
Overcompensation of CoA Trapping by Di(2-ethylhexyl) Phthalate (DEHP) Metabolites in Livers of Wistar Rats


Abstract
:1. Introduction
2. Results
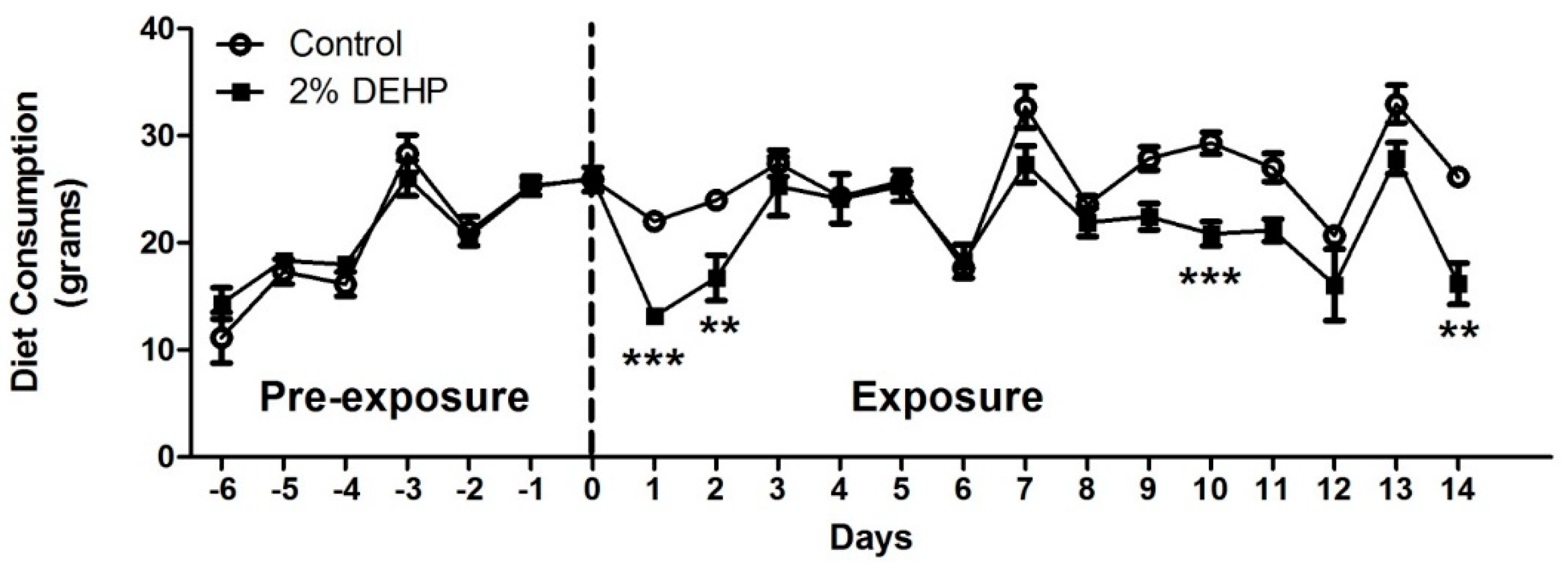
2.1. Effects of DEHP on Diet Consumption
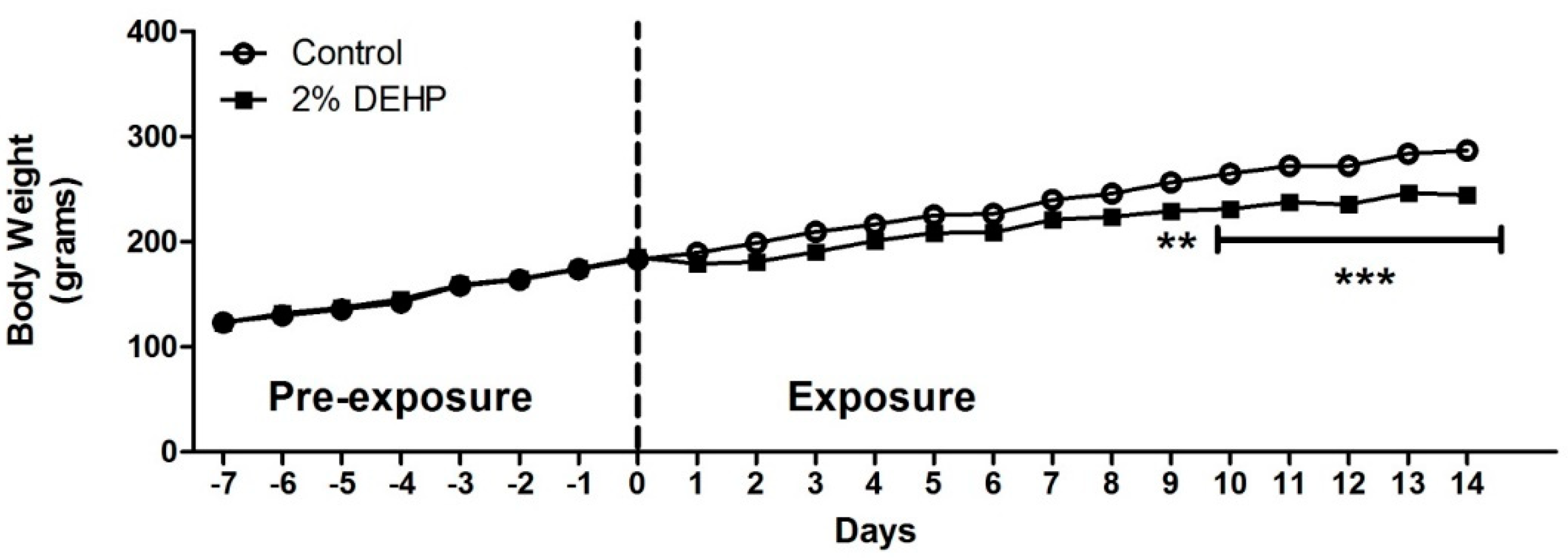
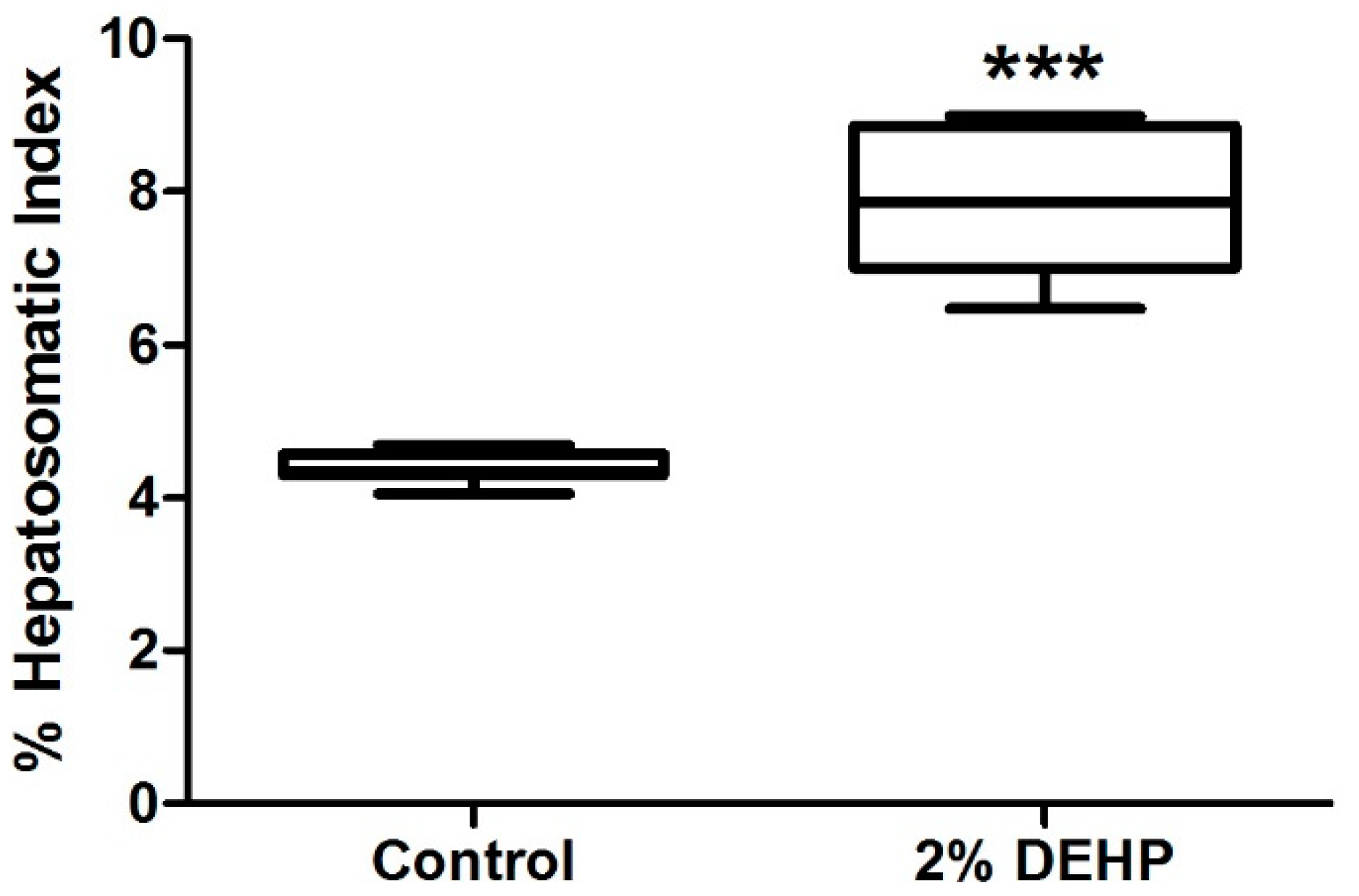
2.2. Effects of DEHP on Rodent Growth and Liver Weight
2.3. Effects of DEHP on CoA Ester Production
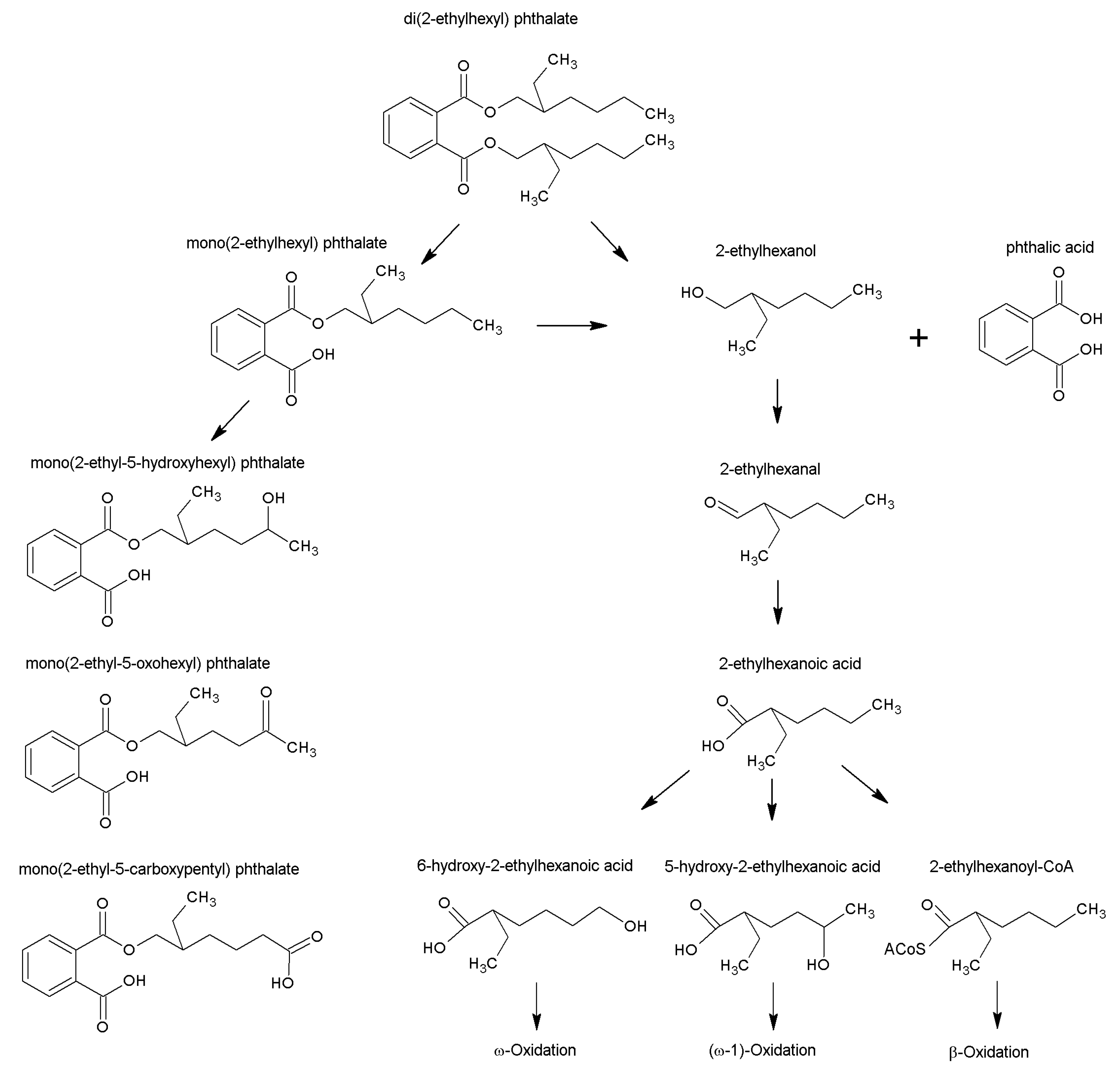
3. Discussion
3.1. DEHP Feeding Study
3.2. Physiological CoA Esters
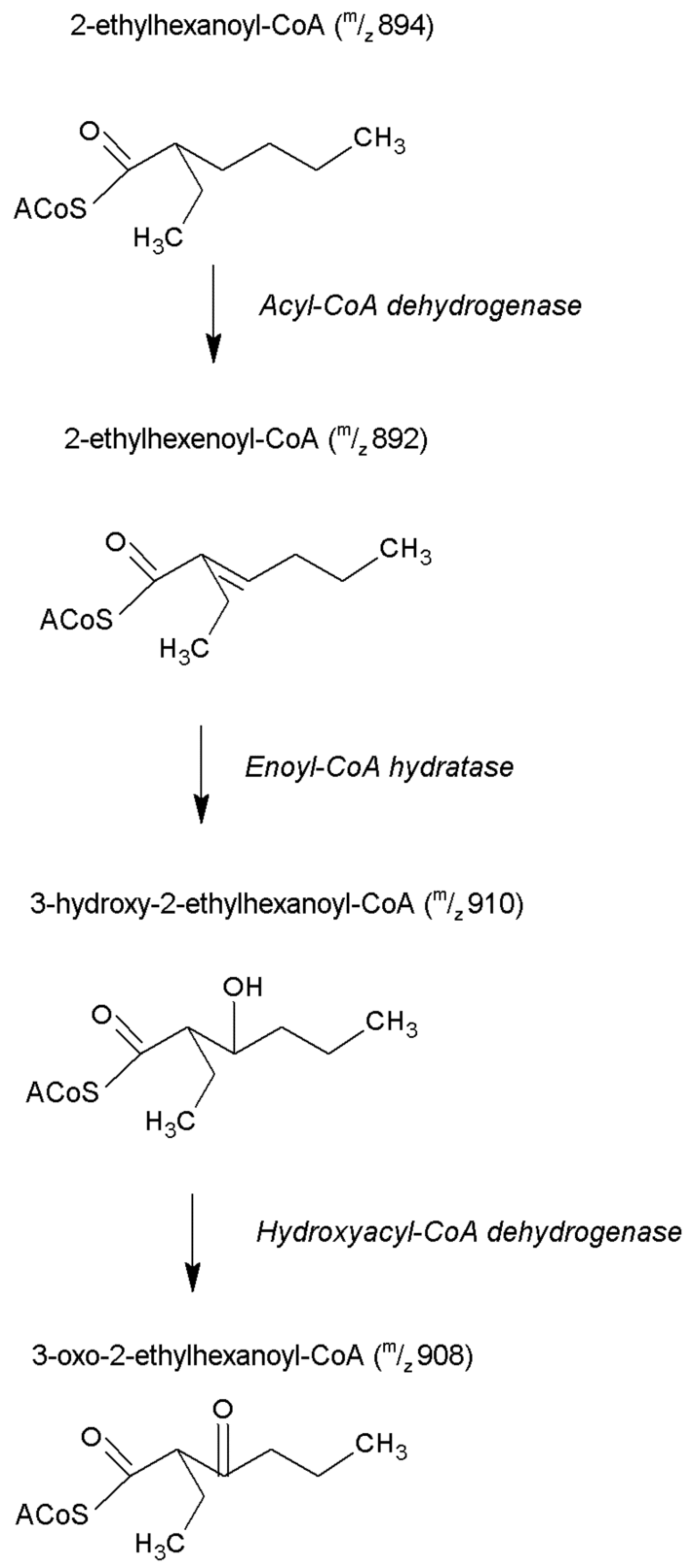
3.3. DEHP-Derived CoA Esters Which Are Isobars of Physiological CoA Esters
3.4. Unidentified CoA Esters
3.5. Constraints on the LC-MS/MS Analyses of CoA Esters
4. Materials and Methods
4.1. Materials
4.2. DEHP Diet Preparation
4.3. GCMS Quantification of DEHP in Diet
4.4. In Vivo Experiment
4.5. LC-MS/MS Quantification of Free CoA and CoA Esters
4.6. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
References
- Koch, H.M.; Preuss, R.; Angerer, J. Di(2-ethylhexyl)phthalate (DEHP): Human metabolism and internal exposure—An update and latest results. Int. J. Androl. 2006, 29, 155–165. [Google Scholar] [CrossRef]
- Hatch, E.E.; Nelson, J.W.; Stahlhut, R.W.; Webster, T.F. Association of endocrine disruptors and obesity: Perspectives from epidemiological studies. Int. J. Androl. 2010, 33, 324–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schecter, A.; Lorber, M.; Guo, Y.; Wu, Q.; Yun, S.H.; Kannan, K.; Hommel, M.; Imran, N.; Hynan, L.S.; Cheng, D.; et al. Phthalate concentrations and dietary exposure from food purchased in New York State. Environ. Health Perspect. 2013, 121, 473–494. [Google Scholar] [CrossRef] [Green Version]
- Dewalque, L.; Charlier, C.; Pirard, C. Estimated daily intake and cumulative risk assessment of phthalate diesters in a Belgian general population. Toxicol. Lett. 2014, 231, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Park, M.J. Phthalate exposure and childhood obesity. Ann. Pediatr. Endocrinol. Metab. 2014, 19, 69–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Meng, X.; Chen, L.; Li, D.; Zhao, L.; Zhao, Y.; Li, L.; Shi, H. Age and sex-specific relationships between phthalate exposures and obesity in Chinese children at puberty. PLoS ONE 2014, 9, e104852. [Google Scholar] [CrossRef]
- Grindler, N.M.; Allsworth, J.E.; Macones, G.A.; Kannan, K.; Roehl, K.A.; Cooper, A.R. Persistent organic pollutants and early menopause in U.S. women. PLoS ONE 2015, 10, e0116057. [Google Scholar] [CrossRef]
- Calafat, A.M.; McKee, R.H. Integrating biomonitoring exposure data into the risk assessment process: Phthalates [diethyl phthalate and di(2-ethylhexyl) phthalate] as a case study. Environ. Health Perspect. 2006, 114, 1783–1789. [Google Scholar] [CrossRef] [Green Version]
- Blount, B.C.; Silva, M.J.; Caudill, S.P.; Needham, L.L.; Pirkle, J.L.; Sampson, E.J.; Lucier, G.W.; Jackson, R.J.; Brock, J.W. Levels of seven urinary phthalate metabolites in a human reference population. Environ. Health Perspect. 2000, 108, 979–982. [Google Scholar] [CrossRef]
- Koch, H.M.; Drexler, H.; Angerer, J. An estimation of the daily intake of di(2-ethylhexyl)phthalate (DEHP) and other phthalates in the general population. Int. J. Hyg. Environ. Health 2003, 206, 77–83. [Google Scholar] [CrossRef]
- Silva, M.J.; Samandar, E.; Preau, J.L., Jr.; Needham, L.L.; Calafat, A.M. Urinary oxidative metabolites of di(2-ethylhexyl) phthalate in humans. Toxicology 2006, 219, 22–32. [Google Scholar] [CrossRef]
- Guo, Y.; Wu, Q.; Kannan, K. Phthalate metabolites in urine from China, and implications for human exposures. Environ. Int. 2011, 37, 893–898. [Google Scholar] [CrossRef]
- Goen, T.; Dobler, L.; Koschorreck, J.; Muller, J.; Wiesmuller, G.A.; Drexler, H.; Kolossa-Gehring, M. Trends of the internal phthalate exposure of young adults in Germany—Follow-up of a retrospective human biomonitoring study. Int. J. Hyg. Environ. Health 2011, 215, 36–45. [Google Scholar] [CrossRef]
- Song, N.R.; On, J.W.; Lee, J.; Park, J.D.; Kwon, H.J.; Yoon, H.J.; Pyo, H. Biomonitoring of urinary di(2-ethylhexyl) phthalate metabolites of mother and child pairs in South Korea. Environ. Int. 2013, 54, 65–73. [Google Scholar] [CrossRef]
- Colon, I.; Caro, D.; Bourdony, C.J.; Rosario, O. Identification of phthalate esters in the serum of young Puerto Rican girls with premature breast development. Environ. Health Perspect. 2000, 108, 895–900. [Google Scholar] [PubMed] [Green Version]
- Swan, S.H. Environmental phthalate exposure in relation to reproductive outcomes and other health endpoints in humans. Environ. Res. 2008, 108, 177–184. [Google Scholar] [CrossRef] [Green Version]
- Pan, G.; Hanaoka, T.; Yoshimura, M.; Zhang, S.; Wang, P.; Tsukino, H.; Inoue, K.; Nakazawa, H.; Tsugane, S.; Takahashi, K. Decreased serum free testosterone in workers exposed to high levels of di-n-butyl phthalate (DBP) and di-2-ethylhexyl phthalate (DEHP): A cross-sectional study in China. Environ. Health Perspect. 2006, 114, 1643–1648. [Google Scholar] [CrossRef] [Green Version]
- Joensen, U.N.; Frederiksen, H.; Blomberg, J.M.; Lauritsen, M.P.; Olesen, I.A.; Lassen, T.H.; Andersson, A.M.; Jorgensen, N. Phthalate excretion pattern and testicular function: A study of 881 healthy Danish men. Environ. Health Perspect. 2012, 120, 1397–1403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lind, P.M.; Zethelius, B.; Lind, L. Circulating levels of phthalate metabolites are associated with prevalent diabetes in the elderly. Diabetes Care 2012, 35, 1519–1524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Issemann, I.; Green, S. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature 1990, 347, 645–650. [Google Scholar] [CrossRef]
- Gottlicher, M.; Widmark, E.; Li, Q.; Gustafsson, J.A. Fatty acids activate a chimera of the clofibric acid-activated receptor and the glucocorticoid receptor. Proc. Natl. Acad. Sci. USA 1992, 89, 4653–4657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Issemann, I.; Prince, R.A.; Tugwood, J.D.; Green, S. The peroxisome proliferator-activated receptor: Retinoid X receptor heterodimer is activated by fatty acids and fibrate hypolipidaemic drugs. J. Mol. Endocrinol. 1993, 11, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Roberts, R.A. Peroxisome proliferators: Mechanisms of adverse effects in rodents and molecular basis for species differences. Arch. Toxicol. 1999, 73, 413–418. [Google Scholar] [CrossRef]
- Feige, J.N.; Gelman, L.; Rossi, D.; Zoete, V.; Metivier, R.; Tudor, C.; Anghel, S.I.; Grosdidier, A.; Lathion, C.; Engelborghs, Y.; et al. The endocrine disruptor monoethyl-hexyl-phthalate is a selective peroxisome proliferator-activated receptor gamma modulator that promotes adipogenesis. J. Biol. Chem. 2007, 282, 19152–19166. [Google Scholar] [CrossRef] [Green Version]
- Feige, J.N.; Gerber, A.; Casals-Casas, C.; Yang, Q.; Winkler, C.; Bedu, E.; Bueno, M.; Gelman, L.; Auwerx, J.; Gonzalez, F.J.; et al. The pollutant diethylhexyl phthalate regulates hepatic energy metabolism via species-specific PPARalpha-dependent mechanisms. Environ. Health Perspect. 2010, 118, 234–241. [Google Scholar] [CrossRef]
- Berger, J.; Moller, D.E. The mechanisms of action of PPARs. Annu. Rev. Med. 2002, 53, 409–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rakhshandehroo, M.; Knoch, B.; Muller, M.; Kersten, S. Peroxisome proliferator-activated receptor alpha target genes. PPAR Res. 2010, 2010, 612089. [Google Scholar] [CrossRef] [Green Version]
- Eveillard, A.; Lasserre, F.; de Tayrac, M.; Polizzi, A.; Claus, S.; Canlet, C.; Mselli-Lakhal, L.; Gotardi, G.; Paris, A.; Guillou, H.; et al. Identification of potential mechanisms of toxicity after di-(2-ethylhexyl)-phthalate (DEHP) adult exposure in the liver using a systems biology approach. Toxicol. Appl. Pharmacol. 2009, 236, 282–292. [Google Scholar] [CrossRef]
- Knights, K.M. Role of hepatic fatty acid: Coenzyme A ligases in the metabolism of xenobiotic carboxylic acids. Clin. Exp. Pharmacol. Physiol. 1998, 25, 776–782. [Google Scholar] [CrossRef]
- Martinez, D.L.; Tsuchiya, Y.; Gout, I. Coenzyme A biosynthetic machinery in mammalian cells. Biochem. Soc. Trans. 2014, 42, 1112–1117. [Google Scholar] [CrossRef]
- Lock, E.A.; Mitchell, A.M.; Elcombe, C.R. Biochemical mechanisms of induction of hepatic peroxisome proliferation. Annu. Rev. Pharmacol. Toxicol. 1989, 29, 145–163. [Google Scholar] [CrossRef]
- Brass, E.P. Overview of coenzyme A metabolism and its role in cellular toxicity. Chem. Biol. Interact. 1994, 90, 203–214. [Google Scholar] [CrossRef]
- Knights, K.M.; Drew, R. The effects of ibuprofen enantiomers on hepatocyte intermediary metabolism and mitochondrial respiration. Biochem. Pharmacol. 1992, 44, 1291–1296. [Google Scholar] [CrossRef]
- Silva, M.F.; Aires, C.C.; Luis, P.B.; Ruiter, J.P.; IJlst, L.; Duran, M.; Wanders, R.J.; Tavares de Almeida, I. Valproic acid metabolism and its effects on mitochondrial fatty acid oxidation: A review. J. Inherit. Metab. Dis. 2008, 31, 205–216. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, T.; Miyazawa, S.; Hashimoto, T. Effects of di-(2-ethylhexyl)phthalate administration on carbohydrate and fatty acid metabolism in rat liver. J. Biochem. 1978, 83, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Albro, P.W. The metabolism of 2-ethylhexanol in rats. Xenobiotica 1975, 5, 625–636. [Google Scholar] [CrossRef]
- Walker, V.; Mills, G.A. Urine 4-heptanone: A beta-oxidation product of 2-ethylhexanoic acid from plasticisers. Clin. Chim. Acta 2001, 306, 51–61. [Google Scholar] [CrossRef]
- Stingel, D.; Feldmeier, P.; Richling, E.; Kempf, M.; Elss, S.; Labib, S.; Schreier, P. Urinary 2-ethyl-3-oxohexanoic acid as major metabolite of orally administered 2-ethylhexanoic acid in human. Mol. Nutr. Food Res. 2007, 51, 301–306. [Google Scholar] [CrossRef]
- Deisinger, P.J.; Boatman, R.J.; Guest, D. Metabolism of 2-ethylhexanol administered orally and dermally to the female Fischer 344 rat. Xenobiotica 1994, 24, 429–440. [Google Scholar] [CrossRef]
- Rusyn, I.; Peters, J.M.; Cunningham, M.L. Modes of action and species-specific effects of di-(2-ethylhexyl)phthalate in the liver. Crit. Rev. Toxicol. 2006, 36, 459–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, J.K.; Mannaerts, G.P. Peroxisomal lipid metabolism. Annu. Rev. Nutr. 1994, 14, 343–370. [Google Scholar] [CrossRef]
- David, R.M.; Moore, M.R.; Cifone, M.A.; Finney, D.C.; Guest, D. Chronic peroxisome proliferation and hepatomegaly associated with the hepatocellular tumorigenesis of di(2-ethylhexyl)phthalate and the effects of recovery. Toxicol. Sci. Off. J. Soc. Toxicol. 1999, 50, 195–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- English, J.C.; Deisinger, P.J.; Guest, D. Metabolism of 2-ethylhexanoic acid administered orally or dermally to the female Fischer 344 rat. Xenobiotica 1998, 28, 699–714. [Google Scholar] [CrossRef] [PubMed]
- Bentley, P.; Calder, I.; Elcombe, C.; Grasso, P.; Stringer, D.; Wiegand, H.J. Hepatic peroxisome proliferation in rodents and its significance for humans. Food Chem. Toxicol. 1993, 31, 857–907. [Google Scholar] [CrossRef]
- Sui, H.X.; Zhang, L.; Wu, P.G.; Song, Y.; Yong, L.; Yang, D.J.; Jiang, D.G.; Liu, Z.P. Concentration of di(2-ethylhexyl) phthalate (DEHP) in foods and its dietary exposure in China. Int. J. Hyg. Environ. Health 2014, 217, 695–701. [Google Scholar] [CrossRef] [PubMed]
- Heinemeyer, G.; Sommerfeld, C.; Springer, A.; Heiland, A.; Lindtner, O.; Greiner, M.; Heuer, T.; Krems, C.; Conrad, A. Estimation of dietary intake of bis(2-ethylhexyl)phthalate (DEHP) by consumption of food in the German population. Int. J. Hyg. Environ. Health 2013, 216, 472–480. [Google Scholar] [CrossRef] [PubMed]
- CPSC. United States Consumer Product Safety Commission: Toxicity Review of Di(2-ethylhexyl) Phthalate (DEHP). Available online: https://www.cpsc.gov/s3fs-public/ToxicityReviewOfDEHP.pdf (accessed on 12 September 2021).
- CSTEE. Committee on Toxicity, Ecotoxicity and the Environment: Opinion on Phthalate Migration from Soft PVC Toys and Child-Care Articles. Available online: https://ec.europa.eu/health/scientific_committees/environmental_risks/opinions/sctee/sct_out19_en.htm (accessed on 12 September 2021).
- Rock, C.O.; Calder, R.B.; Karim, M.A.; Jackowski, S. Pantothenate kinase regulation of the intracellular concentration of coenzyme A. J. Biol. Chem. 2000, 275, 1377–1383. [Google Scholar] [CrossRef] [Green Version]
- Ramaswamy, G.; Karim, M.A.; Murti, K.G.; Jackowski, S. PPARalpha controls the intracellular coenzyme A concentration via regulation of PANK1alpha gene expression. J. Lipid Res. 2004, 45, 17–31. [Google Scholar] [CrossRef] [Green Version]
- Skrede, S.; Halvorsen, O. Increased biosynthesis of CoA in the liver of rats treated with clofibrate. Eur. J. Biochem. 1979, 98, 223–229. [Google Scholar] [CrossRef]
- Reilly, S.J.; Tillander, V.; Ofman, R.; Alexson, S.E.; Hunt, M.C. The nudix hydrolase 7 is an Acyl-CoA diphosphatase involved in regulating peroxisomal coenzyme A homeostasis. J. Biochem. 2008, 144, 655–663. [Google Scholar] [CrossRef]
- Theodoulou, F.L.; Sibon, O.C.; Jackowski, S.; Gout, I. Coenzyme A and its derivatives: Renaissance of a textbook classic. Biochem. Soc. Trans. 2014, 42, 1025–1032. [Google Scholar] [CrossRef]
- Schalkwijk, J.; Jansen, P. Chemical biology tools to study pantetheinases of the vanin family. Biochem. Soc. Trans. 2014, 42, 1052–1055. [Google Scholar] [CrossRef] [PubMed]
- Leighton, F.; Bergseth, S.; Rørtveit, T.; Christiansen, E.N.; Bremer, J. Free acetate production by rat hepatocytes during peroxisomal fatty acid and dicarboxylic acid oxidation. J. Biol. Chem. 1989, 264, 10347–10350. [Google Scholar] [CrossRef]
- Himms-Hagen, J.; Harper, M.E. Physiological role of UCP3 may be export of fatty acids from mitochondria when fatty acid oxidation predominates: An hypothesis. Exp. Biol. Med. 2001, 226, 78–84. [Google Scholar] [CrossRef]
- Foster, D.W. Malonyl-CoA: The regulator of fatty acid synthesis and oxidation. J. Clin. Investig. 2012, 122, 1958–1959. [Google Scholar] [CrossRef] [Green Version]
- McGarry, J.D.; Mannaerts, G.P.; Foster, D.W. A possible role for malonyl-CoA in the regulation of hepatic fatty acid oxidation and ketogenesis. J. Clin. Investig. 1977, 60, 265–270. [Google Scholar] [CrossRef]
- Ussher, J.R.; Lopaschuk, G.D. The malonyl CoA axis as a potential target for treating ischaemic heart disease. Cardiovasc. Res. 2008, 79, 259–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zammit, V.A. Regulation of hepatic fatty acid oxidation and ketogenesis. Proc. Nutr. Soc. 1983, 42, 289–302. [Google Scholar] [CrossRef] [Green Version]
- Jenkins, B.; West, J.A.; Koulman, A. A review of odd-chain fatty acid metabolism and the role of pentadecanoic Acid (c15:0) and heptadecanoic Acid (c17:0) in health and disease. Molecules 2015, 20, 2425–2444. [Google Scholar] [CrossRef] [Green Version]
- Mazumder, R.; Sasakawa, T.; Ochoa, S. Metabolism of propionic acid in animal tissues. X. Methylmalonyl co-enzyme A mutase holoenzyme. J. Biol. Chem. 1963, 238, 50–53. [Google Scholar] [CrossRef]
- Kasumov, T.; Martini, W.Z.; Reszko, A.E.; Bian, F.; Pierce, B.A.; David, F.; Roe, C.R.; Brunengraber, H. Assay of the concentration and (13)C isotopic enrichment of propionyl-CoA, methylmalonyl-CoA, and succinyl-CoA by gas chromatography-mass spectrometry. Anal. Biochem. 2002, 305, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Kasumov, T.; Cendrowski, A.V.; David, F.; Jobbins, K.A.; Anderson, V.E.; Brunengraber, H. Mass isotopomer study of anaplerosis from propionate in the perfused rat heart. Arch. Biochem. Biophys. 2007, 463, 110–117. [Google Scholar] [CrossRef] [Green Version]
- Badr, M.Z.; Handler, J.A.; Whittaker, M.; Kauffman, F.C.; Thurman, R.G. Interactions between plasticizers and fatty acid metabolism in the perfused rat liver and in vivo. Inhibition of ketogenesis by 2-ethylhexanol. Biochem. Pharmacol. 1990, 39, 715–721. [Google Scholar] [CrossRef]
- Manninen, A.; Kroger, S.; Liesivuori, J.; Savolainen, H. 2-Ethylhexanoic acid inhibits urea synthesis and stimulates carnitine acetyltransferase activity in rat liver mitochondria. Arch. Toxicol. 1989, 63, 160–161. [Google Scholar] [CrossRef]
- Olowe, Y.; Schulz, H. Regulation of thiolases from pig heart. Control of fatty acid oxidation in heart. Eur. J. Biochem. 1980, 109, 425–429. [Google Scholar] [CrossRef] [PubMed]
- Dalluge, J.J.; Gort, S.; Hobson, R.; Selifonova, O.; Amore, F.; Gokarn, R. Separation and identification of organic acid-coenzyme A thioesters using liquid chromatography/electrospray ionization-mass spectrometry. Anal. Bioanal. Chem. 2002, 374, 835–840. [Google Scholar] [CrossRef]
- Zhang, G.F.; Kombu, R.S.; Kasumov, T.; Han, Y.; Sadhukhan, S.; Zhang, J.; Sayre, L.M.; Ray, D.; Gibson, K.M.; Anderson, V.A.; et al. Catabolism of 4-hydroxyacids and 4-hydroxynonenal via 4-hydroxy-4-phosphoacyl-CoAs. J. Biol. Chem. 2009, 284, 33521–33534. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Zhang, S.; Berthiaume, J.M.; Simons, B.; Zhang, G.F. Novel approach in LC-MS/MS using MRM to generate a full profile of acyl-CoAs: Discovery of acyl-dephospho-CoAs. J. Lipid Res. 2014, 55, 592–602. [Google Scholar] [CrossRef] [Green Version]
- Corkey, B.E.; Hale, D.E.; Glennon, M.C.; Kelley, R.I.; Coates, P.M.; Kilpatrick, L.; Stanley, C.A. Relationship between unusual hepatic acyl coenzyme A profiles and the pathogenesis of Reye syndrome. J. Clin. Investig. 1988, 82, 782–788. [Google Scholar] [CrossRef]
- Grosse, S.D.; Khoury, M.J.; Greene, C.L.; Crider, K.S.; Pollitt, R.J. The epidemiology of medium chain acyl-CoA dehydrogenase deficiency: An update. Genet. Med. 2006, 8, 205–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchell, G.A.; Gauthier, N.; Lesimple, A.; Wang, S.P.; Mamer, O.; Qureshi, I. Hereditary and acquired diseases of acyl-coenzyme A metabolism. Mol. Genet. Metab. 2008, 94, 4–15. [Google Scholar] [CrossRef] [PubMed]
- Dansie, L.E.; Reeves, S.; Miller, K.; Zano, S.P.; Frank, M.; Pate, C.; Wang, J.; Jackowski, S. Physiological roles of the pantothenate kinases. Biochem.Soc. Trans. 2014, 42, 1033–1036. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Retention Time (min) | Control nmol/g | 2% DEHP nmol/g | [DEHP]/[Control] 1 | |
|---|---|---|---|---|
| Free CoA and physiological CoA esters | ||||
| m/z 768: Free CoA | 4.6 | 36.1 ± 3.1 | 364.3 ± 64.5 *** | 10 |
| m/z 810: Acetyl-CoA | 8.1 | 97.7 ± 4.0 | 190.6 ± 18.0 *** | 2 |
| m/z 824: Propionyl-CoA | 8.9 | 7.9 ± 1.1 | 18.0 ± 1.7 *** | 2 |
| m/z 838: Butyryl-CoA | 9.6 | 19.8 ± 4.0 | 29.2 ± 2.4 | 1.5 |
| m/z 852: Pentanoyl-CoA | 10.3 | 7.7 ± 0.7 | 11.0 ± 0.4 ** | 1.4 |
| m/z 854: Malonyl-CoA | 3.2 | 0.2 ± 0.03 | 2.8 ± 0.7 ** | 18 |
| m/z 854: β-hydroxybutyryl-CoA | 8.3 | 9.0 ± 1.3 | 9.1 ± 1.1 | 1.0 |
| m/z 866: Hexanoyl-CoA | 11.0 | 12.0 ± 3.1 | 13.1 ± 1.8 | 1.1 |
| m/z 868: Succinyl-CoA | 6.1 | 3.3 ± 0.5 | 9.2 ± 1.4 ** | 3 |
| m/z 868: Methylmalonyl-CoA | 5.1 | 0.4 ± 0.1 | 1.1 ± 0.2 * | 3 |
| m/z 880: Heptanoyl-CoA | 11.5 | 3.4 ± 0.3 | 4.0 ± 0.3 | 1.2 |
| m/z 912: 3-Hydroxy-3-methylglutaryl-CoA | 7.3 | 5.0 ± 0.7 | 3.2 ± 0.6 | 0.6 |
| m/z 922: Decanoyl-CoA | 12.8 | 0.5 ± 0.1 | 0.3 ± 0.04 | 0.6 |
| m/z 1006: Palmitoyl-CoA | 14.7 | 2.2 ± 0.4 | 3.4 ± 1.0 | 2 |
| Identified DEHP-derived CoA esters which are isobars of physiological CoA esters | ||||
| m/z 892: 3-Octenoyl-CoA 2-Ethyl-2-hexenoyl-CoA 2-Ethyl-5-hexenoyl-CoA | 11.5 | 4.5 ± 0.4 | 53.4 ± 8.8 *** | 12 |
| m/z 894: Octanoyl-CoA 2-Ethylhexanoyl-CoA | 11.6 | 11.8 ± 2.6 | 438.7 ± 59.1 *** | 37 |
| m/z 908: 3-Ketooctanoyl-CoA 3-Keto-2-ethylhexanoyl-CoA | 10.7 | 0.1 ± 0.1 | 12.5 ± 1.7 ** | 98 |
| m/z 910: 3-Hydroxyoctanoyl-CoA 3-Hydroxy-2-ethylhexanoyl-CoA 6-Hydroxy-2-ethylhexanoyl-CoA 5-Hydroxy-2-ethylhexanoyl-CoA | 10.3 | 1.0 ± 0.1 | 23.2 ± 3.9 ** | 22 |
| Unidentified CoA esters | ||||
| m/z 861: Unknown 1 | 9.4 | 0.1 ± 0.02 | 2.0 ± 0.3 *** | 14 |
| m/z 862: Unknown 2 | 7.9 | 0.2 ± 0.03 | 0.7 ± 0.1 *** | 4 |
| m/z 872: Unknown 3 | 10.2 | 0.1 ± 0.03 | 1.5 ± 0.2 *** | 14 |
| m/z 875: Unknown 4 | 9.9 | 0.2 ± 0.03 | 2.9 ± 0.5 *** | 16 |
| m/z 888: Unknown 5 | 9.7 | 0.6 ± 0.1 | 2.6 ± 0.6 *** | 4 |
| m/z 904: Unknown 6 | 9.0 | 0.04 ± 0.04 | 7.1 ± 1.6 ** | 190 |
| m/z 929: Unknown 7 | 9.3 | 0.02 ± 0.01 | 0.5 ± 0.1 ** | 23 |
| m/z 947: Unknown 8 | 10.4 | 0.02 ± 0.01 | 0.6 ± 0.1 ** | 32 |
| m/z 968: Unknown 9 | 9.5 | ND | 0.4 ± 0.1 * | - |
| m/z 989: Unknown 10 | 10.9 | 0.01 ± 0.01 | 0.1 ± 0.1 | 17 |
| m/z 996: Unknown 11 | 11.8 | ND | 0.2 ± 0.1 * | - |
| m/z 998: Unknown 12 | 11.9 | 0.03 ± 0.02 | 0.3 ± 0.1 ** | 11 |
| m/z 1008: Unknown 13 | 10.4 | ND | 0.4 ± 0.1 * | - |
| m/z 1026: Unknown 14 | 8.2 | ND | 0.5 ± 0.1 * | - |
| m/z 1030: Unknown 15 | 9.8 | 0.1 ± 0.1 | 9.6 ± 1.6 *** | 80 |
| m/z 1052: Unknown 16 | 10.5 | ND | 0.3 ± 0.1 * | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hala, D.; Petersen, L.H.; Huggett, D.B.; Puchowicz, M.A.; Brunengraber, H.; Zhang, G.-F. Overcompensation of CoA Trapping by Di(2-ethylhexyl) Phthalate (DEHP) Metabolites in Livers of Wistar Rats. Int. J. Mol. Sci. 2021, 22, 13489. https://doi.org/10.3390/ijms222413489
Hala D, Petersen LH, Huggett DB, Puchowicz MA, Brunengraber H, Zhang G-F. Overcompensation of CoA Trapping by Di(2-ethylhexyl) Phthalate (DEHP) Metabolites in Livers of Wistar Rats. International Journal of Molecular Sciences. 2021; 22(24):13489. https://doi.org/10.3390/ijms222413489
Chicago/Turabian StyleHala, David, Lene H. Petersen, Duane B. Huggett, Michelle A. Puchowicz, Henri Brunengraber, and Guo-Fang Zhang. 2021. "Overcompensation of CoA Trapping by Di(2-ethylhexyl) Phthalate (DEHP) Metabolites in Livers of Wistar Rats" International Journal of Molecular Sciences 22, no. 24: 13489. https://doi.org/10.3390/ijms222413489