
Chronic Administrations of Guanfacine on Mesocortical Catecholaminergic and Thalamocortical Glutamatergic Transmissions


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Effects of Acute and Chronic Administration of Therapeutic-Relevant Dose of Guanfacine on Extracellular Levels of Norepinephrine and Dopamine in the OFC and VTA (Study 1)
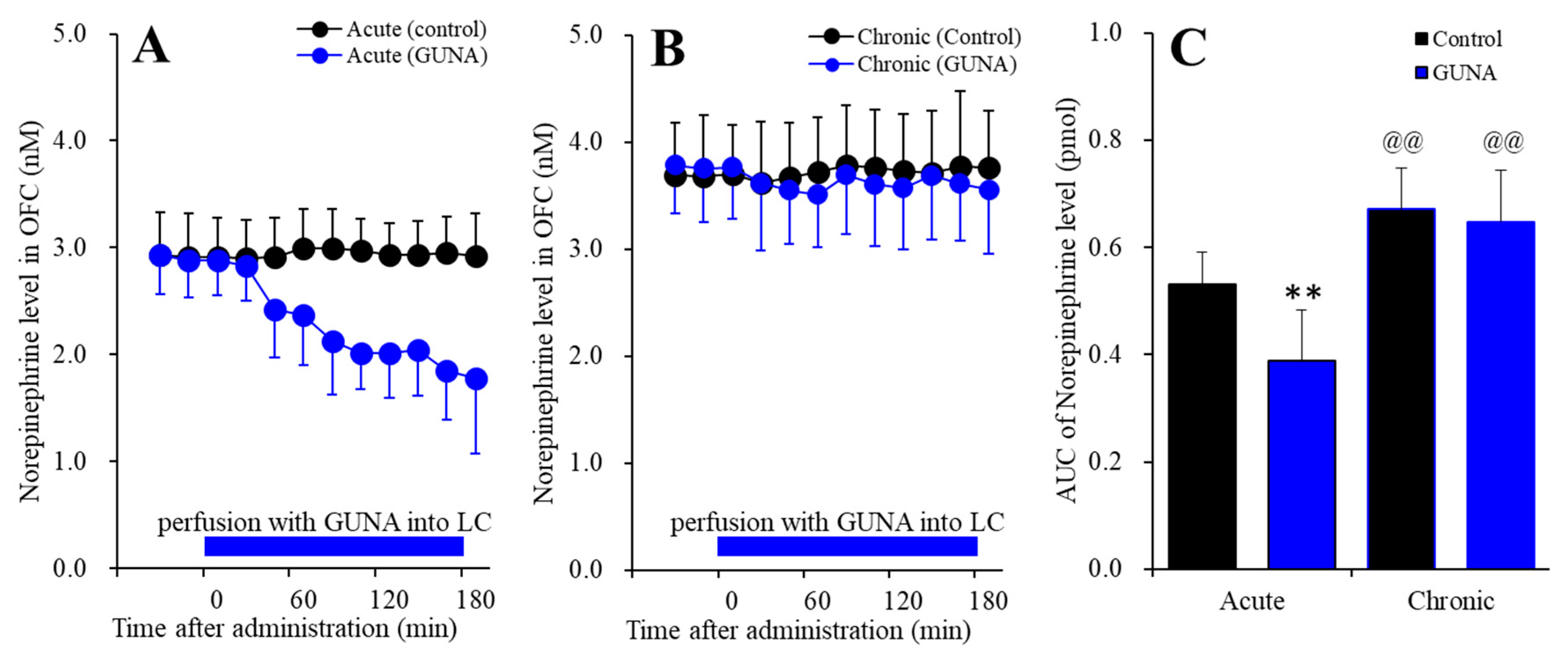
2.1.1. Effects of Acute and Chronic Administration of Therapeutic-Relevant Dose of Guanfacine on Extracellular Norepinephrine Level in the OFC
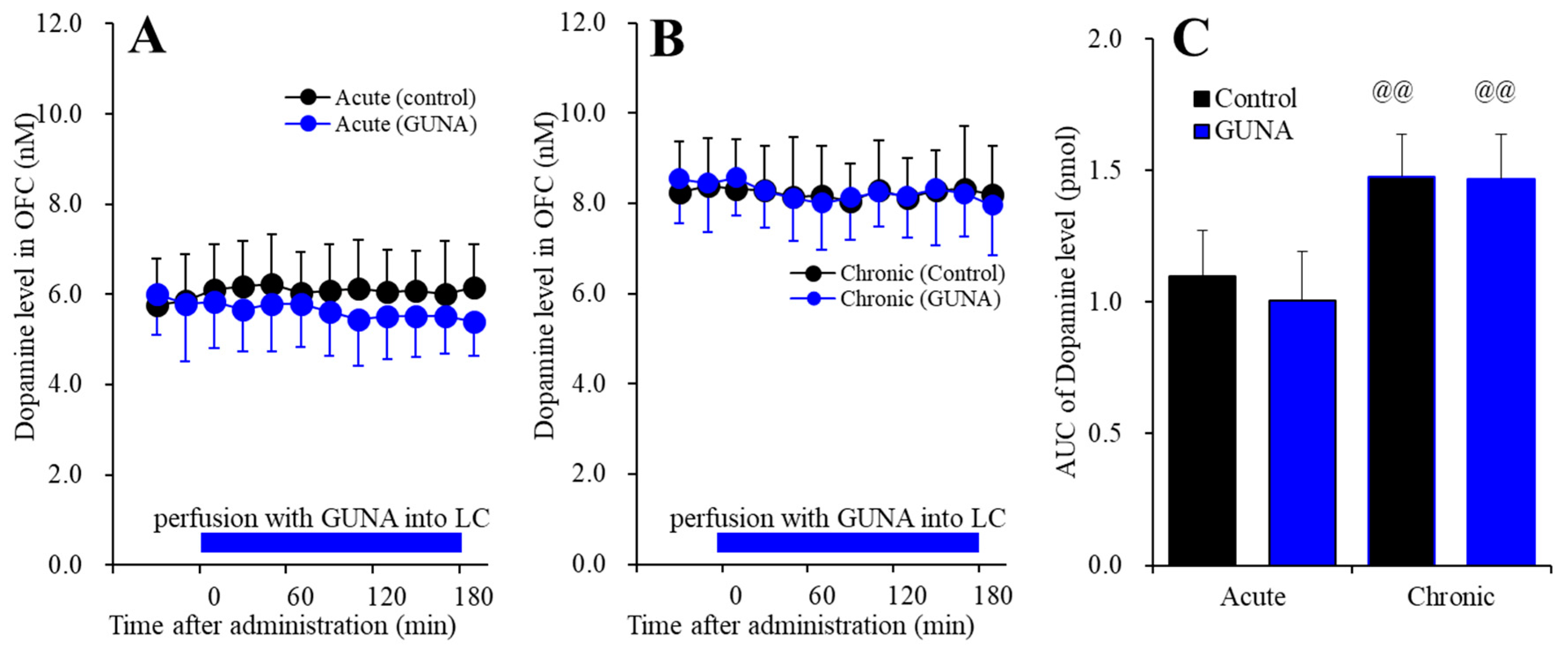
2.1.2. Effects of Acute and Chronic Administration of Therapeutic-Relevant Dose of Guanfacine on Extracellular Dopamine Level in the OFC
2.1.3. Effects of Acute and Chronic Administration of Therapeutic-Relevant Dose of Guanfacine on Extracellular Norepinephrine Level in the VTA
2.1.4. Effects of Acute and Chronic Administration of Therapeutic-Relevant Dose of Guanfacine on Extracellular Norepinephrine Level in the RTN
2.1.5. Effects of Acute and Chronic Administration of Therapeutically Relevant Dose of Guanfacine on Extracellular GABA Level in the MDTN
2.2. Effects of Local Administration of Prazosine into the VTA on Extracellular Levels of Dopamine in the OFC of Chronic Guanfacine Administration (Study 2)
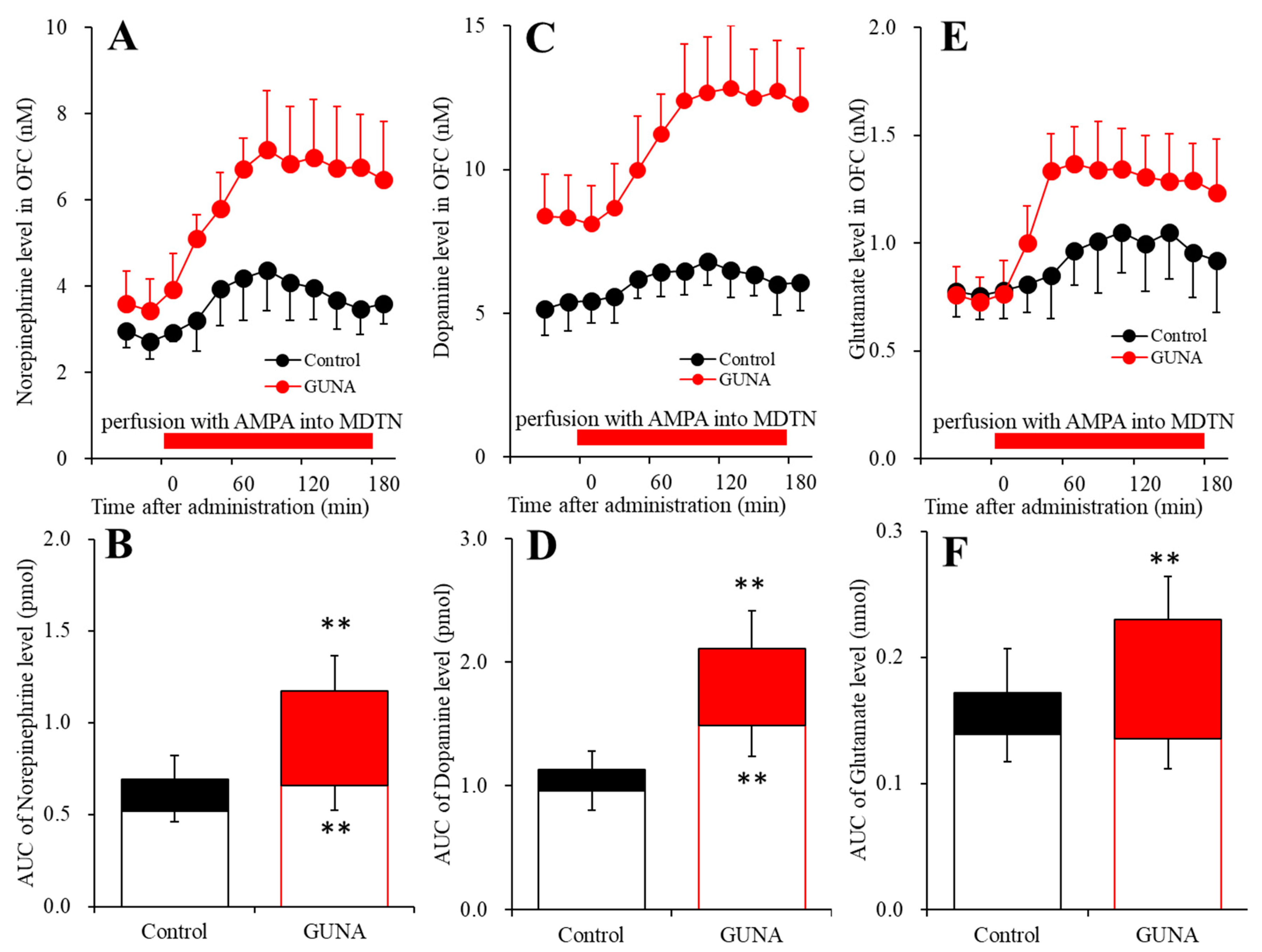
2.3. Effects of Local Administration of AMPA into the MDTN on Extracellular Levels of Norepinephrine, Dopamine and L-glutamate in the OFC of Chronic Guanfacine Administration (Study 3)
2.4. Effects of Subacute and Chronic Guanfacine Administration on α2A Adrenoceptor Expression in the Plasma Membrane of LC, OFC and VTA
3. Discussion
4. Materials and Methods
4.1. Chemical Agents and Drug Administration
4.2. Preparation of Microdialysis
4.3. Determination of Levels of Dopamine, Norepinephrine and L-Glutamate
4.4. Capillary Immunoblotting Analysis
4.5. Data Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Willcutt, E.G. The prevalence of DSM-IV attention-deficit/hyperactivity disorder: A meta-analytic review. Neurotherapeutics 2012, 9, 490–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortese, S. Pharmacologic Treatment of Attention Deficit-Hyperactivity Disorder. N. Engl. J. Med. 2020, 383, 1050–1056. [Google Scholar] [CrossRef]
- Wolraich, M.L.; Hagan, J.F., Jr.; Allan, C.; Chan, E.; Davison, D.; Earls, M.; Evans, S.W.; Flinn, S.K.; Froehlich, T.; Frost, J.; et al. Clinical Practice Guideline for the Diagnosis, Evaluation, and Treatment of Attention-Deficit/Hyperactivity Disorder in Children and Adolescents. Pediatrics 2019, 144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolea-Alamanac, B.; Nutt, D.J.; Adamou, M.; Asherson, P.; Bazire, S.; Coghill, D.; Heal, D.; Muller, U.; Nash, J.; Santosh, P.; et al. Evidence-based guidelines for the pharmacological management of attention deficit hyperactivity disorder: Update on recommendations from the British Association for Psychopharmacology. J. Psychopharmacol. 2014, 28, 179–203. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Couture, J. A review of the pathophysiology, etiology, and treatment of attention-deficit hyperactivity disorder (ADHD). Ann. Pharmacol. 2014, 48, 209–225. [Google Scholar] [CrossRef] [PubMed]
- Mattes, J.A. Treating ADHD in Prison: Focus on Alpha-2 Agonists (Clonidine and Guanfacine). J. Am. Acad. Psychiatry Law 2016, 44, 151–157. [Google Scholar]
- Huss, M.; Chen, W.; Ludolph, A.G. Guanfacine Extended Release: A New Pharmacological Treatment Option in Europe. Clin. Drug Investig. 2016, 36, 1–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pringsheim, T.; Hirsch, L.; Gardner, D.; Gorman, D.A. The pharmacological management of oppositional behaviour, conduct problems, and aggression in children and adolescents with attention-deficit hyperactivity disorder, oppositional defiant disorder, and conduct disorder: A systematic review and meta-analysis. Part 1: Psychostimulants, alpha-2 agonists, and atomoxetine. Can J. Psychiatry 2015, 60, 42–51. [Google Scholar] [CrossRef] [Green Version]
- Bangs, M.E.; Tauscher-Wisniewski, S.; Polzer, J.; Zhang, S.; Acharya, N.; Desaiah, D.; Trzepacz, P.T.; Allen, A.J. Meta-analysis of suicide-related behavior events in patients treated with atomoxetine. J. Am. Acad. Child. Adolesc. Psychiatry 2008, 47, 209–218. [Google Scholar] [CrossRef]
- Strawn, J.R.; Compton, S.N.; Robertson, B.; Albano, A.M.; Hamdani, M.; Rynn, M.A. Extended Release Guanfacine in Pediatric Anxiety Disorders: A Pilot, Randomized, Placebo-Controlled Trial. J. Child. Adolesc. Psychopharmacol. 2017, 27, 29–37. [Google Scholar] [CrossRef]
- Uhlen, S.; Muceniece, R.; Rangel, N.; Tiger, G.; Wikberg, J.E. Comparison of the binding activities of some drugs on alpha 2A, alpha 2B and alpha 2C-adrenoceptors and non-adrenergic imidazoline sites in the guinea pig. Pharmacol. Toxicol. 1995, 76, 353–364. [Google Scholar] [CrossRef]
- Jasper, J.R.; Lesnick, J.D.; Chang, L.K.; Yamanishi, S.S.; Chang, T.K.; Hsu, S.A.; Daunt, D.A.; Bonhaus, D.W.; Eglen, R.M. Ligand efficacy and potency at recombinant alpha2 adrenergic receptors: Agonist-mediated [35S]GTPgammaS binding. Biochem. Pharmacol. 1998, 55, 1035–1043. [Google Scholar] [CrossRef]
- Ramos, B.P.; Arnsten, A.F. Adrenergic pharmacology and cognition: Focus on the prefrontal cortex. Pharmacol. Ther. 2007, 113, 523–536. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Ramos, B.P.; Paspalas, C.D.; Shu, Y.; Simen, A.; Duque, A.; Vijayraghavan, S.; Brennan, A.; Dudley, A.; Nou, E.; et al. Alpha2A-adrenoceptors strengthen working memory networks by inhibiting cAMP-HCN channel signaling in prefrontal cortex. Cell 2007, 129, 397–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, E.M.; Pomerleau, F.; Huettl, P.; Gerhardt, G.A.; Glaser, P.E. Aberrant glutamate signaling in the prefrontal cortex and striatum of the spontaneously hypertensive rat model of attention-deficit/hyperactivity disorder. Psychopharmacology 2014, 231, 3019–3029. [Google Scholar] [CrossRef]
- Saito, Y.; Inoue, T.; Zhu, G.; Kimura, N.; Okada, M.; Nishimura, M.; Kimura, N.; Murayama, S.; Kaneko, S.; Shigemoto, R. Hyperpolarization-activated cyclic nucleotide gated channels: A potential molecular link between epileptic seizures and Aβ generation in Alzheimer’s disease. Mol. Neurodegener. 2012, 7, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawaura, K.; Karasawa, J.; Chaki, S.; Hikichi, H. Stimulation of postsynapse adrenergic alpha2A receptor improves attention/cognition performance in an animal model of attention deficit hyperactivity disorder. Behav. Brain Res. 2014, 270, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Franowicz, J.S.; Kessler, L.E.; Borja, C.M.; Kobilka, B.K.; Limbird, L.E.; Arnsten, A.F. Mutation of the alpha2A-adrenoceptor impairs working memory performance and annuls cognitive enhancement by guanfacine. J. Neurosci. 2002, 22, 8771–8777. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.; McAnulty, G.; Hamoda, H.M.; Sarill, K.; Karmacharya, S.; Gagoski, B.; Ning, L.; Grant, P.E.; Shenton, M.E.; Waber, D.P.; et al. Detecting microstructural white matter abnormalities of frontal pathways in children with ADHD using advanced diffusion models. Brain Imaging. Behav. 2020, 14, 981–997. [Google Scholar] [CrossRef]
- Sethi, A.; Voon, V.; Critchley, H.D.; Cercignani, M.; Harrison, N.A. A neurocomputational account of reward and novelty processing and effects of psychostimulants in attention deficit hyperactivity disorder. Brain 2018, 141, 1545–1557. [Google Scholar] [CrossRef] [Green Version]
- Barbas, H.; Zikopoulos, B.; Timbie, C. Sensory pathways and emotional context for action in primate prefrontal cortex. Biol. Psychiatry 2011, 69, 1133–1139. [Google Scholar] [CrossRef]
- Sara, S.J.; Bouret, S. Orienting and reorienting: The locus coeruleus mediates cognition through arousal. Neuron 2012, 76, 130–141. [Google Scholar] [CrossRef] [Green Version]
- Okada, M.; Kawano, Y.; Fukuyama, K.; Motomura, E.; Shiroyama, T. Candidate Strategies for Development of a Rapid-Acting Antidepressant Class That Does Not Result in Neuropsychiatric Adverse Effects: Prevention of Ketamine-Induced Neuropsychiatric Adverse Reactions. Int. J. Mol. Sci. 2020, 21, 7951. [Google Scholar] [CrossRef]
- Okada, M.; Fukuyama, K.; Kawano, Y.; Shiroyama, T.; Suzuki, D.; Ueda, Y. Effects of acute and sub-chronic administrations of guanfacine on catecholaminergic transmissions in the orbitofrontal cortex. Neuropharmacology 2019, 156, 107547. [Google Scholar] [CrossRef] [PubMed]
- Devoto, P.; Flore, G.; Saba, P.; Fa, M.; Gessa, G.L. Stimulation of the locus coeruleus elicits noradrenaline and dopamine release in the medial prefrontal and parietal cortex. J. Neurochem. 2005, 92, 368–374. [Google Scholar] [CrossRef] [PubMed]
- Devoto, P.; Flore, G.; Vacca, G.; Pira, L.; Arca, A.; Casu, M.A.; Pani, L.; Gessa, G.L. Co-release of noradrenaline and dopamine from noradrenergic neurons in the cerebral cortex induced by clozapine, the prototype atypical antipsychotic. Psychopharmacology 2003, 167, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Yamamura, S.; Ohoyama, K.; Hamaguchi, T.; Kashimoto, K.; Nakagawa, M.; Kanehara, S.; Suzuki, D.; Matsumoto, T.; Motomura, E.; Shiroyama, T.; et al. Effects of quetiapine on monoamine, GABA, and glutamate release in rat prefrontal cortex. Psychopharmacology 2009, 206, 243–258. [Google Scholar] [CrossRef]
- Yamamura, S.; Ohoyama, K.; Hamaguchi, T.; Nakagawa, M.; Suzuki, D.; Matsumoto, T.; Motomura, E.; Tanii, H.; Shiroyama, T.; Okada, M. Effects of zotepine on extracellular levels of monoamine, GABA and glutamate in rat prefrontal cortex. Br. J. Pharmacol. 2009, 157, 656–665. [Google Scholar] [CrossRef] [PubMed]
- McCormick, D.A.; Wang, Z. Serotonin and noradrenaline excite GABAergic neurones of the guinea-pig and cat nucleus reticularis thalami. J. Physiol. 1991, 442, 235–255. [Google Scholar] [CrossRef]
- Nishitomi, K.; Yano, K.; Kobayashi, M.; Jino, K.; Kano, T.; Horiguchi, N.; Shinohara, S.; Hasegawa, M. Systemic administration of guanfacine improves food-motivated impulsive choice behavior primarily via direct stimulation of postsynaptic alpha2A-adrenergic receptors in rats. Behav. Brain Res. 2018, 345, 21–29. [Google Scholar] [CrossRef]
- Yamamura, S.; Ohoyama, K.; Nagase, H.; Okada, M. Zonisamide enhances delta receptor-associated neurotransmitter release in striato-pallidal pathway. Neuropharmacology 2009, 57, 322–331. [Google Scholar] [CrossRef]
- Tanahashi, S.; Yamamura, S.; Nakagawa, M.; Motomura, E.; Okada, M. Dopamine D2 and serotonin 5-HT1A receptors mediate the actions of aripiprazole in mesocortical and mesoaccumbens transmission. Neuropharmacology 2012, 62, 765–774. [Google Scholar] [CrossRef]
- Fukuyama, K.; Hasegawa, T.; Okada, M. Cystine/Glutamate Antiporter and Aripiprazole Compensate NMDA Antagonist-Induced Dysfunction of Thalamocortical L-Glutamatergic Transmission. Int. J. Mol. Sci. 2018, 19, 3645. [Google Scholar] [CrossRef] [Green Version]
- Asanuma, C. Noradrenergic innervation of the thalamic reticular nucleus: A light and electron microscopic immunohistochemical study in rats. J. Comp. Neurol. 1992, 319, 299–311. [Google Scholar] [CrossRef]
- Okada, M.; Fukuyama, K. Interaction between Mesocortical and Mesothalamic Catecholaminergic Transmissions Associated with NMDA Receptor in the Locus Coeruleus. Biomolecules 2020, 10, 990. [Google Scholar] [CrossRef] [PubMed]
- Yamamura, S.; Hamaguchi, T.; Ohoyama, K.; Sugiura, Y.; Suzuki, D.; Kanehara, S.; Nakagawa, M.; Motomura, E.; Matsumoto, T.; Tanii, H.; et al. Topiramate and zonisamide prevent paradoxical intoxication induced by carbamazepine and phenytoin. Epilepsy Res. 2009, 84, 172–186. [Google Scholar] [CrossRef] [PubMed]
- Ohoyama, K.; Yamamura, S.; Hamaguchi, T.; Nakagawa, M.; Motomura, E.; Shiroyama, T.; Tanii, H.; Okada, M. Effect of novel atypical antipsychotic, blonanserin, on extracellular neurotransmitter level in rat prefrontal cortex. Eur. J. Pharm. 2011, 653, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Kielbinski, M.; Bernacka, J.; Solecki, W.B. Differential regulation of phasic dopamine release in the forebrain by the VTA noradrenergic receptor signaling. J. Neurochem. 2019, 149, 747–759. [Google Scholar] [CrossRef]
- Fukuyama, K.; Fukuzawa, M.; Shiroyama, T.; Okada, M. Pathogenesis and pathophysiology of autosomal dominant sleep-related hypermotor epilepsy with S284L-mutant alpha4 subunit of nicotinic ACh receptor. Br. J. Pharmacol. 2020, 177, 2143–2162. [Google Scholar] [CrossRef] [PubMed]
- Aoki, C.; Go, C.G.; Venkatesan, C.; Kurose, H. Perikaryal and synaptic localization of alpha 2A-adrenergic receptor-like immunoreactivity. Brain Res. 1994, 650, 181–204. [Google Scholar] [CrossRef]
- Qin, K.; Sethi, P.R.; Lambert, N.A. Abundance and stability of complexes containing inactive G protein-coupled receptors and G proteins. FASEB J. 2008, 22, 2920–2927. [Google Scholar] [CrossRef]
- Okubo, R.; Hasegawa, T.; Fukuyama, K.; Shiroyama, T.; Okada, M. Current limitations and candidate potential of 5-HT7 receptor antagonism in psychiatric pharmacotherapy. Front. Psychiatry 2021, 12, 623684. [Google Scholar] [CrossRef]
- Okada, M.; Matsumoto, R.; Yamamoto, Y.; Fukuyama, K. Effects of Subchronic Administrations of Vortioxetine, Lurasidone, and Escitalopram on Thalamocortical Glutamatergic Transmission Associated with Serotonin 5-HT7 Receptor. Int. J. Mol. Sci. 2021, 22, 1351. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Fukuyama, K.; Shiroyama, T.; Murata, M. A Working Hypothesis Regarding Identical Pathomechanisms between Clinical Efficacy and Adverse Reaction of Clozapine via the Activation of Connexin43. Int. J. Mol. Sci. 2020, 21, 7019. [Google Scholar] [CrossRef] [PubMed]
- Porrino, L.J.; Crane, A.M.; Goldman-Rakic, P.S. Direct and indirect pathways from the amygdala to the frontal lobe in rhesus monkeys. J. Comp. Neurol. 1981, 198, 121–136. [Google Scholar] [CrossRef] [PubMed]
- Russchen, F.T.; Amaral, D.G.; Price, J.L. The afferent input to the magnocellular division of the mediodorsal thalamic nucleus in the monkey, Macaca fascicularis. J. Comp. Neurol. 1987, 256, 175–210. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, A.S.; Browning, P.G.; Baxter, M.G. Neurotoxic lesions of the medial mediodorsal nucleus of the thalamus disrupt reinforcer devaluation effects in rhesus monkeys. J. Neurosci. 2007, 27, 11289–11295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izquierdo, A.; Murray, E.A. Selective bilateral amygdala lesions in rhesus monkeys fail to disrupt object reversal learning. J. Neurosci. 2007, 27, 1054–1062. [Google Scholar] [CrossRef] [Green Version]
- Okada, M.; Okubo, R.; Fukuyama, K. Vortioxetine Subchronically Activates Serotonergic Transmission via Desensitization of Serotonin 5-HT1A Receptor with 5-HT3 Receptor Inhibition in Rats. Int. J. Mol. Sci. 2019, 20, 6235. [Google Scholar] [CrossRef] [Green Version]
- Okada, M.; Fukuyama, K.; Ueda, Y. Lurasidone inhibits NMDA receptor antagonist-induced functional abnormality of thalamocortical glutamatergic transmission via 5-HT7 receptor blockade. Br J. Pharmacol. 2019, 176, 4002–4018. [Google Scholar] [CrossRef]
- Okada, M.; Fukuyama, K.; Okubo, R.; Shiroyama, T.; Ueda, Y. Lurasidone Sub-Chronically Activates Serotonergic Transmission via Desensitization of 5-HT1A and 5-HT7 Receptors in Dorsal Raphe Nucleus. Pharmaceuticals 2019, 12, 149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okada, M.; Fukuyama, K.; Nakano, T.; Ueda, Y. Pharmacological Discrimination of Effects of MK801 on Thalamocortical, Mesothalamic, and Mesocortical Transmissions. Biomolecules 2019, 9, 746. [Google Scholar] [CrossRef] [Green Version]
- Okada, M.; Fukuyama, K.; Kawano, Y.; Shiroyama, T.; Ueda, Y. Memantine protects thalamocortical hyper-glutamatergic transmission induced by NMDA receptor antagonism via activation of system xc−. Pharm. Res. Perspect. 2019, 7, e00457. [Google Scholar] [CrossRef] [Green Version]
- Fukuyama, K.; Kato, R.; Murata, M.; Shiroyama, T.; Okada, M. Clozapine Normalizes a Glutamatergic Transmission Abnormality Induced by an Impaired NMDA Receptor in the Thalamocortical Pathway via the Activation of a Group III Metabotropic Glutamate Receptor. Biomolecules 2019, 9, 234. [Google Scholar] [CrossRef] [Green Version]
- Fukuyama, K.; Okada, M. Age-Dependent and Sleep/Seizure-Induced Pathomechanisms of Autosomal Dominant Sleep-Related Hypermotor Epilepsy. Int. J. Mol. Sci. 2020, 21, 8142. [Google Scholar] [CrossRef]
- Fukuyama, K.; Fukuzawa, M.; Shiroyama, T.; Okada, M. Pathomechanism of nocturnal paroxysmal dystonia in autosomal dominant sleep-related hypermotor epilepsy with S284L-mutant alpha4 subunit of nicotinic ACh receptor. Biomed. Pharm. 2020, 126, 110070. [Google Scholar] [CrossRef]
- Fukuyama, K.; Fukuzawa, M.; Okubo, R.; Okada, M. Upregulated Connexin 43 Induced by Loss-of-Functional S284L-Mutant alpha4 Subunit of Nicotinic ACh Receptor Contributes to Pathomechanisms of Autosomal Dominant Sleep-Related Hypermotor Epilepsy. Pharmaceuticals 2020, 13, 58. [Google Scholar] [CrossRef] [Green Version]
- Fukuyama, K.; Fukuzawa, M.; Okada, M. Upregulated and Hyperactivated Thalamic Connexin 43 Plays Important Roles in Pathomechanisms of Cognitive Impairment and Seizure of Autosomal Dominant Sleep-Related Hypermotor Epilepsy with S284L-Mutant alpha4 Subunit of Nicotinic ACh Receptor. Pharmaceuticals 2020, 13, 99. [Google Scholar] [CrossRef]
- Liebe, T.; Li, M.; Colic, L.; Munk, M.H.J.; Sweeney-Reed, C.M.; Woelfer, M.; Kretzschmar, M.A.; Steiner, J.; von During, F.; Behnisch, G.; et al. Ketamine influences the locus coeruleus norepinephrine network, with a dependency on norepinephrine transporter genotype—A placebo controlled fMRI study. Neuroimage Clin. 2018, 20, 715–723. [Google Scholar] [CrossRef] [PubMed]
- Sarkisyan, G.; Hedlund, P.B. The 5-HT7 receptor is involved in allocentric spatial memory information processing. Behav. Brain Res. 2009, 202, 26–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okada, M. Can rodent models manifest pathomechanisms of genetic epilepsy? Br. J. Pharmacol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Oka, T.; Nakamoto, M.; Fukuyama, K.; Shiroyama, T. Astroglial Connexin43 as a Potential Target for a Mood Stabiliser. Int. J. Mol. Sci. 2020, 22, 339. [Google Scholar] [CrossRef] [PubMed]
- Mustafa, M.R.; Stolz, J.F.; Luscombe, D.K. Effect of long-term rolipram administration on the sensitivity of alpha 2-adrenoceptors in rat brain. Eur. J. Pharm. 1988, 148, 171–177. [Google Scholar] [CrossRef]
- Verrotti, A.; Moavero, R.; Panzarino, G.; Di Paolantonio, C.; Rizzo, R.; Curatolo, P. The Challenge of Pharmacotherapy in Children and Adolescents with Epilepsy-ADHD Comorbidity. Clin. Drug Investig. 2018, 38, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Cortese, S.; Adamo, N.; Del Giovane, C.; Mohr-Jensen, C.; Hayes, A.J.; Carucci, S.; Atkinson, L.Z.; Tessari, L.; Banaschewski, T.; Coghill, D.; et al. Comparative efficacy and tolerability of medications for attention-deficit hyperactivity disorder in children, adolescents, and adults: A systematic review and network meta-analysis. Lancet. Psychiatry 2018, 5, 727–738. [Google Scholar] [CrossRef] [Green Version]
- Elliott, J.; Johnston, A.; Husereau, D.; Kelly, S.E.; Eagles, C.; Charach, A.; Hsieh, S.C.; Bai, Z.; Hossain, A.; Skidmore, B.; et al. Pharmacologic treatment of attention deficit hyperactivity disorder in adults: A systematic review and network meta-analysis. PLoS ONE 2020, 15, e0240584. [Google Scholar] [CrossRef] [PubMed]
- Padilha, S.; Virtuoso, S.; Tonin, F.S.; Borba, H.H.L.; Pontarolo, R. Efficacy and safety of drugs for attention deficit hyperactivity disorder in children and adolescents: A network meta-analysis. Eur. Child Adolesc. Psychiatry 2018, 27, 1335–1345. [Google Scholar] [CrossRef]
- Sadacca, B.F.; Wikenheiser, A.M.; Schoenbaum, G. Toward a theoretical role for tonic norepinephrine in the orbitofrontal cortex in facilitating flexible learning. Neuroscience 2017, 345, 124–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lammel, S.; Lim, B.K.; Ran, C.; Huang, K.W.; Betley, M.J.; Tye, K.M.; Deisseroth, K.; Malenka, R.C. Input-specific control of reward and aversion in the ventral tegmental area. Nature 2012, 491, 212–217. [Google Scholar] [CrossRef] [Green Version]
- Vander Weele, C.M.; Siciliano, C.A.; Matthews, G.A.; Namburi, P.; Izadmehr, E.M.; Espinel, I.C.; Nieh, E.H.; Schut, E.H.S.; Padilla-Coreano, N.; Burgos-Robles, A.; et al. Dopamine enhances signal-to-noise ratio in cortical-brainstem encoding of aversive stimuli. Nature 2018, 563, 397–401. [Google Scholar] [CrossRef]
- Weele, C.M.V.; Siciliano, C.A.; Tye, K.M. Dopamine tunes prefrontal outputs to orchestrate aversive processing. Brain Res. 2019, 1713, 16–31. [Google Scholar] [CrossRef]
- Koda, K.; Ago, Y.; Cong, Y.; Kita, Y.; Takuma, K.; Matsuda, T. Effects of acute and chronic administration of atomoxetine and methylphenidate on extracellular levels of noradrenaline, dopamine and serotonin in the prefrontal cortex and striatum of mice. J. Neurochem. 2010, 114, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Caldji, C.; Sharma, S.; Plotsky, P.M.; Meaney, M.J. Influence of neonatal rearing conditions on stress-induced adrenocorticotropin responses and norepinepherine release in the hypothalamic paraventricular nucleus. J Neuroendocr. 2000, 12, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Clayton, E.C.; Williams, C.L. Adrenergic activation of the nucleus tractus solitarius potentiates amygdala norepinephrine release and enhances retention performance in emotionally arousing and spatial memory tasks. Behav. Brain Res. 2000, 112, 151–158. [Google Scholar] [CrossRef]
- Williams, C.L.; Men, D.; Clayton, E.C. The effects of noradrenergic activation of the nucleus tractus solitarius on memory and in potentiating norepinephrine release in the amygdala. Behav. Neurosci. 2000, 114, 1131–1144. [Google Scholar] [CrossRef] [PubMed]
- Strobel, C.; Hunt, S.; Sullivan, R.; Sun, J.; Sah, P. Emotional regulation of pain: The role of noradrenaline in the amygdala. Sci. China Life Sci. 2014, 57, 384–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okada, M.; Hirano, T.; Mizuno, K.; Kawata, Y.; Wada, K.; Murakami, T.; Tasaki, H.; Kaneko, S. Effects of carbamazepine on hippocampal serotonergic system. Epilepsy Res. 1998, 31, 187–198. [Google Scholar] [CrossRef]
- Okada, M.; Kawata, Y.; Murakami, T.; Wada, K.; Mizuno, K.; Kaneko, S. Interaction between purinoceptor subtypes on hippocampal serotonergic transmission using in vivo microdialysis. Neuropharmacology 1999, 38, 707–715. [Google Scholar] [CrossRef]
- Paxinos, G.; Watson, C. The Rat Brain: In Stereotoxic Coordinates, 6th ed.; Academic Press: San Dieg, CA, USA, 2007. [Google Scholar]
- Yoshida, S.; Okada, M.; Zhu, G.; Kaneko, S. Effects of zonisamide on neurotransmitter exocytosis associated with ryanodine receptors. Epilepsy Res 2005, 67, 153–162. [Google Scholar] [CrossRef]
- Okada, M.; Zhu, G.; Yoshida, S.; Hirose, S.; Kaneko, S. Protein kinase associated with gating and closing transmission mechanisms in temporoammonic pathway. Neuropharmacology 2004, 47, 485–504. [Google Scholar] [CrossRef]
- Fukuyama, K.; Ueda, Y.; Okada, M. Effects of Carbamazepine, Lacosamide and Zonisamide on Gliotransmitter Release Associated with Activated Astroglial Hemichannels. Pharmaceuticals 2020, 13, 117. [Google Scholar] [CrossRef]
- Fukuyama, K.; Okubo, R.; Murata, M.; Shiroyama, T.; Okada, M. Activation of Astroglial Connexin is Involved in Concentration-Dependent Double-Edged Sword Clinical Action of Clozapine. Cells 2020, 9, 414. [Google Scholar] [CrossRef] [Green Version]
- Fukuyama, K.; Okada, M. Effects of levetiracetam on astroglial release of kynurenine-pathway metabolites. Br. J. Pharmacol. 2018, 175, 4253–4265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamura, S.; Hoshikawa, M.; Dai, K.; Saito, H.; Suzuki, N.; Niwa, O.; Okada, M. ONO-2506 inhibits spikewave discharges in a genetic animal model without affecting traditional convulsive tests via gliotransmission regulation. Br. J. Pharmacol. 2013, 168, 1088–1100. [Google Scholar] [CrossRef] [PubMed]
- Tanahashi, S.; Yamamura, S.; Nakagawa, M.; Motomura, E.; Okada, M. Clozapine, but not haloperidol, enhances glial D-serine and L-glutamate release in rat frontal cortex and primary cultured astrocytes. Br. J. Pharmacol. 2012, 165, 1543–1555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanahashi, S.; Ueda, Y.; Nakajima, A.; Yamamura, S.; Nagase, H.; Okada, M. Novel delta1-receptor agonist KNT-127 increases the release of dopamine and L-glutamate in the striatum, nucleus accumbens and median pre-frontal cortex. Neuropharmacology 2012, 62, 2057–2067. [Google Scholar] [CrossRef] [PubMed]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fukuyama, K.; Nakano, T.; Shiroyama, T.; Okada, M. Chronic Administrations of Guanfacine on Mesocortical Catecholaminergic and Thalamocortical Glutamatergic Transmissions. Int. J. Mol. Sci. 2021, 22, 4122. https://doi.org/10.3390/ijms22084122
Fukuyama K, Nakano T, Shiroyama T, Okada M. Chronic Administrations of Guanfacine on Mesocortical Catecholaminergic and Thalamocortical Glutamatergic Transmissions. International Journal of Molecular Sciences. 2021; 22(8):4122. https://doi.org/10.3390/ijms22084122
Chicago/Turabian StyleFukuyama, Kouji, Tomosuke Nakano, Takashi Shiroyama, and Motohiro Okada. 2021. "Chronic Administrations of Guanfacine on Mesocortical Catecholaminergic and Thalamocortical Glutamatergic Transmissions" International Journal of Molecular Sciences 22, no. 8: 4122. https://doi.org/10.3390/ijms22084122