
Migraine: Calcium Channels and Glia


Abstract
:1. Review Criteria
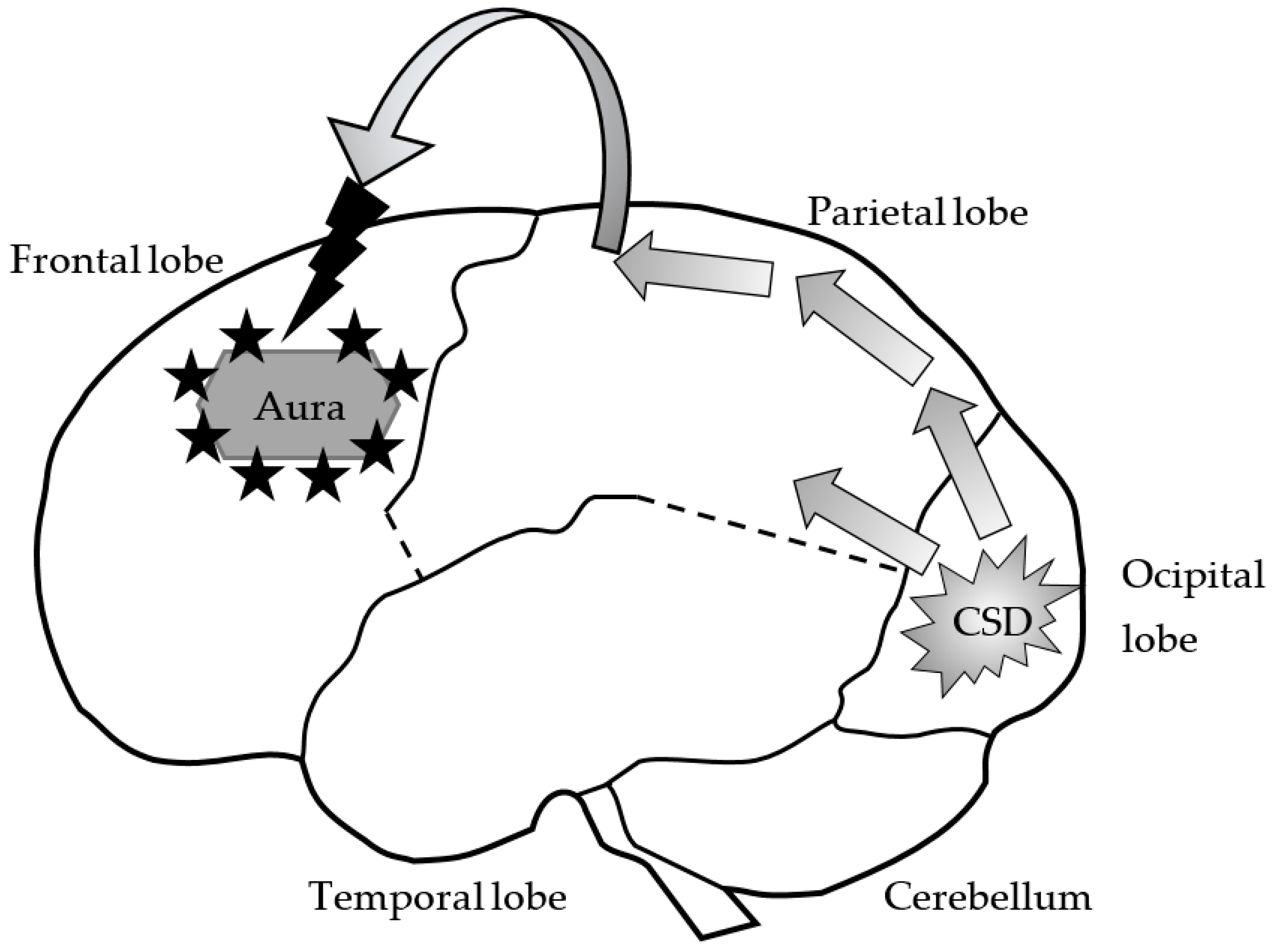
2. Migraine
2.1. The Pathogenesis of Migraine
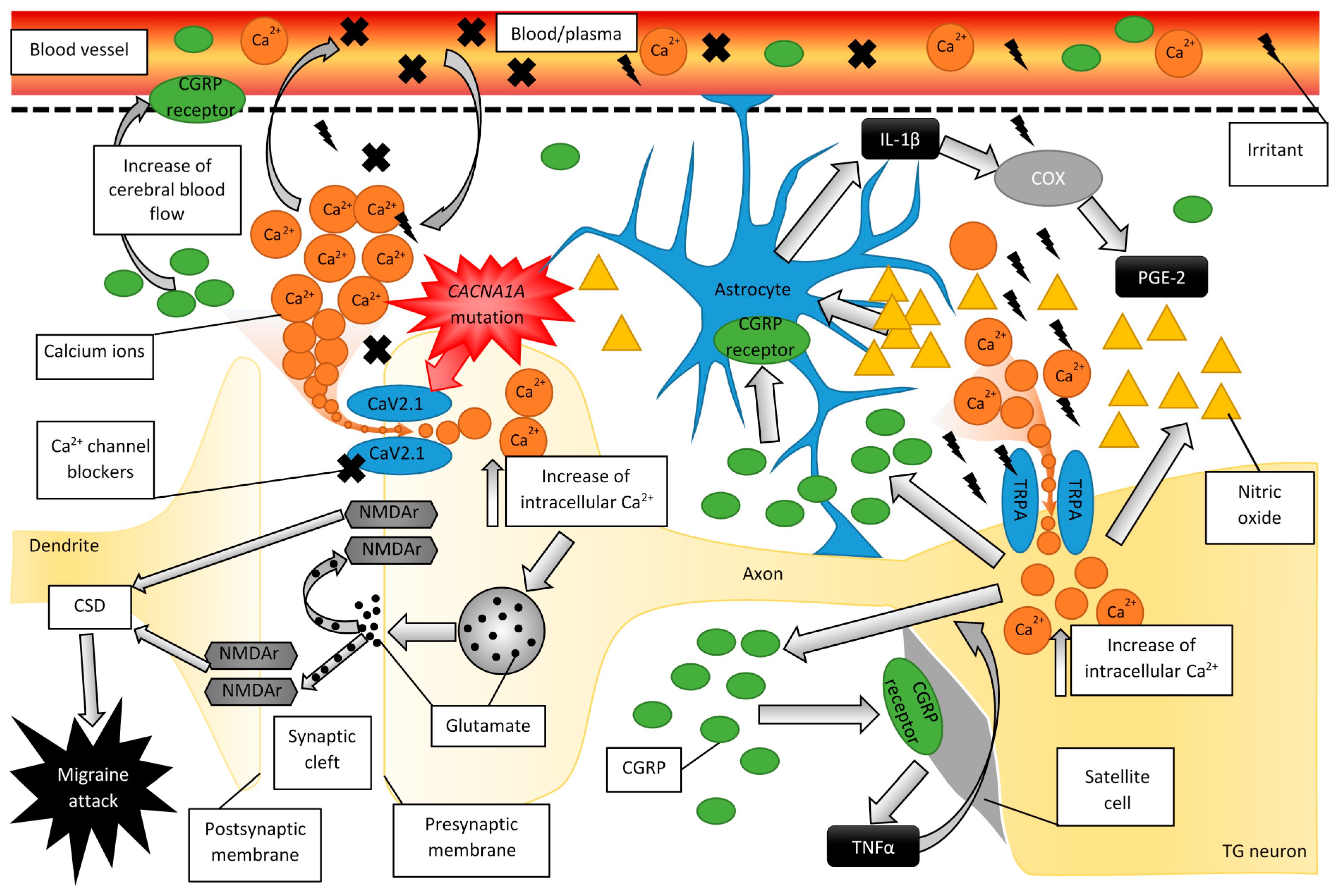
2.2. Migraine Disturbs Calcium Homeostasis
2.3. Experimental Evidence for the Role of CGRP in Migraine
2.4. The Role of CGRP and Glial Cells in Migraine Pathogenesis
2.5. Familial Hemiplegic Migraine
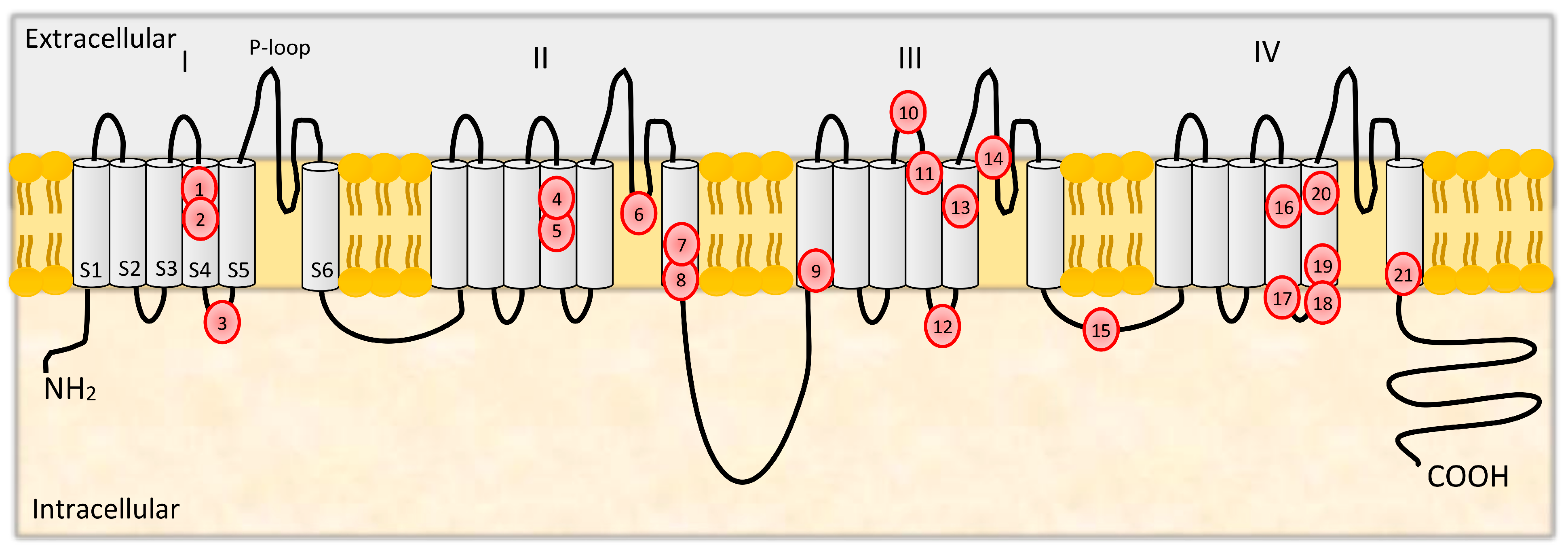
3. Structure and Functions of CaV2.1
3.1. Mutations in CACNA1A
3.2. Calcium-Related Therapeutics in Migraine
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bolay, H.; Ozge, A.; Saginc, P.; Orekici, G.; Uludüz, D.; Yalın, O.; Siva, A.; Bıçakçi, S.; Karakurum, B.; Öztürk, M. Gender influences headache characteristics with increasing age in migraine patients. Cephalalgia 2015, 35, 792–800. [Google Scholar] [CrossRef]
- Silberstein, S.D. Migraine. Lancet 2004, 363, 381–391. [Google Scholar] [CrossRef]
- World Health Organization. Atlas of Headache Disorders and Resources in the World 2011; World Health Organisation: Geneva, Switzerland, 2011.
- Vos, T.; Flaxman, A.D.; Naghavi, M.; Lozano, R.; Michaud, C.; Ezzati, M.; Shibuya, K.A.; Salomon, J.; Abdalla, S.; Aboyans, V.; et al. Years lived with disability (YLDs) for 1160 sequelae of 289 diseases and injuries 1990–2010: A systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012, 380, 2163–2196. [Google Scholar] [CrossRef]
- Simon, R.P.; Aminoff, M.J.; Greenberg, D.A. Clinical Neurology, 7th ed.; McGraw-Hill Professional Publishing: New York, NY, USA, 2009; Available online: http://public.ebookcentral.proquest.com/choice/publicfullrecord.aspx?p=4667887 (accessed on 30 October 2020).
- Viana, M.; Sprenger, T.; Andelova, M.; Goadsby, P.J. The typical duration of migraine aura: A systematic review. Cephalalgia 2013, 33, 483–490. [Google Scholar] [CrossRef]
- Headache Classification Committee of the International Headache Society (IHS). The International Classification of Headache Disorders; (beta version), 3rd ed. Cephalalgia 2013, 33, 629–808. [Google Scholar] [CrossRef] [Green Version]
- Headache Classification Committee of the International Headache Society (IHS). The International Classification Of Headache Disorders. 3rd edition. Cephalalgia 2018, 38, 1–211. [Google Scholar] [CrossRef]
- Menken, M.; Munsat, T.L.; Toole, J.F. The global burden of disease study: Implications for neurology. Arch. Neurol. 2000, 57, 418. [Google Scholar] [CrossRef]
- Pietrobon, D. Calcium channels and migraine. Biochim. Biophys. Acta (BBA) Biomembr. 2013, 1828, 1655–1665. [Google Scholar] [CrossRef]
- May, A.; Goadsby, P.J. The Trigeminovascular System in Humans: Pathophysiologic Implications for Primary Headache Syndromes of the Neural Influences on the Cerebral Circulation. Br. J. Pharmacol. 1999, 19, 115–127. [Google Scholar] [CrossRef] [Green Version]
- Eftekhari, S.; Salvatore, C.A.; Johansson, S.; Chen, T.-B.; Zeng, Z.; Edvinsson, L. Localization of CGRP, CGRP receptor, PACAP and glutamate in trigeminal ganglion. Relation to the blood–brain barrier. Brain Res. 2015, 1600, 93–109. [Google Scholar] [CrossRef]
- Leao, A.A.P. Spreading depression of activity in the cerebral cortex. J. Neurophysiol. 1944, 7, 359–390. [Google Scholar] [CrossRef]
- Charles, A.C.; Baca, S.M. Cortical spreading depression and migraine. Nat. Rev. Neurol. 2013, 9, 637–644. [Google Scholar] [CrossRef] [PubMed]
- Hansen, J.M.; Baca, S.M.; VanValkenburgh, P.; Charles, A. Distinctive anatomical and physiological features of migraine aura revealed by 18 years of recording. Brain 2013, 136, 3589–3595. [Google Scholar] [CrossRef] [Green Version]
- Goadsby, P.J.; Edvinsson, L.; Ekman, R. Release of vasoactive peptides in the extracerebral circulation of hu-mans and the cat during activation of the trigeminovascular system. Ann. Neurol. 1988, 23, 193–196. [Google Scholar] [CrossRef] [PubMed]
- Bolay, H.; Reuter, U.; Dunn, A.K.; Huang, Z.; Boas, D.A.; Moskowitz, M.A. Intrinsic brain activity triggers trigeminal meningeal afferents in a migraine model. Nat. Med. 2002, 8, 136–142. [Google Scholar] [CrossRef]
- Zhang, X.; Levy, D.; Kainz, V.; Noseda, R.; Jakubowski, M.; Burstein, R. Activation of central trigeminovascular neurons by cortical spreading depression. Ann. Neurol. 2010, 69, 855–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gargus, J.J. Genetic Calcium Signaling Abnormalities in the Central Nervous System: Seizures, Migraine, and Autism. Ann. N. Y. Acad. Sci. 2008, 1151, 133–156. [Google Scholar] [CrossRef] [PubMed]
- Ducros, A.; Tournier-Lasserve, E.; Bousser, M.-G. The genetics of migraine. Lancet Neurol. 2002, 1, 285–293. [Google Scholar] [CrossRef]
- Montagna, P. Migraine: A genetic disease? Neurol. Sci. 2008, 29, 47–51. [Google Scholar] [CrossRef]
- Mulley, J.C.; Scheffer, I.E.; Petrou, S.; Dibbens, L.M.; Berkovic, S.F.; Harkin, L.A. SCN1A mutations and epilepsy. Hum. Mutat. 2005, 25, 535–542. [Google Scholar] [CrossRef]
- Talavera, K.; Startek, J.B.; Alvarez-Collazo, J.; Boonen, B.; Alpizar, Y.A.; Sanchez, A.; Naert, R.; Nilius, B. Mammalian transient receptor potential trpa1 channels: From structure to disease. Physiol. Rev. 2020, 100, 725–803. [Google Scholar] [CrossRef]
- Piekut, T.; Wong, Y.Y.; Senatore, A.; Walker, E.S.; Smith, C.L.; Gauberg, J.; Harracksingh, A.N.; Lowden, C.; Novogradac, B.B.; Cheng, H.-Y.M.; et al. Early metazoan origin and multiple losses of a novel clade of rim presynaptic calcium channel scaffolding protein homologs. Genome Biol. Evol. 2020, 12, 1217–1239. [Google Scholar] [CrossRef]
- Pietrobon, D. CaV2.1 channelopathies. Pflügers Archiv Eur. J. Physiol. 2010, 460, 375–393. [Google Scholar] [CrossRef]
- Wild, V.; Messlinger, K.; Fischer, M.J. Hydrogen sulfide determines HNO-induced stimulation of trigeminal afferents. Neurosci. Lett. 2015, 602, 104–109. [Google Scholar] [CrossRef]
- Kunkler, P.E.; Ballardl, C.J.; Oxford, G.S.; Hurley, J.H. TRPA1 receptors mediate environmental irritant-induced meningeal vasodilatation. Pain 2011, 152, 38–44. [Google Scholar] [CrossRef] [Green Version]
- Nanou, E.; Catterall, W.A. Calcium channels, synaptic plasticity, and neuropsychiatric disease. Neuron 2018, 98, 466–481. [Google Scholar] [CrossRef] [Green Version]
- Catterall, W.A. Voltage-gated calcium channels. Handb. Cell Sign. 2010, 3, 897–909. [Google Scholar] [CrossRef] [Green Version]
- Amrutkar, D.; Ploug, K.; Olesen, J.; Jansen-Olesen, I. Role for voltage gated calcium channels in calcitonin gene-related peptide release in the rat trigeminovascular system. Neuroscience 2011, 172, 510–517. [Google Scholar] [CrossRef] [PubMed]
- Yin, P.; Anttila, V.; Siewert, K.M.; Palotie, A.; Smith, G.D.; Voight, B.F. Serum calcium and risk of migraine: A Mendelian randomization study. Hum. Mol. Genet. 2016, 26, 820–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rau, J.C.; Dodick, D.W. Other preventive anti-migraine treatments: Ace inhibitors, arbs, calcium channel blockers, serotonin antagonists, and nmda receptor antagonists. Curr. Treat. Options Neurol. 2019, 21, 17. [Google Scholar] [CrossRef] [PubMed]
- Edvinsson, L. Functional role of perivascular peptides in the control of cerebral circulation. Trends Neurosci. 1985, 8, 126–131. [Google Scholar] [CrossRef]
- Eftekhari, S.; Salvatore, C.; Calamari, A.; Kane, S.; Tajti, J.; Edvinsson, L. Differential distribution of calcitonin gene-related peptide and its receptor components in the human trigeminal ganglion. Neuroscience 2010, 169, 683–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulderry, P.K.; Ghatei, M.A.; Spokes, R.A.; Jones, P.M.; Pierson, A.M.; Hamid, Q.A.; Kanse, S.; Amara, S.G.; Burrin, J.M.; Legon, S.; et al. Differential expression of a-CGRP and b-CGRP by primary sensory neurons and enteric au-tonomic neurons of the rat. Neuroscience 1988, 25, 195–205. [Google Scholar]
- Thomsen, L.; Kruuse, C.; Iversen, H.K.; Olesen, J. A nitric oxide donor (nitroglycerin) triggers genuine migraine attacks. Eur. J. Neurol. 1994, 1, 73–80. [Google Scholar] [CrossRef]
- Weidner, C.; Klede, M.; Rukwied, R.; Lischetzki, G.; Neisius, U.; Schmelz, M.; Skov, P.S.; Petersen, L.J. Acute effects of substance p and calcitonin gene-related peptide in human skin—A microdialysis study. J. Investig. Dermatol. 2000, 115, 1015–1020. [Google Scholar] [CrossRef]
- Guo, S.; Vollesen, A.L.; Olesen, J.; Ashina, M. Premonitory and nonheadache symptoms induced by CGRP and PACAP38 in patients with migraine. Pain 2016, 157, 2773–2781. [Google Scholar] [CrossRef] [Green Version]
- Goadsby, P.J.; Edvinsson, L.; Ekman, R. Vasoactive peptide release in the extracerebral circulation of humans during migraine headache. Ann. Neurol. 1990, 28, 183–187. [Google Scholar] [CrossRef]
- Goadsby, P.J.; Edvinsson, L. The trigeminovascular system and migraine: Studies characterizing cerebrovascular and neuropeptide changes seen in humans and cats. Ann. Neurol. 1993, 33, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Cernuda-Morollón, E.; Larrosa, D.; Ramón, C.; Vega, J.; Martínez-Camblor, P.; Pascual, J. Interictal increase of CGRP levels in peripheral blood as a biomarker for chronic migraine. Neurology 2013, 81, 1191–1196. [Google Scholar] [CrossRef]
- Juhasz, G.; Zsombok, T.; Jakab, B.; Nemeth, J.; Szolcsanyi, J.; Bagdy, G. Sumatriptan causes parallel decrease in plasma calcitonin gene-related peptide (Cgrp) Concentration and migraine headache during nitroglycerin induced migraine attack. Cephalalgia 2005, 25, 179–183. [Google Scholar] [CrossRef]
- Cady, R.K.; Vause, C.V.; Ho, T.W.; Bigal, M.E.; Durham, P.L. Elevated saliva calcitonin gene-related peptide levels during acute migraine predict therapeutic response to rizatriptan. Headache: J. Head Face Pain 2009, 49, 1258–1266. [Google Scholar] [CrossRef]
- Ho, T.W.; Olesen, J.; Dodick, D.W.; Kost, J.; Lines, C.; Ferrari, M.D. Antimigraine efficacy of telcagepant based on patient’s historical triptan response. Headache 2011, 51, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, J.; Goadsby, P.J. New agents for acute treatment of migraine: CGRP receptor antagonists, iNOS inhibitors. Curr. Treat. Options Neurol. 2011, 14, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Olesen, J.; Diener, H.-C.; Husstedt, I.W.; Goadsby, P.J.; Hall, D.; Meier, U.; Pollentier, S.; Lesko, L.M. Calcitonin gene–related peptide receptor antagonist bibn 4096 BS for the acute treatment of migraine. N. Engl. J. Med. 2004, 350, 1104–1110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silberstein, S.D. Emerging target-based paradigms to prevent and treat migraine. Clin. Pharmacol. Ther. 2013, 93, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Voss, T.; Lipton, R.B.; Dodick, D.W.; Dupre, N.; Ge, J.Y.; Bachman, R.; Assaid, C.; Aurora, S.K.; Michelson, D.A. phase IIb randomized, double-blind, placebo-controlled trial of ubrogepant for the acute treatment of mi-graine. Cephalalgia 2016, 36, 887–898. [Google Scholar] [CrossRef]
- Diener, H.-C.; Barbanti, P.; Dahlöf, C.; Reuter, U.; Habeck, J.; Podhorna, J. BI 44370 TA, an oral CGRP antagonist for the treatment of acute migraine attacks: Results from a phase II study. Cephalalgia 2011, 31, 573–584. [Google Scholar] [CrossRef]
- Marcus, R.; Goadsby, P.J.; Dodick, D.; Stock, D.; Manos, G.; Fischer, T.Z. BMS-927711 for the acute treatment of migraine: A double-blind, randomized, placebo controlled, dose-ranging trial. Cephalalgia 2014, 34, 114–125. [Google Scholar] [CrossRef]
- Stauffer, V.L.; Dodick, D.W.; Zhang, Q.; Carter, J.N.; Ailani, J.; Conley, R.R. Evaluation of galcanezumab for the prevention of episodic migraine: The EVOLVE-1 randomized clinical trial. JAMA Neurol. 2018, 75, 1080–1088. [Google Scholar] [CrossRef]
- Skljarevski, V.; Matharu, M.; Millen, A.B.; Ossipov, M.H.; Kim, B.-K.; Yang, J.Y. Efficacy and safety of galcanezumab for the prevention of episodic migraine: Results of the EVOLVE-2 Phase 3 randomized controlled clinical trial. Cephalalgia 2018, 38, 1442–1454. [Google Scholar] [CrossRef]
- Dodick, D.W.; Goadsby, P.J.; Spierings, E.L.H.; Scherer, J.C.; Sweeney, S.P.; Grayzel, D.S. Safety and efficacy of LY2951742, a monoclonal antibody to calcitonin gene-related peptide, for the prevention of migraine: A phase 2, randomised, double-blind, placebo-controlled study. Lancet Neurol. 2014, 13, 885–892. [Google Scholar] [CrossRef]
- Detke, H.C.; Goadsby, P.J.; Wang, S.; Friedman, D.I.; Selzler, K.J.; Aurora, S.K. Galcanezumab in chronic migraine: The randomized, double-blind, placebo-controlled REGAIN study. Neurology 2018, 91, e2211–e2221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bigal, E.M.; Dodick, D.W.; Rapoport, A.M.; Silberstein, S.D.; Ma, Y.; Yang, R.; Loupe, P.S.; Burstein, R.; Newman, L.C.; Lipton, R.B. Safety, tolerability, and efficacy of TEV-48125 for preventive treatment of high-frequency episodic migraine: A multicentre, randomised, double-blind, placebo-controlled, phase 2b study. Lancet Neurol. 2015, 14, 1081–1090. [Google Scholar] [CrossRef]
- Dodick, D.W.; Goadsby, P.J.; Silberstein, S.D.; Lipton, R.B.; Olesen, J.; Ashina, M.; Wilks, K.; Kudrow, D.; Kroll, R.; Kohrman, B.; et al. Safety and efficacy of ALD403, an antibody to calcitonin gene-related peptide, for the prevention of frequent episodic migraine: A randomised, double-blind, placebo-controlled, exploratory phase 2 trial. Lancet Neurol. 2014, 13, 1100–1107. [Google Scholar] [CrossRef]
- Sun, H.; Dodick, D.W.; Silberstein, S.; Goadsby, P.J.; Reuter, U.; Ashina, M.; Saper, J.; Cady, R.; Chon, Y.; Dietrich, J.; et al. Safety and efficacy of AMG 334 for prevention of episodic migraine: A randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Neurol. 2016, 15, 382–390. [Google Scholar] [CrossRef]
- Tepper, S.; Ashina, M.; Reuter, U.; Brandes, J.L.; Doležil, D.; Silberstein, S.; Winner, P.; Leonardi, D.; Mikol, D.; Lenz, R. Safety and efficacy of erenumab for preventive treatment of chronic migraine: A randomised, double-blind, placebo-controlled phase 2 trial. Lancet Neurol. 2017, 16, 425–434. [Google Scholar] [CrossRef]
- Messlinger, K.; Balcziak, L.K.; Russo, A.F. Cross-talk signaling in the trigeminal ganglion: Role of neuropeptides and other mediators. J. Neural Transm. 2020, 127, 431–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dieterle, A.; Fischer, M.J.; Link, A.S.; Neuhuber, W.L.; Messlinger, K. Increase in CGRP- and nNOS-immunoreactive neurons in the rat trigeminal ganglion after infusion of an NO donor. Cephalalgia 2011, 31, 31–42. [Google Scholar] [CrossRef]
- Iyengar, S.; Johnson, K.W.; Ossipov, M.H.; Aurora, S.K. CGRP and the trigeminal system in migraine. Headache J. Head Face Pain 2019, 59, 659–681. [Google Scholar] [CrossRef] [Green Version]
- Capuano, A.; De Corato, A.; Lisi, L.; Tringali, G.; Navarra, P.; Russo, C.D. Proinflammatory-activated trigeminal satellite cells promote neuronal sensitization: Relevance for migraine pathology. Mol. Pain 2009, 5, 43. [Google Scholar] [CrossRef] [Green Version]
- De Corato, A.; Lisi, L.; Capuano, A.; Tringali, G.; Tramutola, A.; Navarra, P.; Russo, C.D. Trigeminal satellite cells express functional calcitonin gene-related peptide receptors, whose activation enhances interleukin-1beta pro-inflammatory effects. J. Neuroimmunol. 2011, 237, 39–46. [Google Scholar] [CrossRef]
- Thalakoti, S.; Patil, V.V.; Damodaram, S.; Vause, C.V.; Langford, L.E.; Freeman, S.E.; Durham, P.L. Neuron-glia signaling in trigeminal ganglion: Implications for migraine pathology. Headache 2007, 47, 1008–1023. [Google Scholar] [CrossRef] [Green Version]
- Bowen, E.J.; Schmidt, T.W.; Firm, C.S.; Russo, A.F.; Durham, P.L. Tumor necrosis factor-alpha stimulation of calcitonin gene-related peptide expression and secretion from rat trigeminal ganglion neurons. J. Neurochem. 2005, 96, 65–77. [Google Scholar] [CrossRef] [Green Version]
- Kwok, Y.H.; Swift, J.E.; Gazerani, P.; Rolan, P. A double-blind, randomized, placebo-controlled pilot trial to determine the efficacy and safety of ibudilast, a potential glial attenuator, in chronic migraine. J. Pain Res. 2016, 9, 899–907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durham, Z.L.; Hawkins, J.L.; Durham, P.L. Tumor necrosis factor-Alpha stimulates cytokine expression and transient sensitization of trigeminal nociceptive neurons. Arch. Oral Biol. 2017, 75, 100–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durham, P.L. Diverse physiological roles of calcitonin gene-related peptide in migraine pathology: Modulation of neuronal-glial-immune cells to promote peripheral and central sensitization. Curr. Pain Headache Rep. 2016, 20, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cady, R.J.; Glenn, J.R.; Smith, K.M.; Durham, P.L. Calcitonin gene-related peptide promotes cellular changes in trigeminal neurons and glia implicated in peripheral and central sensitization. Mol. Pain. 2011, 7. [Google Scholar] [CrossRef] [Green Version]
- Cornelison, L.E.; Hawkins, J.L.; Durham, P.L. Elevated levels of calcitonin gene-related peptide in upper spinal cord promotes sensitization of primary trigeminal nociceptive neurons. Neuroscience 2016, 339, 491–501. [Google Scholar] [CrossRef] [Green Version]
- Weir, A.G.; Cader, M.Z. New directions in migraine. BMC Med. 2011, 9, 116. [Google Scholar] [CrossRef] [Green Version]
- Spraya, D.C.; Hananib, M. Gap junctions, pannexins and pain. Neurosci. Lett. 2019, 695, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Hansen, J.M.; Thomsen, L.L.; Olesen, J.; Ashina, M. Calcitonin gene-related peptide does not cause the familial hemiplegic migraine phenotype. Neurology 2008, 71, 841–847. [Google Scholar] [CrossRef]
- Ptáček, L.J. Channelopathies: Ion channel disorders of muscle as a paradigm for paroxysmal disorders of the nervous system. Neuromuscul. Disord. 1997, 7, 250–255. [Google Scholar] [CrossRef]
- Ducros, A.; Denier, C.; Joutel, A.; Cecillon, M.; Lescoat, C.; Vahedi, K.; Darcel, F.; Vicaut, E.; Bousser, M.-G.; Tournier-Lasserve, E. The clinical spectrum of familial hemiplegic migraine associated with mutations in a neuronal calcium channel. N. Engl. J. Med. 2001, 345, 17–24. [Google Scholar] [CrossRef]
- Goadsby, P.J.; Holland, P.R.; Martins-Oliveira, M.; Hoffmann, J.; Schankin, C.; Akerman, S. Pathophysiology of migraine: A disorder of sensory processing. Physiol. Rev. 2017, 97, 553–622. [Google Scholar] [CrossRef]
- Ophoff, A.R.; Terwindt, G.M.; Haan, J.; Lindhout, D.; van Ommen, G.-J.B.; Hofker, M.H.; Ferrari, M.D.; Frants, R.R.; Vergouwe, M.N.; van Eijk, R.; et al. Familial hemiplegic migraine and episodic ataxia type-2 are caused by mutations in the CA2+ channel gene CACNL1a4. Cell 1996, 87, 543–552. [Google Scholar] [CrossRef] [Green Version]
- De Fusco, M.; Marconi, R.; Silvestri, L.; Atorino, L.; Rampoldi, L.; Morgante, L.; Ballabio, A.; Aridon, P.; Casari, G. Haploinsufficiency of ATP1A2 encoding the Na+/K+ pump α2 subunit associated with familial hemiplegic migraine type 2. Nat. Genet. 2003, 33, 192–196. [Google Scholar] [CrossRef] [PubMed]
- Dichgans, M.; Freilinger, T.; Eckstein, G.; Babini, E.; Lorenz-Depiereux, B.; Biskup, S.; Ferrari, M.D.; Herzog, J.; Maagdenberg, A.M.J.M.V.D.; Pusch, M.; et al. Mutation in the neuronal voltage-gated sodium channel SCN1A in familial hemiplegic migraine. Lancet 2005, 366, 371–377. [Google Scholar] [CrossRef]
- Leo, L.; Gherardini, L.; Barone, V.; De Fusco, M.; Pietrobon, D.; Pizzorusso, T.; Casari, G. Increased susceptibility to cortical spreading depression in the mouse model of familial hemiplegic migraine type 2. PLoS Genet. 2011, 7, e1002129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Xiao, H.; Qin, X.; Nong, Y.; Zou, D.; Wu, Y. The genetic relationship between epilepsy and hemiplegic migraine. Neuropsychiatr. Dis. Treat. 2017, 13, 1175–1179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urbano, F.J.; Pagani, M.R.; Uchitel, O.D. Calcium channels, neuromuscular synaptic transmission and neurological diseases. J. Neuroimmunol. 2008, 201-202, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Westenbroek, R.; Sakurai, T.; Elliott, E.; Hell, J.; Starr, T.; Snutch, T.; Catterall, W. Immunochemical identification and subcellular distribution of the alpha 1A subunits of brain calcium channels. J. Neurosci. 1995, 15, 6403–6418. [Google Scholar] [CrossRef] [Green Version]
- Akerman, S.; Williamson, D.J.; Goadsby, P.J. Voltage-dependent calcium channels are involved in neurogenic dural vasodilatation via a presynaptic transmitter release mechanism: Voltage-dependent calcium channels. Br. J. Pharmacol. 2003, 140, 558–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pietrobon, D. Function and dysfunction of synaptic calcium channels: Insights from mouse models. Curr. Opin. Neurobiol. 2005, 15, 257–265. [Google Scholar] [CrossRef]
- Santafé, M.M.; Salon, I.; Garcia, N.; Lanuza, M.A.; Uchitel, O.D.; Tomàs, J. Modulation of ACh release by presynaptic muscarinic autoreceptors in the neuromuscular junction of the newborn and adult rat: Muscarinic autoreceptors in neuromuscular transmission. Eur. J. Neurosci. 2003, 17, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Santafé, M.M.; Lanuza, M.A.; Garcia, N.; Tomàs, J. Muscarinic autoreceptors modulate transmitter release through protein kinase C and protein kinase A in the rat motor nerve terminal. Eur. J. Neurosci. 2006, 23, 2048–2056. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.Y.; Yong, T.F.; Yu, C.Y.; Liang, M.C.; Pletnikova, O.; Troncoso, J.C.; Burgunder, J.-M.; Soong, T.W. Age and gender-dependent alternative splicing of P/Q-type calcium channel EF-hand. Neuroscience 2007, 145, 1026–1036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Travaglini, L.; Nardella, M.; Bellacchio, E.; D’Amico, A.; Capuano, A.; Frusciante, R.; Di Capua, M.; Cusmai, R.; Barresi, S.; Morlino, S.; et al. Missense mutations of CACNA1A are a frequent cause of autosomal dominant nonprogressive congenital ataxia. Eur. J. Paediatr. Neurol. 2017, 21, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Kors, E.E.; Terwindt, G.M.; Vermeulen, F.L.M.G.; Fitzsimons, R.B.; Jardine, P.E.; Heywood, P.; Love, S.; Maagdenberg, A.M.J.M.V.D.; Haan, J.; Frants, R.R.; et al. Delayed cerebral edema and fatal coma after minor head trauma: Role of the CACNA1A calcium channel subunit gene and relationship with familial hemiplegic migraine. Ann. Neurol. 2001, 49, 753–760. [Google Scholar] [CrossRef] [PubMed]
- Chan, Y.-C.; Burgunder, J.-M.; Wilder-Smith, E.; Chew, S.-E.; Lam-Mok-Sing, K.M.; Sharma, V.; Ong, B.K. Electroencephalographic changes and seizures in familial hemiplegic migraine patients with the CACNA1A gene S218L mutation. J. Clin. Neurosci. 2008, 15, 891–894. [Google Scholar] [CrossRef]
- Freilinger, T.; Ackl, N.; Ebert, A.; Schmidt, C.; Rautenstrauss, B.; Dichgans, M.; Danek, A. A novel mutation in CACNA1A associated with hemiplegic migraine, cerebellar dysfunction and late-onset cognitive decline. J. Neurol. Sci. 2011, 300, 160–163. [Google Scholar] [CrossRef] [PubMed]
- Battistini, S.; Stenirri, S.; Piatti, M.; Gelfi, C.; Righetti, P.G.; Rocchi, R.; Giannini, F.; Battistini, N.; Guazzi, G.C.; Ferrari, M.; et al. A new CACNA1A gene mutation in acetazolamide-responsive familial hemiplegic migraine and ataxia. Neurology 1999, 53, 38. [Google Scholar] [CrossRef] [PubMed]
- Terwindt, G.; Kors, E.; Haan, J.; Vermeulen, F.; Maagdenberg, A.V.D.; Frants, R.; Ferrari, M. Mutation analysis of the CACNA1A calcium channel subunit gene in 27 patients with sporadic hemiplegic migraine. Arch. Neurol. 2002, 59, 1016–1018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alonso, I.; Barros, J.; Tuna, A.; Coelho, J.; Sequeiros, J.; Silveira, I.; Coutinho, P. Phenotypes of spinocerebellar ataxia type 6 and familial hemiplegic migraine caused by a unique CACNA1a missense mutation in patients from a large family. Arch. Neurol. 2003, 60, 610–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Vries, B.; Freilinger, T.; Vanmolkot, K.R.; Koenderink, J.B.; Stam, A.H.; Terwindt, G.M.; Babini, E.; van den Boogerd, E.H.; van den Heuvel, J.J.; Frants, R.R.; et al. Systematic analysis of three FHM genes in 39 sporadic patients with hemiplegic migraine. Neurology 2007, 69, 2170–2176. [Google Scholar] [CrossRef] [Green Version]
- Friend, K.L.; Crimmins, D.; Phan, T.G.; Sue, C.M.; Colley, A.; Fung, V.S.; Morris, J.G.; Sutherland, G.R.; Richards, R.I. Detection of a novel missense mutation and second recurrent mutation in the CACNA1A gene in individuals with EA-2 and FHM. Hum. Genet. 1999, 105, 261–265. [Google Scholar] [CrossRef] [PubMed]
- Ducros, A.; Denier, C.; Hirsch, E.; Chedru, F.; Bisgård, C.; Lucotte, G.; Després, P.; Billard, C.; Barthez, M.; Ponsot, G.; et al. Recurrence of the T666M calcium channel CACNA1a gene mutation in familial hemiplegic migraine with progressive cerebellar ataxia. Am. J. Hum. Genet. 1999, 64, 89–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kors, E.E.; Haan, J.; Giffin, N.J.; Pazdera, L.; Schnittger, C.; Lennox, G.G.; Terwindt, G.M.; Vermeulen, F.L.; Van den Maagdenberg, A.M.; Frants, R.R.; et al. Expanding the phenotypic spectrum of the CACNA1A gene T666M mutation: A description of 5 families with familial hemiplegic migraine. Arch. Neurol. 2003, 60, 684–688. [Google Scholar] [CrossRef] [Green Version]
- Stam, A.; Vanmolkot, K.; Kremer, H.; Gärtner, J.; Brown, J.; Leshinsky-Silver, E.; Gilad, R.; Kors, E.; Frankhuizen, W.; Ginjaar, H.; et al. CACNA1A R1347Q: A frequent recurrent mutation in hemiplegic migraine. Clin. Genet. 2008, 74, 481–485. [Google Scholar] [CrossRef] [PubMed]
- Vahedi, K.; Denier, C.; Ducros, A.; Bousson, V.; Levy, C.; Chabriat, H.; Haguenau, M.; Tournier-Lasserve, E.; Bousser, M.G. CACNA1A gene de novo mutation causing hemiplegic migraine, coma, and cerebellar atrophy. Neurology 2000, 55, 1040–1042. [Google Scholar] [CrossRef]
- Carrera, P.; Piatti, M.; Stenirri, S.; Grimaldi, L.M.E.; Marchioni, E.; Curcio, M.; Righetti, P.G.; Ferrari, M.; Gelfi, C. Genetic heterogeneity in Italian families with familial hemiplegic migraine. Neurology 1999, 53, 26. [Google Scholar] [CrossRef]
- Kors, E.E.; Melberg, A.; Vanmolkot, K.R.J.; Kumlien, E.; Haan, J.; Raininko, R.; Flink, R.; Ginjaar, H.B.; Frants, R.R.; Ferrari, M.D.; et al. Childhood epilepsy, familial hemiplegic migraine, cerebellar ataxia, and a new CACNA1A mutation. Neurology 2004, 63, 1136–1137. [Google Scholar] [CrossRef]
- Labrum, R.W.; Rajakulendran, S.; Graves, T.D.; Eunson, L.H.; Bevan, R.; Sweeney, M.G.; Hammans, S.R.; Tubridy, N.; Britton, T.; Carr, L.J.; et al. Large scale calcium channel gene rearrangements in episodic ataxia and hemiplegic migraine: Implications for diagnostic testing. J. Med Genet. 2009, 46, 786–791. [Google Scholar] [CrossRef] [Green Version]
- Kraus, R.L.; Sinnegger, M.J.; Glossmann, H.; Hering, S.; Striessnig, J. Familial hemiplegic migraine mutations change α 1A Ca 2+ Channel Kinetics. J. Biol. Chem. 1998, 273, 5586–5590. [Google Scholar] [CrossRef] [Green Version]
- Hans, M.; Luvisetto, S.; Williams, M.E.; Spagnolo, M.; Urrutia, A.; Tottene, A.; Brust, P.F.; Johnson, E.C.; Harpold, M.M.; Stauderman, K.A.; et al. Functional consequences of mutations in the human α 1a calcium channel subunit linked to familial hemiplegic migraine. J. Neurosci. 1999, 19, 1610–1619. [Google Scholar] [CrossRef] [Green Version]
- Stenirri, S.; Carrera, P.; Kraus, R.L.; Sinnegger, M.J.; Koschak, A.; Glossmann, H.; Striessnig, J. Three new familial hemiplegic migraine mutants affect P/Q-type CA2+ channel kinetics. J. Biol. Chem. 2000, 275, 9239–9243. [Google Scholar] [CrossRef] [Green Version]
- Tottene, A.; Fellin, T.; Pagnutti, S.; Luvisetto, S.; Striessnig, J.; Fletcher, C.; Pietrobon, D. Familial hemiplegic mi-graine mutations increase Ca2+ influx through single human CaV2.1 channels and decrease maximal CaV2.1 current density in neurons. Proc. Natl. Acad. Sci. USA 2002, 99, 13284–13289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melliti, K.; Grabner, M.; Seabrook, G.R. The familial hemiplegic migraine mutation R192q reduces G-protein-mediated inhibition of p/q-type (Ca v 2.1) calcium channels expressed in human embryonic kidney cells. J. Physiol. 2003, 546, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Müllner, C.; Broos, L.A.; van den Maagdenberg, A.M.; Striessnig, J. Familial hemiplegic migraine type 1 mutations k1336e, w1684r, and v1696i alter CA V 2.1 CA 2+ channel gating: Evidence for β-subunit isoform-specific effects. J. Biol. Chem. 2004, 279, 51844–51850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tottene, A.; Pivotto, F.; Fellin, T.; Cesetti, T.; van den Maagdenberg, A.M.; Pietrobon, D. Specific kinetic alterations of human CA V 2.1 calcium channels produced by mutation s218l causing familial hemiplegic migraine and delayed cerebral edema and coma after minor head trauma. J. Biol. Chem. 2005, 280, 17678–17686. [Google Scholar] [CrossRef] [Green Version]
- Barrett, C.F.; Cao, Y.-Q.; Tsien, R.W. Gating deficiency in a familial hemiplegic migraine type 1 mutant P/Q-type calcium channel. J. Biol. Chem. 2005, 280, 24064–24071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, N.; Sandoval, A.; Felix, R.; Van den Maagdenberg, A.; De Waard, M. The S218L familial hemiplegic mi-graine mutation promotes deinhibition of Cav2.1 calcium channels during direct G-protein regulation. Pflüg Arch. 2008, 457, 315–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, P.J.; Garcia, E.; David, L.S.; Mulatz, K.J.; Spacey, S.D.; Snutch, T.P. Ca V 2.1 P/Q-type calcium channel alter-native splicing affects the functional impact of familial hemiplegic migraine mutations: Implications for calcium channelopathies. Channels 2009, 3, 110–121. [Google Scholar] [CrossRef] [Green Version]
- Eikermann-Haerter, K.; Dileköz, E.; Kudo, C.; Savitz, S.I.; Waeber, C.; Baum, M.J.; Ferrari, M.D.; van den Maagden-berg, A.M.; Moskowitz, M.A.; Ayata, C. Genetic and hormonal factors modulate spreading depression and tran-sient hemiparesis in mouse models of familial hemiplegic migraine type 1. J. Clin. Invest. 2009, 119, 99–109. [Google Scholar]
- Serra, S.A.; Fernàndez-Castillo, N.; Macaya, A.; Cormand, B.; Valverde, M.A.; Fernández-Fernández, J.M. The hem-iplegic migraine-associated Y1245C mutation in CACNA1A results in a gain of channel function due to its effect on the voltage sensor and G-protein-mediated inhibition. Pflüg Arch. 2009, 458, 489–502. [Google Scholar] [CrossRef] [PubMed]
- Khennouf, L.; Gesslein, B.; Lind, B.L.; van den Maagdenberg, A.M.; Lauritzen, M. Activity-dependent calcium, oxygen, and vascular responses in a mouse model of familial hemiplegic migraine type 1: Calcium, oxygen and vascular responses in migraine. Ann. Neurol. 2016, 80, 219–232. [Google Scholar] [CrossRef]
- Cao, Y.Q.; Piedras-Rentería, E.S.; Smith, G.B.; Chen, G.; Harata, N.C.; Tsien, R.W. Presynaptic Ca2+ Channels Com-pete for Channel Type-Preferring Slots in Altered Neurotransmission Arising from Ca2+ Channelopathy. Neuron 2004, 43, 387–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Y.-Q.; Tsien, R.W. Effects of familial hemiplegic migraine type 1 mutations on neuronal P/Q-type Ca2+ channel activity and inhibitory synaptic transmission. Proc. Natl. Acad. Sci. USA 2005, 102, 2590–2595. [Google Scholar] [CrossRef] [Green Version]
- Tottene, A.; Conti, R.; Fabbro, A.; Vecchia, D.; Shapovalova, M.; Santello, M.; Maagdenberg, A.M.V.D.; Ferrari, M.D.; Pietrobon, D. Enhanced excitatory transmission at cortical synapses as the basis for facilitated spreading depression in CAV2.1 knockin migraine mice. Neuron 2009, 61, 762–773. [Google Scholar] [CrossRef] [PubMed]
- Haan, J.; Kors, E.E.; Vanmolkot, K.R.; van den Maagdenberg, A.M.; Frants, R.R.; Ferrari, M.D. Migraine genetics: An up-date. Curr. Pain Headache Rep. 2005, 9, 213–220. [Google Scholar] [CrossRef]
- van den Maagdenberg, A.M.; Pietrobon, D.; Pizzorusso, T.; Kaja, S.; Broos, L.A.; Cesetti, T.; van de Ven, R.C.; Tottene, A.; van der Kaa, J. A cacna1a knockin migraine mouse model with increased susceptibil-ity to cortical spreading depression. Neuron 2004, 41, 701–710. [Google Scholar] [CrossRef]
- Smitherman, T.A.; Burch, R.; Sheikh, H.; Loder, E. The prevalence, impact, and treatment of migraine and SE-vere headaches in the United States: A review of statistics from national surveillance studies. Headache 2013, 53, 427–436. [Google Scholar] [CrossRef]
- Antonaci, F.; Ghiotto, N.; Wu, S.; Pucci, E.; Costa, A. Recent advances in migraine therapy. SpringerPlus 2016, 5, 637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diener, H.-C.; Dodick, D.W.; Goadsby, P.J.; Lipton, R.B.; Olesen, J.; Silberstein, S.D. Chronic migraine—classification, characteristics and treatment. Nat. Rev. Neurol. 2012, 8, 162–171. [Google Scholar] [CrossRef]
- Burton, W.N.; Landy, S.H.; Downs, K.E.; Runken, M.C. The impact of migraine and the effect of migraine treat-ment on workplace productivity in the united states and suggestions for future research. Mayo Clin. Proc. 2009, 84, 436–445. [Google Scholar] [CrossRef] [Green Version]
- Nimmrich, V.; Gross, G. P/Q-type calcium channel modulators: P/Q-type calcium channel blockers. Br. J. Pharmacol. 2012, 167, 741–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beswick, P. Progress in the discovery of Ca channel blockers for the treatment of pain. In Comprehensive Medicinal Chemistry III; Elsevier BV: Amsterdam, The Netherlands, 2017; pp. 65–130. [Google Scholar]
- Dussor, G. ASICs as therapeutic targets for migraine. Neuropharmacology 2015, 94, 64–71. [Google Scholar] [CrossRef] [Green Version]
- Mazzuca, M.; Heurteaux, C.; Alloui, A.; Diochot, S.; Baron, A.; Voilley, N.; Blondeau, N.; Escoubas, P.; Gélot, A.; Cupo, A.; et al. A tarantula peptide against pain via ASIC1a channels and opioid mechanisms. Nat. Neurosci. 2007, 10, 943–945. [Google Scholar] [CrossRef] [PubMed]
- Sarchielli, P.; Alberti, A.; Floridi, A.; Gallai, V. Levels of nerve growth factor in cerebrospinal fluid of chronic daily headache patients. Neurology 2001, 57, 132–134. [Google Scholar] [CrossRef]
- Kowalska, M.; Prendecki, M.; Kozubski, W.; Lianeri, M.; Dorszewska, J. Molecular factors in migraine. Oncotarget 2016, 7, 50708–50718. [Google Scholar] [CrossRef] [Green Version]
- Holland, P.R.; Akerman, S.; Andreou, A.P.; Karsan, N.; Wemmie, J.A.; Goadsby, P.J. Acid-sensing ion channel 1: A novel therapeutic target for migraine with aura. Ann. Neurol. 2012, 72, 559–563. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Mutation | Location | Population | Phenotypic Spectrum | References |
|---|---|---|---|---|
| R192Q rs121908211 | exon 4 | Italian family | No data | Ophoff et al. [77] |
| R195K rs121908222 | exon 4 | French family | FHM without cerebellar signs | Ducros et al. [75] |
| S218L rs121908225 | exon 5 | British and Australian families, Malaysian family | Minor head trauma–triggered delayed severe cerebral edema and coma; childhood seizures | Kors et al. [90] Chan et al. [91] |
| V581M | exon 13 | German family | FHM with cerebellar dysfunction and late-onset cognitive decline | Freilinger et al. [92] |
| R583Q rs121908217 | exon 13 | Italian family, Dutch patient, Portuguese family, Dutch patient | FHM with consciousness and fever lasting several days, late-onset cerebellar ataxia and cerebellar atrophy; symptoms triggered by minor head trauma; sporadic hemiplegic migraine without cerebellar signs, age at onset 13 years | Battistini et al. [93] Terwindt et al. [94] Alonso et al. [95] de Vries et al. [96] |
| T666M rs121908212 | exon 16 | American family, Australian family, French families, Dutch patient, Dutch families | Age at onset between 2 and 22 years; FHM with progressive cerebellar ataxia; sporadic hemiplegic migraine; progressive cognitive dysfunction | Ophoff et al. [77] Friend et al. [97] Ducros et al. [98] Terwindt et al. [94] Kors et al. [99] |
| V714A rs121908213 | exon 17 | British family | Age at onset between 10 and 21 years | Ophoff et al. [77] |
| D715E rs121908218 | exon 17 | French family | FHM with progressive cerebellar ataxia | Ducros et al. [98] |
| K1336E | exon 25 | French family | FHM without cerebellar signs | Ducros et al. [75] |
| R1347Q rs121908230 | exon 25 | Dutch families | Wide clinical spectrum ranging from (trauma triggered) hemiplegic migraine with and without ataxia, loss of consciousness and epilepsy, early age at onset (usually before the age of 3) | Stam et al. [100] |
| Y1385C rs121908219 | exon 26 | French patients | FHM with cerebellar signs, coma, hyperthermia, meningeal signs, and partial seizure, | Vahedi et al. [101] Ducros et al. [75] |
| V1457L rs121908237 | exon 27 | Italian family | Mean age at onset 34 years, various degrees of aphasia congruent with the hemispheric dominance, without cerebellar ataxia or coma | Carrera et al. [102] |
| R1668W | exon 32 | French family | FHM with or without cerebellar signs | Ducros et al. [75] |
| W1684R | exon 32 | French family | FHM with cerebellar signs | Ducros et al. [75] |
| V1696I | exon 33 | French family | FHM without cerebellar signs | Ducros et al. [75] |
| I1710T rs121909326 | exon 33 | Dutch family | FHM with childhood-onset of cerebellar ataxia (SCA6), childhood complex partial and generalized tonic-clonic seizures that occurred independently of the FHM attacks | Kors et al. [103] |
| I1811L rs121908214 | exon 36 | Dutch and American families | Only one family displayed cerebellar atrophy and in that family only some members were affected | Ophoff et al. [77] |
| 18.2-kb deletion | exons 39–47 | Irish, Indian, and Danish patients | FHM with or without ataxia, no seizures, no family history | Labrum et al. [104] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kowalska, M.; Prendecki, M.; Piekut, T.; Kozubski, W.; Dorszewska, J. Migraine: Calcium Channels and Glia. Int. J. Mol. Sci. 2021, 22, 2688. https://doi.org/10.3390/ijms22052688
Kowalska M, Prendecki M, Piekut T, Kozubski W, Dorszewska J. Migraine: Calcium Channels and Glia. International Journal of Molecular Sciences. 2021; 22(5):2688. https://doi.org/10.3390/ijms22052688
Chicago/Turabian StyleKowalska, Marta, Michał Prendecki, Thomas Piekut, Wojciech Kozubski, and Jolanta Dorszewska. 2021. "Migraine: Calcium Channels and Glia" International Journal of Molecular Sciences 22, no. 5: 2688. https://doi.org/10.3390/ijms22052688