, N.; Covaci, A.; Perovich, L.J.; Brody, J.G.; Rudel, R.A. Urinary biomonitoring of phosphate flame retardants: Levels in california adults and recommendations for future studies. Environ. Sci. Technol. 2014, 48, 13625–13633. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Ji, S.; Chai, L.; Yang, F.; Zhao, M.; Liu, W.; Schu, G.; Ji, L. Metabolic Mechanism of Aryl Phosphorus Flame Retardants by Cytochromes P450: A Combined Experimental and Computational Study on Triphenyl Phosphate. Environ. Sci. Technol. 2018, 52, 14411–14421. [Google Scholar] [CrossRef]
- Zota, A.R.; Mitro, S.D.; Robinson, J.F.; Hamilton, E.G.; Park, J.S.; Parry, E.; Zoeller, R.T.; Woodruff, T.J. Polybrominated diphenyl ethers (PBDEs) and hydroxylated PBDE metabolites (OH-PBDEs) in maternal and fetal tissues, and associations with fetal cytochrome P450 gene expression. Environ. Int. 2018, 112, 269–278. [Google Scholar] [CrossRef]
- Schattenberg, J.M.; Czaja, M.J. Regulation of the effects of CYP2E1-induced oxidative stress by JNK signaling. Redox Biol. 2014, 3, 7–15. [Google Scholar] [CrossRef] [Green Version]
- Aubert, J.; Begriche, K.; Knockaert, L.; Robin, M.A.; Fromenty, B. Increased expression of cytochrome P450 2E1 in nonalcoholic fatty liver disease: Mechanisms and pathophysiological role. Clin. Res. Hepatol. Gastroenterol. 2011, 35, 630–637. [Google Scholar] [CrossRef] [PubMed]
- Fery, Y.; Buschauer, I.; Salzig, C.; Lang, P.; Schrenk, D. Technical pentabromodiphenyl ether and hexabromocyclododecane as activators of the pregnane-X-receptor (PXR). Toxicology 2009, 264, 45–51. [Google Scholar] [CrossRef]
- Pacyniak, E.K.; Cheng, X.; Cunningham, M.L.; Crofton, K.; Klaassen, C.D.; Guo, G.L. The flame retardants, polybrominated diphenyl ethers, are pregnane X receptor activators. Toxicol. Sci. 2007, 97, 94–102. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.K.; Pak, Y.K. Persistent Organic Pollutants, Mitochondrial Dysfunction, and Metabolic Syndrome. In Mitochondrial Dysfunction Caused by Drugs and Environmental Toxicants; John Wiley & Sons Inc.: Hoboken, NJ, USA, 2018; Volume 2, pp. 691–707. [Google Scholar]
- Zhou, P.K.; Huang, R.X. Targeting of the respiratory chain by toxicants: Beyond the toxicities to mitochondrial morphology. Toxicol. Res. Viewp. Toxicol. Res. 2018, 7, 1008–1011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, J.N.; Leung, M.C.K.; Rooney, J.P.; Sendoel, A.; Hengartner, M.O.; Kisby, G.E.; Bess, A.S. Mitochondria as a Target of environmental Toxicants. Toxicol. Sci. 2013, 134, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Waxman, D.J. P450 gene induction by structurally diverse xenochemicals: Central role of nuclear receptors CAR, PXR, and PPAR. Arch. Biochem. Biophys. 1999, 369, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, C.; Hossain, M.; Solanki, J.; Najm, I.M.; Marchi, N.; Janigro, D. Overexpression of pregnane X and glucocorticoid receptors and the regulation of cytochrome P450 in human epileptic brain endothelial cells. Epilepsia 2017, 58, 576–585. [Google Scholar] [CrossRef] [Green Version]
- Guo, Z.; Li, M.; Han, B.; Qi, X. Association of non-alcoholic fatty liver disease with thyroid function: A systematic review and meta-analysis. Dig. Liver Dis. 2018, 50, 1153–1162. [Google Scholar] [CrossRef]
- Tanase, D.M.; Gosav, E.M.; Neculae, E.; Costea, C.F.; Ciocoiu, M.; Hurjui, L.L.; Tarniceriu, C.C.; Floria, M. Hypothyroidism-induced nonalcoholic fatty liver disease (Hin): Mechanisms and emerging therapeutic options. Int. J. Mol. Sci. 2020, 21, 5927. [Google Scholar] [CrossRef]
- Meerts, I.A.T.M.; Van Zanden, J.J.; Luijks, E.A.C.; Van Leeuwen-Bol, I.; Marsh, G.; Jakobsson, E.; Bergman, Å.; Brouwer, A. Potent competitive interactions of some brominated flame retardants and related compounds with human transthyretin in Vitro. Toxicol. Sci. 2000, 56, 95–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Ji, C.; Yin, X.; Yan, L.; Lu, M.; Zhao, M. Thyroid hormone-disrupting activity and ecological risk assessment of phosphorus-containing flame retardants by in vitro, in vivo and in silico approaches. Environ. Pollut. 2016, 210, 27–33. [Google Scholar] [CrossRef]
- Yan, F.; Wang, Q.; Lu, M.; Chen, W.; Song, Y.; Jing, F.; Guan, Y.; Wang, L.; Lin, Y.; Bo, T.; et al. Thyrotropin increases hepatic triglyceride content through upregulation of SREBP-1c activity. J. Hepatol. 2014, 61, 1358–1364. [Google Scholar] [CrossRef]
- Song, Y.; Xu, C.; Shao, S.; Liu, J.; Xing, W.; Xu, J.; Qin, C.; Li, C.; Hu, B.; Yi, S.; et al. Thyroid-stimulating hormone regulates hepatic bile acid homeostasis via SREBP-2/HNF-4α/CYP7A1 axis. J. Hepatol. 2015, 62, 1171–1179. [Google Scholar] [CrossRef]
- Wang, H.; Chen, J.; Hollister, K.; Sowers, L.C.; Forman, B.M. Endogenous bile acids are ligands for the nuclear receptor FXR/BAR. Mol. Cell 1999, 3, 543–553. [Google Scholar] [CrossRef]
- Teodoro, J.S.; Rolo, A.P.; Palmeira, C.M. Hepatic FXR: Key regulator of whole-body energy metabolism. Trends Endocrinol. Metab. 2011, 22, 458–466. [Google Scholar] [CrossRef]
- Mullur, R.; Liu, Y.Y.; Brent, G.A. Thyroid hormone regulation of metabolism. Physiol. Rev. 2014, 94, 355–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.Y.; Brent, G.A. Thyroid hormone crosstalk with nuclear receptor signaling in metabolic regulation. Trends Endocrinol. Metab. 2010, 21, 166–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinken, M. Adverse Outcome Pathways and Drug-Induced Liver Injury Testing. Chem. Res. Toxicol. 2015, 28, 1391–1397. [Google Scholar] [CrossRef] [Green Version]
- Cave, M.C.; Clair, H.B.; Hardesty, J.E.; Falkner, K.C.; Feng, W.; Clark, B.J.; Sidey, J.; Shi, H.; Aqel, B.A.; McClain, C.J.; et al. Nuclear receptors and nonalcoholic fatty liver disease. Biochim. Biophys. Acta BBA Gene Regul. Mech. 2016, 1859, 1083–1099. [Google Scholar] [CrossRef] [Green Version]
- Semple, S. Assessing occupational and environmental exposure. Occup. Med. 2005, 55, 419–424. [Google Scholar] [CrossRef] [Green Version]
- Ingle, M.E.; Watkins, D.; Rosario, Z.; VélezVega, C.M.; Calafat, A.M.; Ospina, M.; Ferguson, K.K.; Cordero, J.F.; Alshawabkeh, A.; Meeker, J.D. An exploratory analysis of urinary organophosphate ester metabolites and oxidative stress among pregnant women in Puerto Rico. Sci. Total Environ. 2020, 703, 134798. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Li, M.; Pan, L.; Duan, Y.; Duan, X.; Li, Y.; Sun, H. Exposure to organophosphate ester flame retardants and plasticizers during pregnancy: Thyroid endocrine disruption and mediation role of oxidative stress. Environ. Int. 2021, 146, 106215. [Google Scholar] [CrossRef]
- Yuan, Y.; Meeker, J.D.; Ferguson, K.K. Serum polybrominated diphenyl ether (PBDE) concentrations in relation to biomarkers of oxidative stress and inflammation: The National Health and Nutrition Examination Survey 2003–2004. Sci. Total Environ. 2017, 575, 400–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsubara, K.; Nakamura, N.; Sanoh, S.; Ohta, S.; Kitamura, S.; Uramaru, N.; Miyagawa, S.; Iguchi, T.; Fujimoto, N. Altered expression of the Olr59, Ethe1, and Slc10a2 genes in the liver of F344 rats by neonatal thyroid hormone disruption. J. Appl. Toxicol. 2017, 37, 1030–1035. [Google Scholar] [CrossRef]
- Walley, S.N.; Krumm, E.A.; Yasrebi, A.; Kwiecinski, J.; Wright, V.; Baker, C.; Roepke, T.A. Maternal organophosphate flame-retardant exposure alters offspring energy and glucose homeostasis in a sexually dimorphic manner in mice. Appl. Toxicol. 2021, 41, 572–586. [Google Scholar] [CrossRef]
- Jurenka, J.S. Anti-inflammatory properties of curcumin, a major constituent of Curcuma longa: A review of preclinical and clinical research. Altern. Med. Rev. 2009, 14, 141–153. [Google Scholar] [PubMed]
- Wree, A.; Broderick, L.; Canbay, A.; Hoffman, H.M.; Feldstein, A.E. From NAFLD to NASH to cirrhosis-new insights into disease mechanisms. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 627–636. [Google Scholar]
- Joshi-Barve, S.; Kirpich, I.; Cave, M.C.; Marsano, L.S.; McClain, C.J. Alcoholic, Nonalcoholic, and Toxicant-Associated Steatohepatitis: Mechanistic Similarities and Differences. Cell. Mol. Gastroenterol. Hepatol. 2015, 1, 356–367. [Google Scholar] [PubMed] [Green Version]
- Treviño, L.S.; Katz, T.A. Endocrine disruptors and developmental origins of nonalcoholic fatty liver disease. Endocrinology 2018, 159, 20–31. [Google Scholar] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Flame Retardants | Inference and Summary | Test System | References |
|---|---|---|---|
| BDE-47 |
| C57BL/6J mice fed HFD | [152] |
| BDE-209, DBDPE |
| Sprague Dawley rats | [153] |
| BDE-47, BDE-32 |
| human hepatocellular carcinoma cells (HepG2 cells) | [154] |
| TDCPP, TCPP, TCEP |
| Human hepatocarcinoma cells (SMMC-7721 cells) | [155] |
| TDCPP |
| Adult zebrafish | [156] |
| TOCP |
| Mouse liver cancer cells (Hepa 1–6) and mice | [157] |
| BDE-153 |
| HepG2 cells | [158] |
| OctaBDE |
| Wistar rat | [159] |
| TCPP |
| Human fetal liver (L02 cells) | [160] |
| DE-71 |
| F344/N rats and B6C3F1 mice | [161] |
| BDE-47 |
| Human fetal liver–derived hematopoietic stem cells | [162] |
| BDE-209 |
| ICR mice | [163] |
| BDE-47 |
| Pregnant CD-1 mice | [164] |
| HBCD |
| Fischer rats | [165] |
| TDCIPP |
| Chicken embryos | [166] |
| TPhP |
| Adult zebrafish | [167] |
| HBCD |
| Male C57BL/6JJcl mice fed HFD | [168] |
| Penta & Deca BDPE |
| Female Wistar rats | [140,169] |
| BDE-47 |
| Pregnant ICR mice | [170] |
| AMEP, ADEP |
| BALB/c mice | [171] |
| Dechlorane Plus |
| Mice | [172] |
| TCEP |
| HepG2 cells | [173] |
| THP |
| L02 cells, mouse hepatocyte (AML12), and C57BL/6 mice | [174] |
| BDE-99 perinatal exposure |
| Sprague Dawley rats | [175] |
| EHDPP |
| L02 cells | [176] |
| TBBPA |
| L02cells | [177] |
| Contribution of the IFNs Signaling in NAFLD | FRs-Mediated IFN Signaling |
|---|---|
| Higher frequencies of IFN-γ+ and/or IL-4+ cells were detected among CD4+ T cells in peripheral blood of NASH patients [197]. Increased IFN-γ in the liver of pediatric (<15 years) NASH patients was observed [198]. IFN-γ induced liver inflammation, hepatocyte injury in the progression of NASH in mice [106]. | DE-71 enhanced IFN-γ in vitro in PBMCs [178]. Prenatal exposure to decabrominated diphenyl ether (DBDE) increased IFN-γ in the bronchoalveolar lavage fluids in offspring mice [199]. |
| IFN-γ contributed to hepatic inflammation in diet-induced NASH in rats, rat macrophage, and hepatocellular carcinoma cell lines [200]. | TBBPA increased IFN-γ in vitro in human PBMCs [180,181]. |
| IFN-γ-treatment activated hepatic stellate cells and increased hepatocyte apoptosis, hepatic inflammation, serum AST and fibrosis in mouse liver [201]. | TCBPA increased secretion of IFN-γ in the serum of mice [186]. |
| STING-IRF3 activation-induced inflammation, hepatocyte injury and apoptosis, and disturbed glucose and lipid metabolism in mice and in LO2 cells [84]. | BDE209 increased IFN-γ in the serum of male offspring [191]. |
| Type I and/or type II IFN signaling was associated with oxidative damage in mouse hepatocytes [202] as well as insulin resistance in mouse adipocytes culture [100]. | TPHP, TDCPP, TNBP, TOCP, TCEP, and TBOEP modulated JAK–STAT signaling in human leukemia monocytic culture [193]. |
| TNF-α and type I IFN production in Kupffer cells and dendritic cells induced hepatic cell death leading to NASH in mice and murine normal hepatocyte cell culture by TLR7-mediated signaling [203]. | TDCIPP upregulated TLR signaling, STAT1, IRF7, and induced inflammation and hepatotoxicity in zebrafish [196]. |
| Upregulation of IL-1β, TNF-α, and IFN-γ in the liver of mice in NASH [204,205]. | TBBPA upregulated hepatic IFN signaling and genes regulating fatty acid metabolism in rats [187]. |
| Increased TLR4 and IRF3 gene expression were observed in patients with NASH and hepatocytes exposed to palmitate and lipopolysaccharides [206]. TLR2, TNF-α, and IFN-γ are up-regulated in livers of rats in NASH [207]. | BDE-209 enhanced TLR4-dependent lipid uptake in vitro in human macrophages [192]. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Negi, C.K.; Khan, S.; Dirven, H.; Bajard, L.; Bláha, L. Flame Retardants-Mediated Interferon Signaling in the Pathogenesis of Nonalcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2021, 22, 4282. https://doi.org/10.3390/ijms22084282
Negi CK, Khan S, Dirven H, Bajard L, Bláha L. Flame Retardants-Mediated Interferon Signaling in the Pathogenesis of Nonalcoholic Fatty Liver Disease. International Journal of Molecular Sciences. 2021; 22(8):4282. https://doi.org/10.3390/ijms22084282
Chicago/Turabian StyleNegi, Chander K., Sabbir Khan, Hubert Dirven, Lola Bajard, and Luděk Bláha. 2021. "Flame Retardants-Mediated Interferon Signaling in the Pathogenesis of Nonalcoholic Fatty Liver Disease" International Journal of Molecular Sciences 22, no. 8: 4282. https://doi.org/10.3390/ijms22084282