
Insights into Interactions between Interleukin-6 and Dendritic Polyglycerols


Abstract
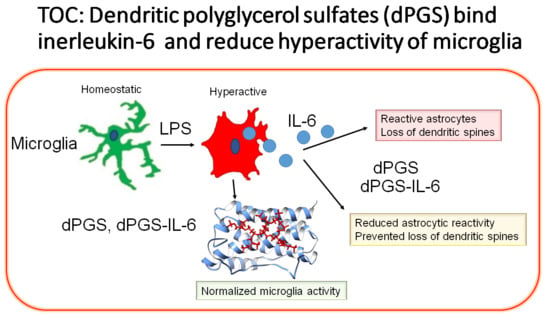
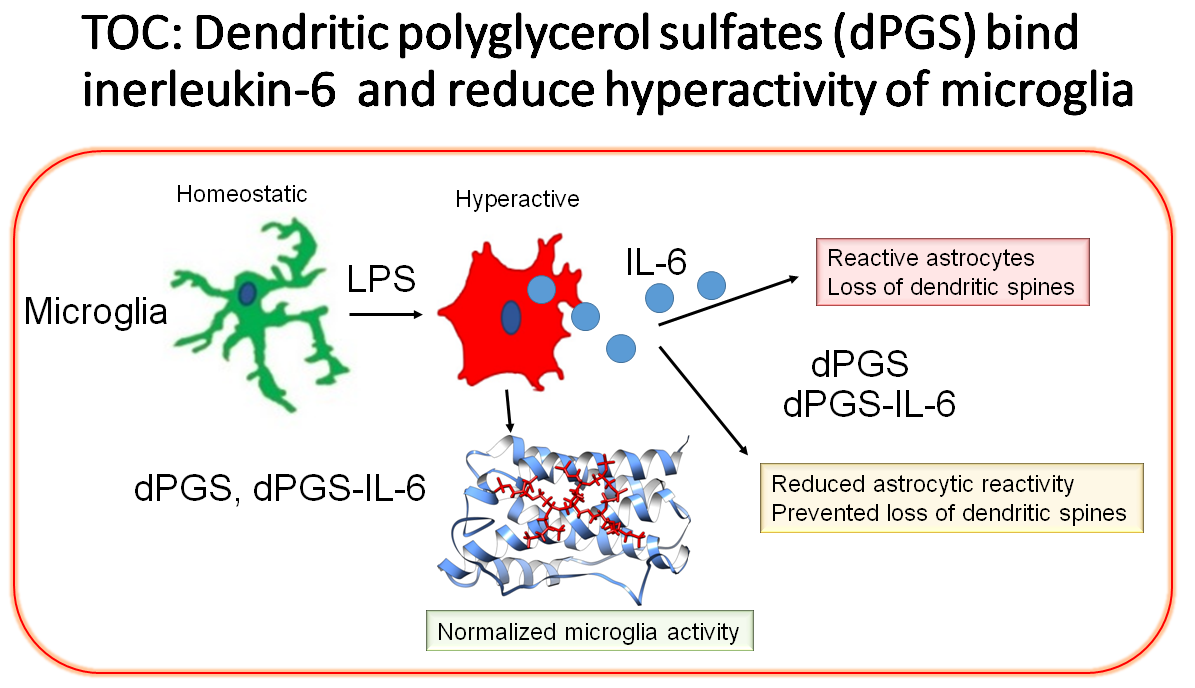
:
1. Introduction
2. Results
2.1. Molecular Modeling Aspects
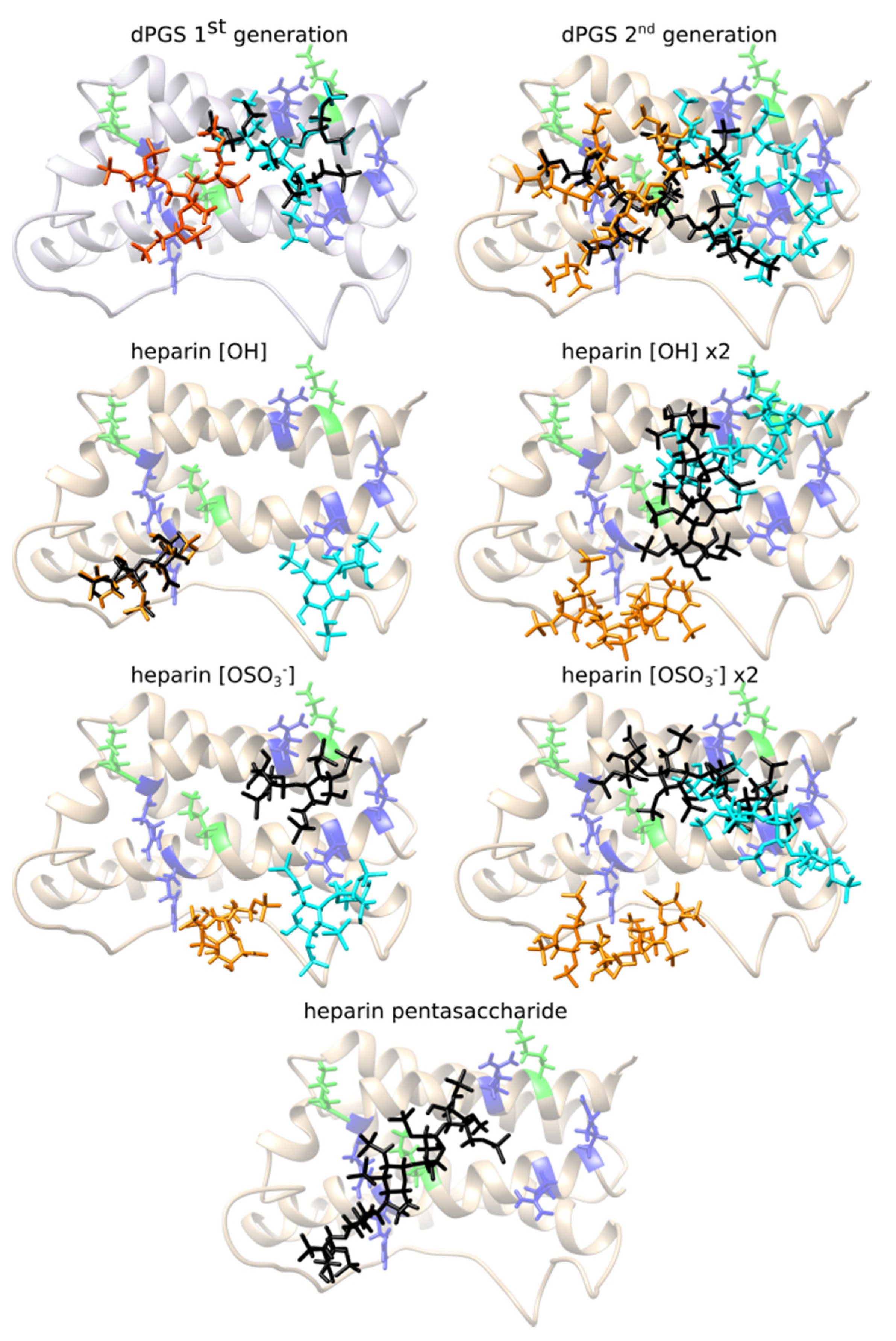
2.2. dPGS Docking to hIL-6
2.3. Heparin Docking to hIL-6
2.4. Molecular Dynamics Simulations of the dPGS and hIL-6
3. Discussion
4. Materials and Methods
4.1. Docking Simulations
4.2. Details of the Molecular Dynamics Simulations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| dPGS | Dendritic Polyglycerol Sulfates |
| hIL-6 | Human Interleukin-6 |
| GAGs | Glucosaminoglycans |
| MD | Molecular Dynamics |
References
- Garbers, C.; Heink, S.; Korn, T.; Rose-John, S. Interleukin-6: Designing specific therapeutics for a complex cytokine. Nat. Rev. Drug Discov. 2018, 17, 395–412. [Google Scholar] [CrossRef]
- Fuster, J.J.; Walsh, K. The good, the bad, and the ugly of interleukin-6 signaling. EMBO J. 2014, 33, 1425–1427. [Google Scholar] [CrossRef] [Green Version]
- Akira, S.; Taga, T.; Kishimoto, T. Interleukin-6 in biology and medicine. Adv. Immunol. 1993, 54, 1–78. [Google Scholar] [CrossRef] [PubMed]
- Somers, W.; Stahl, M.; Seehra, J.S. 1.9 Å crystal structure of interleukin 6: Implications for a novel mode of receptor dimerization and signaling. EMBO J. 1997, 16, 989–997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sumikawa, H.; Suzuki, E. Tertiary structural models of human interleukin-6 and evaluation by comparison with X-ray and NMR structures. Chem. Pharm. Bull. 1998, 46, 136–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, T.; Narazaki, M.; Kishimoto, T. IL-6 in Inflammation, Immunity, and Disease. Cold Spring Harb. Perspect. Biol. 2014, 6. [Google Scholar] [CrossRef] [PubMed]
- Kany, S.; Vollrath, J.T.; Relja, B. Cytokines in Inflammatory Disease. Int. J. Mol. Sci. 2019, 20, 6008. [Google Scholar] [CrossRef] [Green Version]
- Ledeboer, A.; Brevé, J.J.; Poole, S.; Tilders, F.J.; Van Dam, A.M. Interleukin-10, interleukin-4, and transforming growth factor-beta differentially regulate lipopolysaccharide-induced production of pro-inflammatory cytokines and nitric oxide in co-cultures of rat astroglial and microglial cells. Glia 2000, 30, 134–142. [Google Scholar] [CrossRef]
- Alonzi, T.; Fattori, E.; Lazzaro, D.; Costa, P.; Probert, L.; Kollias, G.; De Benedetti, F.; Poli, V.; Ciliberto, G. Interleukin 6 Is Required for the Development of Collagen-induced Arthritis. J. Exp. Med. 1998, 187, 461–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohshima, S.; Saeki, Y.; Mima, T.; Sasai, M.; Nishioka, K.; Nomura, S.; Kopf, M.; Katada, Y.; Tanaka, T.; Suemura, M.; et al. Interleukin 6 plays a key role in the development of antigen-induced arthritis. Proc. Natl. Acad. Sci. USA 1998, 95, 8222–8226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Screpanti, I.; Musiani, P.; Bellavia, D.; Cappelletti, M.; Aiello, F.B.; Maroder, M.; Frati, L.; Modesti, A.; Gulino, A.; Poli, V. Inactivation of the IL-6 gene prevents development of multicentric Castleman’s disease in C/EBP beta-deficient mice. J. Exp. Med. 1996, 184, 1561–1566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomalia, D.A.; Naylor, A.M.; Goddard III, W.A. Starburst Dendrimers: Molecular-Level Control of Size, Shape, Surface Chemistry, Topology, and Flexibility from Atoms to Macroscopic Matter. Angew. Chem. Int. Ed. Engl. 1990, 29, 138–175. [Google Scholar] [CrossRef]
- Tomalia, D.A.; Fréchet, J.M.J. Discovery of dendrimers and dendritic polymers: A brief historical perspective. J. Polym. Sci. Part A Polym. Chem. 2002, 40, 2719–2728. [Google Scholar] [CrossRef]
- Fruchon, S.; Poupot, R. Pro-Inflammatory Versus Anti-Inflammatory Effects of Dendrimers: The Two Faces of Immuno-Modulatory Nanoparticles. Nanomaterials 2017, 7, 251. [Google Scholar] [CrossRef] [PubMed]
- Maysinger, D.; Gröger, D.; Lake, A.; Licha, K.; Weinhart, M.; Chang, P.K.Y.; Mulvey, R.; Haag, R.; McKinney, R.A. Dendritic Polyglycerol Sulfate Inhibits Microglial Activation and Reduces Hippocampal CA1 Dendritic Spine Morphology Deficits. Biomacromolecules 2015, 16, 3073–3082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayder, M.; Fruchon, S.; Fournié, J.-J.; Poupot, M.; Poupot, R. Anti-inflammatory properties of dendrimers per se. Sci. World J. 2011, 11, 1367–1382. [Google Scholar] [CrossRef]
- Türk, H.; Haag, R.; Alban, S. Dendritic Polyglycerol Sulfates as New Heparin Analogues and Potent Inhibitors of the Complement System. Bioconjugate Chem. 2004, 15, 162–167. [Google Scholar] [CrossRef]
- Reimann, S.; Gröger, D.; Kühne, C.; Riese, S.B.; Dernedde, J.; Haag, R. Shell Cleavable Dendritic Polyglycerol Sulfates Show High Anti-Inflammatory Properties by Inhibiting L-Selectin Binding and Complement Activation. Adv. Healthc. Mater. 2015, 4, 2154–2162. [Google Scholar] [CrossRef] [PubMed]
- Maysinger, D.; Ji, J.; Moquin, A.; Hossain, S.; Hancock, M.A.; Zhang, I.; Chang, P.K.Y.; Rigby, M.; Anthonisen, M.; Grütter, P.; et al. Dendritic Polyglycerol Sulfates in the Prevention of Synaptic Loss and Mechanism of Action on Glia. ACS Chem. Neurosci. 2018, 9, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Maysinger, D.; Lalancette-Hébert, M.; Ji, J.; Jabbour, K.; Dernedde, J.; Silberreis, K.; Haag, R.; Kriz, J. Dendritic polyglycerols are modulators of microglia-astrocyte crosstalk. Future Neurol. 2019, 14, FNL31. [Google Scholar] [CrossRef]
- Li, J.P.; Spillmann, D. Heparan sulfate proteoglycans as multifunctional cell regulators: Cell surface receptors. Methods Mol. Biol. (CliftonN.J.) 2012, 836, 239–255. [Google Scholar] [CrossRef]
- Arungundram, S.; Al-Mafraji, K.; Asong, J.; Leach, F.E.; Amster, I.J.; Venot, A.; Turnbull, J.E.; Boons, G.-J. Modular Synthesis of Heparan Sulfate Oligosaccharides for Structure−Activity Relationship Studies. J. Am. Chem. Soc. 2009, 131, 17394–17405. [Google Scholar] [CrossRef] [Green Version]
- Liang, Y.; Kiick, K.L. Heparin-functionalized polymeric biomaterials in tissue engineering and drug delivery applications. Acta Biomater. 2014, 10, 1588–1600. [Google Scholar] [CrossRef] [Green Version]
- Hileman, R.E.; Fromm, J.R.; Weiler, J.M.; Linhardt, R.J. Glycosaminoglycan-protein interactions: Definition of consensus sites in glycosaminoglycan binding proteins. BioEssays 1998, 20, 156–167. [Google Scholar] [CrossRef]
- Loo, B.M.; Kreuger, J.; Jalkanen, M.; Lindahl, U.; Salmivirta, M. Binding of heparin/heparan sulfate to fibroblast growth factor receptor 4. J. Biol. Chem. 2001, 276, 16868–16876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez-Silva, M.; Borojevic, R. GM-CSF and IL-3 activities in schistosomal liver granulomas are controlled by stroma-associated heparan sulfate proteoglycans. J. Leukoc. Biol. 1996, 59, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Clarke, D.; Katoh, O.; Gibbs, R.V.; Griffiths, S.D.; Gordon, M.Y. Interaction of interleukin 7 (IL-7) with glycosaminoglycans and its biological relevance. Cytokine 1995, 7, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Salek-Ardakani, S.; Arrand, J.R.; Shaw, D.; Mackett, M. Heparin and heparan sulfate bind interleukin-10 and modulate its activity. Blood 2000, 96, 1879–1888. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, K.G.; Gillam, F.B.; Hopkins, J.J.; Jayanthi, S.; Gundampati, R.K.; Su, G.; Bear, J.; Pilkington, G.R.; Jalah, R.; Felber, B.K.; et al. Molecular mechanisms of heparin-induced modulation of human interleukin 12 bioactivity. J. Biol. Chem. 2019. [Google Scholar] [CrossRef] [PubMed]
- Ramsden, L.; Rider, C.C. Selective and differential binding of interleukin (IL)-1α, IL-1β, IL-2 and IL-6 to glycosaminoglycans. Eur. J. Immunol. 1992, 22, 3027–3031. [Google Scholar] [CrossRef] [PubMed]
- Mulloy, B.; Forster, M.J.; Jones, C.; Davies, D.B. N.m.r. and molecular-modelling studies of the solution conformation of heparin. Biochem. J. 1993, 293 Pt 3, 849–858. [Google Scholar] [CrossRef] [Green Version]
- Mummery, R.S.; Rider, C.C. Characterization of the Heparin-Binding Properties of IL-6. J. Immunol. 2000, 165, 5671–5679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikam, R.; Xu, X.; Ballauff, M.; Kanduč, M.; Dzubiella, J. Charge and hydration structure of dendritic polyelectrolytes: Molecular simulations of polyglycerol sulphate. Soft Matter 2018, 14, 4300–4310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Ran, Q.; Dey, P.; Nikam, R.; Haag, R.; Ballauff, M.; Dzubiella, J. Counterion-Release Entropy Governs the Inhibition of Serum Proteins by Polyelectrolyte Drugs. Biomacromolecules 2018, 19, 409–416. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Kollman, P.A. Free energy calculations on dimer stability of the HIV protease using molecular dynamics and a continuum solvent model11Edited by B. Honig. J. Mol. Biol. 2000, 303, 567–582. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, B.; Kollman, P.A. Binding of a diverse set of ligands to avidin and streptavidin: An accurate quantitative prediction of their relative affinities by a combination of molecular mechanics and continuum solvent models. J. Med. Chem. 2000, 43, 3786–3791. [Google Scholar] [CrossRef] [PubMed]
- Litov, L.; Petkov, P.; Rangelov, M.; Ilieva, N.; Lilkova, E.; Todorova, N.; Krachmarova, E.; Malinova, K.; Gospodinov, A.; Hristova, R.; et al. Heparin as an Anti-Inflammatory Agent. bioRxiv 2020. [Google Scholar] [CrossRef]
- Kaur, S.; Bansal, Y.; Kumar, R.; Bansal, G. A panoramic review of IL-6: Structure, pathophysiological roles and inhibitors. Bioorganic Med. Chem. 2020, 28, 115327. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.Y.; Zhang, H.X.; Mezei, M.; Cui, M. Molecular docking: A powerful approach for structure-based drug discovery. Curr. Comput. Aided Drug Des. 2011, 7, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Mosier, P.D.; Krishnasamy, C.; Kellogg, G.E.; Desai, U.R. On the Specificity of Heparin/Heparan Sulfate Binding to Proteins. Anion-Binding Sites on Antithrombin and Thrombin Are Fundamentally Different. PLoS ONE 2012, 7, e48632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminformatics 2012, 4, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jubb, H.C.; Higueruelo, A.P.; Ochoa-Montaño, B.; Pitt, W.R.; Ascher, D.B.; Blundell, T.L. Arpeggio: A Web Server for Calculating and Visualising Interatomic Interactions in Protein Structures. J. Mol. Biol. 2017, 429, 365–371. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Hornak, V.; Abel, R.; Okur, A.; Strockbine, B.; Roitberg, A.; Simmerling, C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins 2006, 65, 712–725. [Google Scholar] [CrossRef] [Green Version]
- Berendsen, H.J.C.; Grigera, J.R.; Straatsma, T.P. The missing term in effective pair potentials. J. Phys. Chem. 1987, 91, 6269–6271. [Google Scholar] [CrossRef]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A. Case, “Development and testing of a general amber force field” Journal of Computational Chemistry(2004) 25(9) 1157–1174. J. Comput. Chem. 2005, 26, 114. [Google Scholar] [CrossRef]
- Jakalian, A.; Jack, D.B.; Bayly, C.I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem. 2002, 23, 1623–1641. [Google Scholar] [CrossRef] [PubMed]
- Hess, B.; Bekker, H.; Berendsen, H.; Fraaije, J. LINCS: A Linear Constraint Solver for molecular simulations. J. Comput. Chem. 1998, 18. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- RStudio Team. RStudio: Integrated Development for R. Rstudio; PBC: Boston, MA, USA, 2020. [Google Scholar]
- Kumari, R.; Kumar, R.; Lynn, A. g_mmpbsa—A GROMACS Tool for High-Throughput MM-PBSA Calculations. J. Chem. Inf. Modeling 2014, 54, 1951–1962. [Google Scholar] [CrossRef] [PubMed]
- Baker, N.A.; Sept, D.; Joseph, S.; Holst, M.J.; McCammon, J.A. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA 2001, 98, 10037. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand/Docking Box | RKRK Site | KRRR Site | RKRK&KRRR |
|---|---|---|---|
| dPGS 1st generation 29 torsions | −5.7 kcal/mol | −6.0 kcal/mol | −6.0 kcal/mol |
| dPGS 2nd generation 53 torsions | −5.7 kcal/mol | −5.8 kcal/mol | −5.9 kcal/mol |
| Heparin disaccharide subunit [OH] 14 torsions | −6.3 kcal/mol | −6.6 kcal/mol | −6.5 kcal/mol |
| Heparin disaccharide subunit [OSO3−] 15 torsions | −5.9 kcal/mol | −6.0 kcal/mol | −5.7 kcal/mol |
| Heparin disaccharide subunit [OH] x2 28 torsions | −6.5 kcal/mol | −6.0 kcal/mol | −6.3 kcal/mol |
| Heparin disaccharide subunit [OSO3−] x2 30 torsions | −6.2 kcal/mol | −5.5 kcal/mol | −6.0 kcal/mol |
| Heparin pentasaccharide 37 torsions | / | / | −6.4 kcal/mol |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sanader Maršić, Ž.; Maysinger, D.; Bonačić-Kouteckỳ, V. Insights into Interactions between Interleukin-6 and Dendritic Polyglycerols. Int. J. Mol. Sci. 2021, 22, 2415. https://doi.org/10.3390/ijms22052415
Sanader Maršić Ž, Maysinger D, Bonačić-Kouteckỳ V. Insights into Interactions between Interleukin-6 and Dendritic Polyglycerols. International Journal of Molecular Sciences. 2021; 22(5):2415. https://doi.org/10.3390/ijms22052415
Chicago/Turabian StyleSanader Maršić, Željka, Dušica Maysinger, and Vlasta Bonačić-Kouteckỳ. 2021. "Insights into Interactions between Interleukin-6 and Dendritic Polyglycerols" International Journal of Molecular Sciences 22, no. 5: 2415. https://doi.org/10.3390/ijms22052415