
Correlation between Oxidative Stress and Transforming Growth Factor-Beta in Cancers

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Reactive Oxygen Species
2.1. Biology of ROS
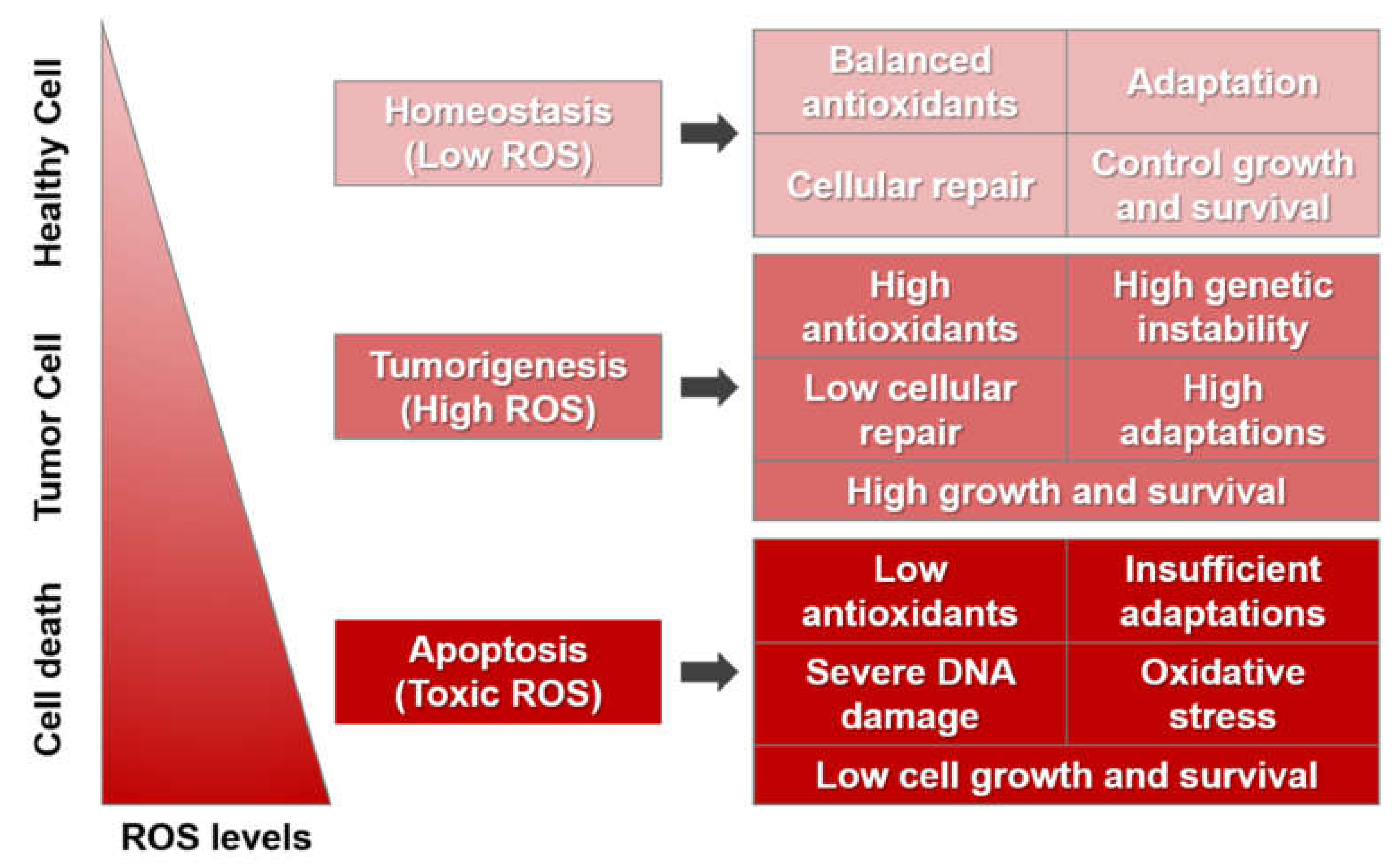
2.2. Roles of ROS in Cancer
3. Transforming Growth Factor-Beta
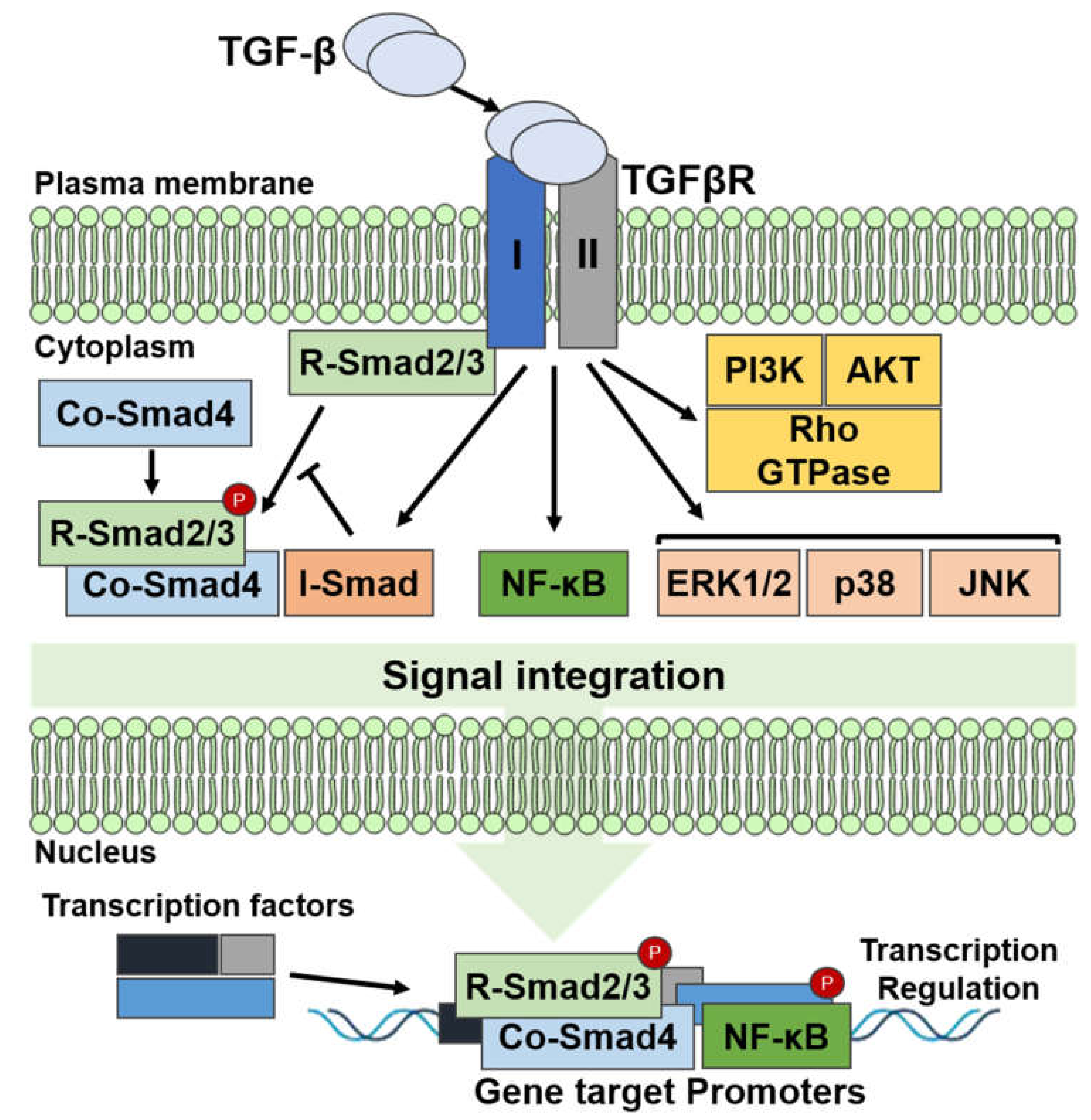
3.1. Transforming Growth Factor-Beta Signaling
3.2. Roles of Transforming Growth Factor-Beta in Cancer
4. Relationship of TGF-β and ROS
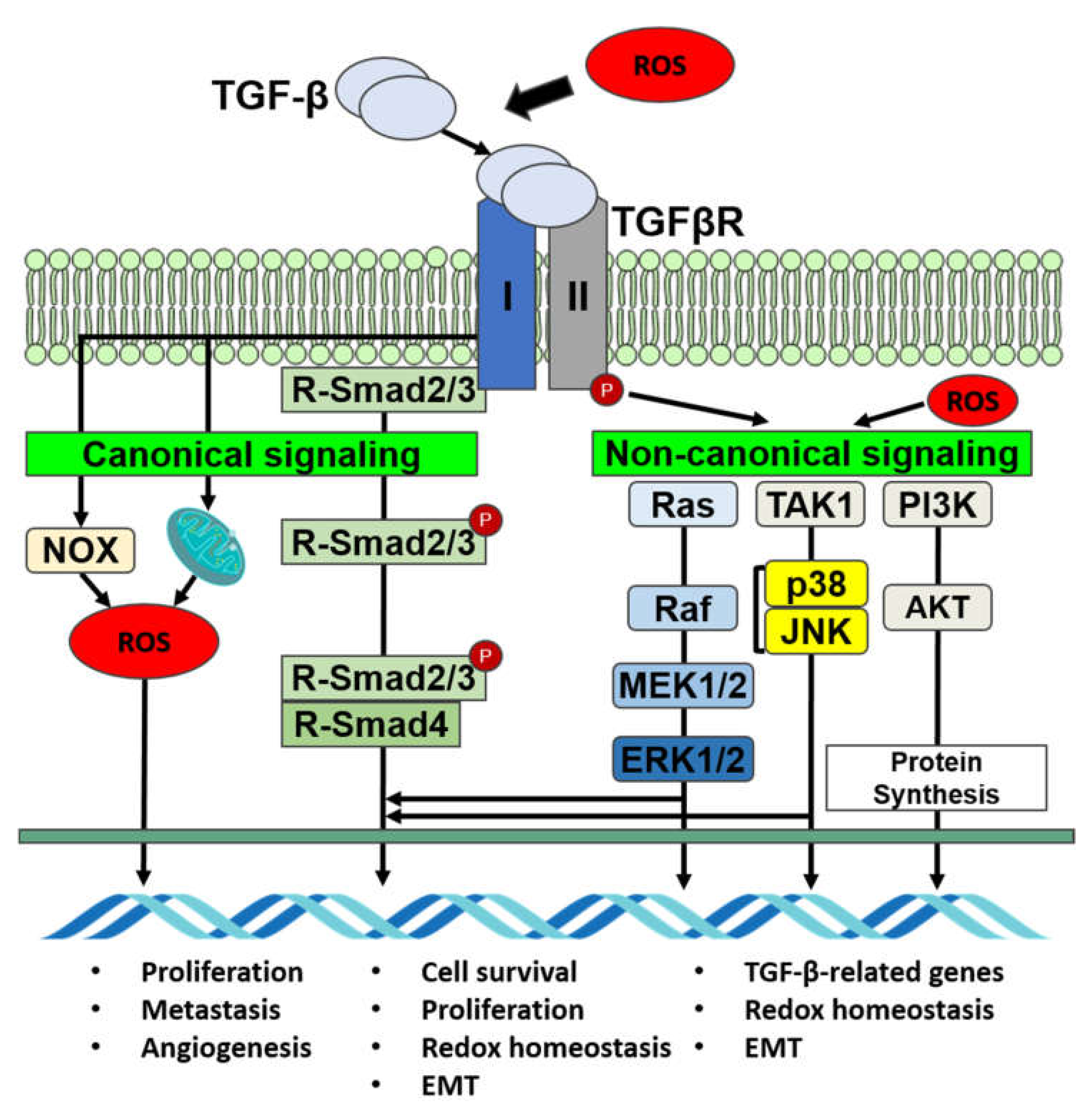
4.1. TGF-β Regulates ROS Activity
4.2. TGF-β Signaling Is Mediated by ROS
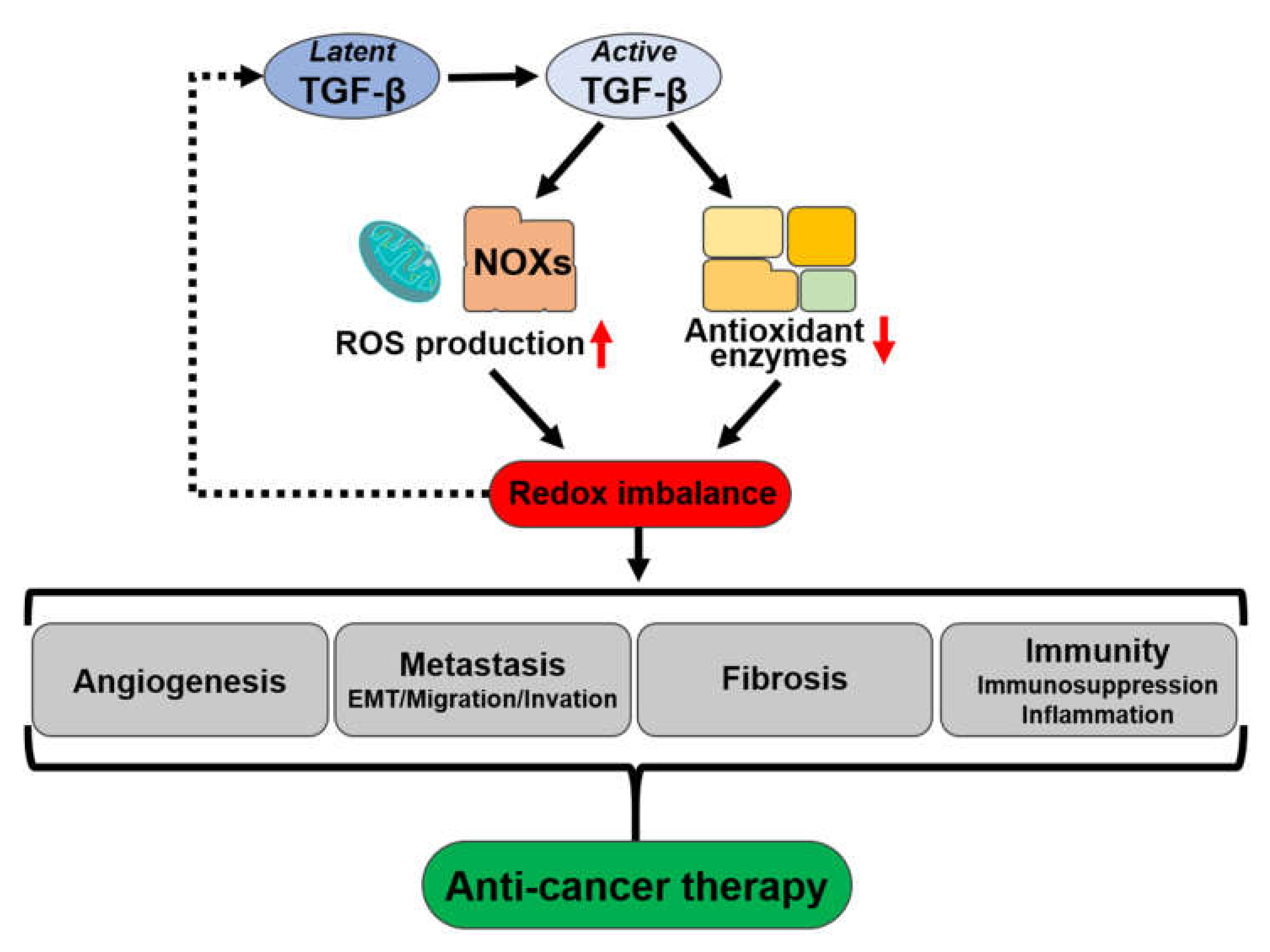
4.3. Association of ROS with the Activation of Latent TGF-β
4.4. TGF-β and ROS Share Downstream Mediators
5. Crosstalk of TGF-β and ROS in Cancers
5.1. Roles of TGF-β and ROS in EMT
5.2. Roles of ROS and TGF-β in Cellular Senescence and Cancer
5.3. Interplay within ROS and TGF-β Signaling in Cancer
5.4. TGF-β and Oxidative Stress Crosstalk in Cancer-Cell Metabolism
5.5. Redox Modulation of TGF-β Signaling
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Roberts, A.B.; Wakefield, L.M. The two faces of transforming growth factor beta in carcinogenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 8621–8623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiery, J.P. Epithelial-mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2002, 2, 442–454. [Google Scholar] [CrossRef] [PubMed]
- Wakefield, L.M.; Roberts, A.B. TGF-beta signaling: Positive and negative effects on tumorigenesis. Curr. Opin. Genet Dev. 2002, 12, 22–29. [Google Scholar] [CrossRef]
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer 2011, 11, 85–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glasauer, A.; Chandel, N.S. Targeting antioxidants for cancer therapy. Biochem. Pharmacol. 2014, 92, 90–101. [Google Scholar] [CrossRef]
- Liu, R.M.; Pravia, K.A.G. Oxidative stress and glutathione in TGF-beta-mediated fibrogenesis. Free Radical. Bio. Med. 2010, 48, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Tochhawng, L.; Deng, S.; Pervaiz, S.; Yap, C.T. Redox regulation of cancer cell migration and invasion. Mitochondrion 2013, 13, 246–253. [Google Scholar] [CrossRef]
- Ishikawa, F.; Kaneko, E.; Sugimoto, T.; Ishijima, T.; Wakamatsu, M.; Yuasa, A.; Sampei, R.; Mori, K.; Nose, K.; Shibanuma, M. A mitochondrial thioredoxin-sensitive mechanism regulates TGF-beta-mediated gene expression associated with epithelial-mesenchymal transition. Biochem. Bioph. Res. Co. 2014, 443, 821–827. [Google Scholar] [CrossRef]
- Annes, J.P.; Munger, J.S.; Rifkin, D.B. Making sense of latent TGF beta activation. J. Cell Sci. 2003, 116, 217–224. [Google Scholar] [CrossRef] [Green Version]
- Auten, R.L.; Davis, J.M. Oxygen Toxicity and Reactive Oxygen Species: The Devil Is in the Details. Pediatr. Res. 2009, 66, 121–127. [Google Scholar] [CrossRef] [Green Version]
- Dickinson, B.C.; Chang, C.J. Chemistry and biology of reactive oxygen species in signaling or stress responses. Nat. Chem. Biol. 2011, 7, 504–511. [Google Scholar] [CrossRef] [Green Version]
- Hayes, J.D.; Dinkova-Kostova, A.T.; Tew, K.D. Oxidative Stress in Cancer. Cancer Cell 2020, 38, 167–197. [Google Scholar] [CrossRef]
- Nikitovic, D.; Corsini, E.; Kouretas, D.; Tsatsakis, A.; Tzanakakis, G. ROS-major mediators of extracellular matrix remodeling during tumor progression. Food Chem. Toxicol. 2013, 61, 178–186. [Google Scholar] [CrossRef]
- Giannoni, E.; Parri, M.; Chiarugi, P. EMT and Oxidative Stress: A Bidirectional Interplay Affecting Tumor Malignancy. Antioxid. Redox Sign. 2012, 16, 1248–1263. [Google Scholar] [CrossRef]
- Krause, K.H. Aging: A revisited theory based on free radicals generated by NOX family NADPH oxidases. Exp. Gerontol. 2007, 42, 256–262. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Wang, Y.; Qi, H.; Liu, Y.; Duan, C.; Liu, X.; Xia, T.; Chen, D.; Piao, H.L.; Liu, H.X. The double-edged roles of ROS in cancer prevention and therapy. Theranostics 2021, 11, 4839–4857. [Google Scholar] [CrossRef]
- Aggarwal, V.; Tuli, H.S.; Varol, A.; Thakral, F.; Yerer, M.B.; Sak, K.; Varol, M.; Jain, A.; Khan, M.A.; Sethi, G. Role of Reactive Oxygen Species in Cancer Progression: Molecular Mechanisms and Recent Advancements. Biomolecules 2019, 9, 735. [Google Scholar] [CrossRef] [Green Version]
- Scialo, F.; Fernandez-Ayala, D.J.; Sanz, A. Role of Mitochondrial Reverse Electron Transport in ROS Signaling: Potential Roles in Health and Disease. Front. Physiol. 2017, 8, 428. [Google Scholar] [CrossRef]
- Dodson, M.; Castro-Portuguez, R.; Zhang, D.D. NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox. Biol. 2019, 23, 101107. [Google Scholar] [CrossRef]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta 2016, 1863, 2977–2992. [Google Scholar] [CrossRef]
- Athreya, K.; Xavier, M.F. Antioxidants in the Treatment of Cancer. Nutr. Cancer 2017, 69, 1099–1104. [Google Scholar] [CrossRef]
- Fukai, T.; Ushio-Fukai, M. Cross-Talk between NADPH Oxidase and Mitochondria: Role in ROS Signaling and Angiogenesis. Cells 2020, 9, 1849. [Google Scholar] [CrossRef]
- Waghela, B.N.; Vaidya, F.U.; Agrawal, Y.; Santra, M.K.; Mishra, V.; Pathak, C. Molecular insights of NADPH oxidases and its pathological consequences. Cell. Biochem. Funct. 2021, 39, 218–234. [Google Scholar] [CrossRef]
- Sabharwal, S.S.; Schumacker, P.T. Mitochondrial ROS in cancer: Initiators, amplifiers or an Achilles’ heel? Nat. Rev. Cancer 2014, 14, 709–721. [Google Scholar] [CrossRef] [Green Version]
- Elchuri, S.; Oberley, T.D.; Qi, W.; Eisenstein, R.S.; Jackson Roberts, L.; Van Remmen, H.; Epstein, C.J.; Huang, T.T. CuZnSOD deficiency leads to persistent and widespread oxidative damage and hepatocarcinogenesis later in life. Oncogene 2005, 24, 367–380. [Google Scholar] [CrossRef] [Green Version]
- Gamcsik, M.P.; Kasibhatla, M.S.; Teeter, S.D.; Colvin, O.M. Glutathione levels in human tumors. Biomarkers 2012, 17, 671–691. [Google Scholar] [CrossRef]
- Harris, I.S.; Treloar, A.E.; Inoue, S.; Sasaki, M.; Gorrini, C.; Lee, K.C.; Yung, K.Y.; Brenner, D.; Knobbe-Thomsen, C.B.; Cox, M.A.; et al. Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer Cell 2015, 27, 211–222. [Google Scholar] [CrossRef] [Green Version]
- Bansal, A.; Simon, M.C. Glutathione metabolism in cancer progression and treatment resistance. J. Cell Biol. 2018, 217, 2291–2298. [Google Scholar] [CrossRef] [Green Version]
- Trachootham, D.; Lu, W.Q.; Ogasawara, M.A.; Valle, N.R.D.; Huang, P. Redox regulation of cell survival. Antioxid. Redox. Sign. 2008, 10, 1343–1374. [Google Scholar] [CrossRef] [Green Version]
- Grimsrud, P.A.; Xie, H.W.; Griffin, T.J.; Bernlohr, D.A. Oxidative stress and covalent modification of protein with bioactive aldehydes. J. Biol. Chem. 2008, 283, 21837–21841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, B.; Chen, Y.M.; Clair, D.K.S. ROS and p53: A versatile partnership. Free Radical. Bio. Med. 2008, 44, 1529–1535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, Y.P.; Lim, T.T.; Chan, Y.L.; Song, A.C.M.; Yeo, B.H.; Vojtesek, B.; Coomber, D.; Rajagopal, G.; Lane, D. The p53 knowledgebase: An integrated information resource for p53 research. Oncogene 2007, 26, 1517–1521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semenza, G.L. Hypoxia-inducible factors: Mediators of cancer progression and targets for cancer therapy. Trends Pharmacol. Sci. 2012, 33, 207–214. [Google Scholar] [CrossRef] [Green Version]
- Chang, T.C.; Huang, C.J.; Tam, K.; Chen, S.F.; Tan, K.T.; Tsai, M.S.; Lin, T.N.; Shyue, S.K. Stabilization of hypoxia-inducible factor-1 alpha by prostacyclin under prolonged hypoxia via reducing reactive oxygen species level in endothelial cells. J. Biol. Chem. 2005, 280, 36567–36574. [Google Scholar] [CrossRef] [Green Version]
- Srinivas, V.; Leshchinsky, I.; Sang, N.; King, M.P.; Minchenko, A.; Caro, J. Oxygen sensing and HIF-1 activation does not require an active mitochondrial respiratory chain electron-transfer pathway. J. Biol. Chem. 2001, 276, 21995–21998. [Google Scholar] [CrossRef] [Green Version]
- Nelson, K.K.; Melendez, J.A. Mitochondrial redox control of matrix metalloproteinases. Free Radical. Bio. Med. 2004, 37, 768–784. [Google Scholar] [CrossRef]
- Diaz, B.; Shani, G.; Pass, I.; Anderson, D.; Quintavalle, M.; Courtneidge, S.A. Tks5-Dependent, Nox-Mediated Generation of Reactive Oxygen Species Is Necessary for Invadopodia Formation. Sci. Signal. 2009, 2, ra53–ra53. [Google Scholar] [CrossRef] [Green Version]
- Vaquero, E.C.; Edderkaoui, M.; Pandol, S.J.; Gukovsky, I.; Gukovskaya, A.S. Reactive oxygen species produced by NAD(P)H oxidase inhibit apoptosis in pancreatic cancer cells. J. Biol. Chem. 2004, 279, 34643–34654. [Google Scholar] [CrossRef] [Green Version]
- Gupta, N.; Verma, K.; Nalla, S.; Kulshreshtha, A.; Lall, R.; Prasad, S. Free Radicals as a Double-Edged Sword: The Cancer Preventive and Therapeutic Roles of Curcumin. Molecules 2020, 25, 5390. [Google Scholar] [CrossRef]
- Attisano, L.; Wrana, J.L. Signal transduction by the TGF-beta superfa).mily. Science 2002, 296, 1646–1647. [Google Scholar] [CrossRef]
- Padua, D.; Massague, J. Roles of TGFbeta in metastasis. Cell Res 2009, 19, 89–102. [Google Scholar] [CrossRef]
- Miah, S.; Banks, C.A.S.; Ogunbolude, Y.; Bagu, E.T.; Berg, J.M.; Saraf, A.; Tettey, T.T.; Hattem, G.; Dayebgadoh, G.; Kempf, C.G.; et al. BRK phosphorylates SMAD4 for proteasomal degradation and inhibits tumor suppressor FRK to control SNAIL, SLUG, and metastatic potential. Sci. Adv. 2019, 5, eaaw3113. [Google Scholar] [CrossRef] [Green Version]
- Hata, A.; Lagna, G.; Massague, J.; Hemmati-Brivanlou, A. Smad6 inhibits BMP/Smad1 signaling by specifically competing with the Smad4 tumor suppressor. Genes. Dev. 1998, 12, 186–197. [Google Scholar] [CrossRef] [Green Version]
- Bernabeu, C.; Lopez-Novoa, J.M.; Quintanilla, M. The emerging role of TGF-beta superfamily coreceptors in cancer. Biochim. Biophys. Acta. 2009, 1792, 954–973. [Google Scholar] [CrossRef]
- Mu, Y.; Gudey, S.K.; Landstrom, M. Non-Smad signaling pathways. Cell Tissue Res. 2012, 347, 11–20. [Google Scholar] [CrossRef]
- Santibanez, J.F.; Kocic, J. Transforming growth factor-beta superfamily, implications in development and differentiation of stem cells. Biomol. Concepts 2012, 3, 429–445. [Google Scholar] [CrossRef]
- Meulmeester, E.; ten Dijke, P. The dynamic roles of TGF-beta in cancer. J. Pathol. 2011, 223, 205–218. [Google Scholar] [CrossRef]
- Park, H.Y.; Wakefield, L.M.; Mamura, M. Regulation of tumor immune surveillance and tumor immune subversion by tgf-Beta. Immune. Netw. 2009, 9, 122–126. [Google Scholar] [CrossRef] [Green Version]
- Teicher, B.A. Transforming growth factor-beta and the immune response to malignant disease. Clin. Cancer Res. 2007, 13, 6247–6251. [Google Scholar] [CrossRef] [Green Version]
- Ivanovic, V.; Todorovic-Rakovic, N.; Demajo, M.; Neskovic-Konstantinovic, Z.; Subota, V.; Ivanisevic-Milovanovic, O.; Nikolic-Vukosavljevic, D. Elevated plasma levels of transforming growth factor-beta 1 (TGF-beta 1) in patients with advanced breast cancer: Association with disease progression. Eur. J. Cancer 2003, 39, 454–461. [Google Scholar] [CrossRef]
- Wunderlich, H.; Steiner, T.; Kosmehl, H.; Junker, U.; Reinhold, D.; Reichelt, O.; Zermann, D.H.; Schubert, J. Increased transforming growth factor beta 1 plasma level in patients with renal cell carcinoma: A tumor-specific marker? Urol. Int. 1998, 60, 205–207. [Google Scholar] [CrossRef] [PubMed]
- Friess, H.; Yamanaka, Y.; Buchler, M.; Ebert, M.; Beger, H.G.; Gold, L.I.; Korc, M. Enhanced Expression of Transforming Growth-Factor-Beta Isoforms in Pancreatic-Cancer Correlates with Decreased Survival. Gastroenterology 1993, 105, 1846–1856. [Google Scholar] [CrossRef]
- Derynck, R.; Turley, S.J.; Akhurst, R.J. TGFbeta biology in cancer progression and immunotherapy. Nat. Rev. Clin. Oncol. 2021, 18, 9–34. [Google Scholar] [CrossRef]
- Derynck, R.; Budi, E.H. Specificity, versatility, and control of TGF-beta family signaling. Sci. Signal. 2019, 12, eaav5183. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.J.; Ren, J.; ten Dijke, P. Targeting TGF beta signal transduction for cancer therapy. Signal Transduct. Tar. 2021, 6, 8. [Google Scholar] [CrossRef]
- David, C.J.; Huang, Y.H.; Chen, M.; Su, J.; Zou, Y.L.; Bardeesy, N.; Iacobuzio-Donahue, C.A.; Massague, J. TGF-beta Tumor Suppression through a Lethal EMT. Cell 2016, 164, 1015–1030. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Liu, Z.; Tan, J.; Dong, H.; Zhang, X. Multispectral imaging reveals hyper active TGF-beta signaling in colorectal cancer. Cancer Biol. Ther. 2018, 19, 105–112. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Kuo, F.; Capistrano, K.J.; Kang, D.; Nixon, B.G.; Shi, W.; Chou, C.; Do, M.H.; Stamatiades, E.G.; Gao, S.; et al. TGF-beta suppresses type 2 immunity to cancer. Nature 2020, 587, 115–120. [Google Scholar] [CrossRef]
- Antony, M.L.; Nair, R.; Sebastian, P.; Karunagaran, D. Changes in expression, and/or mutations in TGF-beta receptors (TGF-beta RI and TGF-beta RII) and Smad 4 in human ovarian tumors. J. Cancer Res. Clin. Oncol. 2010, 136, 351–361. [Google Scholar] [CrossRef]
- Albright, C.D.; Salganik, R.I.; Craciunescu, C.N.; Mar, M.H.; Zeisel, S.H. Mitochondrial and microsomal derived reactive oxygen species mediate apoptosis induced by transforming growth factor-beta 1 in immortalized rat hepatocytes. J. Cell. Biochem. 2003, 89, 254–261. [Google Scholar] [CrossRef]
- Herrera, B.; Murillo, M.M.; Alvarez-Barrientos, A.; Beltran, J.; Fernandez, M.; Fabregat, I. Source of early reactive oxygen species in the apoptosis induced by transforming growth factor-beta in fetal rat hepatocytes. Free Radical. Bio. Med. 2004, 36, 16–26. [Google Scholar] [CrossRef]
- Yoon, Y.S.; Lee, J.H.; Hwang, S.C.; Choi, K.S.; Yoon, G. TGF beta 1 induces prolonged mitochondrial ROS generation through decreased complex IV activity with senescent arrest in Mv1Lu cells. Oncogene 2005, 24, 1895–1903. [Google Scholar] [CrossRef] [Green Version]
- Tobar, N.; Villar, V.; Santibanez, J.F. ROS-NF kappa I’ mediates TGF-beta 1-induced expression of urokinase-type plasminogen activator, matrix metalloproteinase-9 and cell invasion. Mol. Cell. Biochem. 2010, 340, 195–202. [Google Scholar] [CrossRef]
- Boudreau, H.E.; Casterline, B.W.; Rada, B.; Korzeniowska, A.; Leto, T.L. Nox4 involvement in TGF-beta and SMAD3-driven induction of the epithelial-to-mesenchymal transition and migration of breast epithelial cells. Free Radical. Biol. Med. 2012, 53, 1489–1499. [Google Scholar] [CrossRef] [Green Version]
- Boudreau, H.E.; Casterline, B.W.; Burke, D.J.; Leto, T.L. Wild-type and mutant p53 differentially regulate NADPH oxidase 4 in TGF-beta-mediated migration of human lung and breast epithelial cells. Brit. J. Cancer 2014, 110, 2569–2582. [Google Scholar] [CrossRef] [Green Version]
- Hiraga, R.; Kato, M.; Miyagawa, S.; Kamata, T. Nox4-derived ROS Signaling Contributes to TGF-beta-induced Epithelial-mesenchymal Transition in Pancreatic Cancer Cells. Anticancer. Res. 2013, 33, 4431–4438. [Google Scholar]
- Cheung, E.C.; DeNicola, G.M.; Nixon, C.; Blyth, K.; Labuschagne, C.F.; Tuveson, D.A.; Vousden, K.H. Dynamic ROS Control by TIGAR Regulates the Initiation and Progression of Pancreatic Cancer. Cancer Cell. 2020, 37, 168–182 e4. [Google Scholar] [CrossRef] [Green Version]
- Jain, M.; Rivera, S.; Monclus, E.A.; Synenki, L.; Zirk, A.; Eisenbart, J.; Feghali-Bostwick, C.; Mutlu, G.M.; Budinger, G.R.; Chandel, N.S. Mitochondrial reactive oxygen species regulate transforming growth factor-beta signaling. J. Biol. Chem. 2013, 288, 770–777. [Google Scholar] [CrossRef] [Green Version]
- Li, W.Q.; Qureshi, H.Y.; Liacini, A.; Dehnade, F.; Zafarullah, M. Transforming growth factor Beta1 induction of tissue inhibitor of metalloproteinases 3 in articular chondrocytes is mediated by reactive oxygen species. Free Radic. Biol. Med. 2004, 37, 196–207. [Google Scholar] [CrossRef]
- Rhyu, D.Y.; Yang, Y.; Ha, H.; Lee, G.T.; Song, J.S.; Uh, S.T.; Lee, H.B. Role of reactive oxygen species in TGF-beta1-induced mitogen-activated protein kinase activation and epithelial-mesenchymal transition in renal tubular epithelial cells. J. Am. Soc. Nephrol. 2005, 16, 667–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, T.; Quan, T.; Shao, Y.; Voorhees, J.J.; Fisher, G.J. Oxidative exposure impairs TGF-beta pathway via reduction of type II receptor and SMAD3 in human skin fibroblasts. Age 2014, 36, 9623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toledano, M.B.; Leonard, W.J. Modulation of transcription factor NF-kappa B binding activity by oxidation-reduction in vitro. Proc. Natl. Acad. Sci. USA 1991, 88, 4328–4332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, W.; Tsai, W.L.; Shao, R.X.; Wu, G.; Peng, L.F.; Barlow, L.L.; Chung, W.J.; Zhang, L.; Zhao, H.; Jang, J.Y.; et al. Hepatitis C virus regulates transforming growth factor beta1 production through the generation of reactive oxygen species in a nuclear factor kappaB-dependent manner. Gastroenterology 2010, 138, 2509–2518, 2518 e1. [Google Scholar] [CrossRef] [Green Version]
- Bellocq, A.; Azoulay, E.; Marullo, S.; Flahault, A.; Fouqueray, B.; Philippe, C.; Cadranel, J.; Baud, L. Reactive oxygen and nitrogen intermediates increase transforming growth factor-beta1 release from human epithelial alveolar cells through two different mechanisms. Am. J. Respir. Cell Mol. Biol. 1999, 21, 128–136. [Google Scholar] [CrossRef] [Green Version]
- Fukawa, T.; Kajiya, H.; Ozeki, S.; Ikebe, T.; Okabe, K. Reactive oxygen species stimulates epithelial mesenchymal transition in normal human epidermal keratinocytes via TGF-beta secretion. Exp. Cell Res. 2012, 318, 1926–1932. [Google Scholar] [CrossRef]
- Vijayachandra, K.; Higgins, W.; Lee, J.; Glick, A. Induction of p16ink4a and p19ARF by TGFbeta1 contributes to growth arrest and senescence response in mouse keratinocytes. Mol. Carcinog. 2009, 48, 181–186. [Google Scholar] [CrossRef]
- Nogueira, V.; Hay, N. Molecular pathways: Reactive oxygen species homeostasis in cancer cells and implications for cancer therapy. Clin. Cancer Res. 2013, 19, 4309–4314. [Google Scholar] [CrossRef] [Green Version]
- Simeone, D.M.; Zhang, L.Z.; Graziano, K.; Nicke, B.; Pham, T.; Schaefer, C.; Logsdon, C.D. Smad4 mediates activation of mitogen-activated protein kinases by TGF-beta in pancreatic acinar cells. Am. J. Physiol. Cell Ph. 2001, 281, C311–C319. [Google Scholar] [CrossRef]
- Lee, M.K.; Pardoux, C.; Hall, M.C.; Lee, P.S.; Warburton, D.; Qing, J.; Smith, S.M.; Derynck, R. TGF-beta activates Erk MAP kinase signalling through direct phosphorylation of ShcA. Embo. J. 2007, 26, 3957–3967. [Google Scholar] [CrossRef] [Green Version]
- Son, Y.; Kim, S.; Chung, H.T.; Pae, H.O. Reactive oxygen species in the activation of MAP kinases. Methods Enzymol. 2013, 528, 27–48. [Google Scholar]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [Green Version]
- Moustakas, A.; Heldin, C.H. The regulation of TGFbeta signal transduction. Development 2009, 136, 3699–3714. [Google Scholar] [CrossRef] [Green Version]
- Deckers, M.; van Dinther, M.; Buijs, J.; Que, I.; Lowik, C.; van der Pluijm, G.; ten Dijke, P. The tumor suppressor Smad4 is required for transforming growth factor beta-induced epithelial to mesenchymal transition and bone metastasis of breast cancer cells. Cancer Res. 2006, 66, 2202–2209. [Google Scholar] [CrossRef] [Green Version]
- Roberts, A.B.; Tian, F.; Byfield, S.D.; Stuelten, C.; Ooshima, A.; Saika, S.; Flanders, K.C. Smad3 is key to TGF-beta-mediated epithelial-to-mesenchymal transition, fibrosis, tumor suppression and metastasis. Cytokine Growth Factor Rev. 2006, 17, 19–27. [Google Scholar] [CrossRef]
- Hoot, K.E.; Lighthall, J.; Han, G.; Lu, S.L.; Li, A.; Ju, W.; Kulesz-Martin, M.; Bottinger, E.; Wang, X.J. Keratinocyte-specific Smad2 ablation results in increased epithelial-mesenchymal transition during skin cancer formation and progression. J. Clin. Investig. 2008, 118, 2722–2732. [Google Scholar] [CrossRef] [Green Version]
- Oft, M.; Akhurst, R.J.; Balmain, A. Metastasis is driven by sequential elevation of H-ras and Smad2 levels. Nat. Cell. Biol. 2002, 4, 487–494. [Google Scholar] [CrossRef]
- Gorowiec, M.R.; Borthwick, L.A.; Parker, S.M.; Kirby, J.A.; Saretzki, G.C.; Fisher, A.J. Free radical generation induces epithelial-to-mesenchymal transition in lung epithelium via a TGF-beta1-dependent mechanism. Free Radic. Biol. Med. 2012, 52, 1024–1032. [Google Scholar] [CrossRef]
- Roberts, C.K.; Sindhu, K.K. Oxidative stress and metabolic syndrome. Life Sci. 2009, 84, 705–712. [Google Scholar] [CrossRef]
- Tremain, R.; Marko, M.; Kinnimulki, V.; Ueno, H.; Bottinger, E.; Glick, A. Defects in TGF-beta signaling overcome senescence of mouse keratinocytes expressing v-Ha-ras. Oncogene 2000, 19, 1698–1709. [Google Scholar] [CrossRef] [Green Version]
- Hassona, Y.; Cirillo, N.; Lim, K.P.; Herman, A.; Mellone, M.; Thomas, G.J.; Pitiyage, G.N.; Parkinson, E.K.; Prime, S.S. Progression of genotype-specific oral cancer leads to senescence of cancer-associated fibroblasts and is mediated by oxidative stress and TGF-beta. Carcinogenesis 2013, 34, 1286–1295. [Google Scholar] [CrossRef] [Green Version]
- Massague, J. TGFbeta in Cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef] [Green Version]
- Lebrun, J.J. The Dual Role of TGFbeta in Human Cancer: From Tumor Suppression to Cancer Metastasis. ISRN Mol. Biol. 2012, 2012, 381428. [Google Scholar]
- Gorelik, L.; Flavell, R.A. Immune-mediated eradication of tumors through the blockade of transforming growth factor-beta signaling in T cells. Nat. Med. 2001, 7, 1118–1122. [Google Scholar] [CrossRef]
- Sakurai, T.; Kudo, M. Signaling Pathways Governing Tumor Angiogenesis. Oncology 2011, 81, 24–29. [Google Scholar] [CrossRef]
- Zhao, H.; Wei, J.; Sun, J. Roles of TGF-beta signaling pathway in tumor microenvirionment and cancer therapy. Int. Immunopharmacol. 2020, 89, 107101. [Google Scholar] [CrossRef]
- Carmona-Cuenca, I.; Roncero, C.; Sancho, P.; Caja, L.; Fausto, N.; Fernandez, M.; Fabregat, I. Upregulation of the NADPH oxidase NOX4 by TGF-beta in hepatocytes is required for its pro-apoptotic activity. J. Hepatol. 2008, 49, 965–976. [Google Scholar] [CrossRef]
- Kim, Y.M.; Cho, M. Activation of NADPH oxidase subunit NCF4 induces ROS-mediated EMT signaling in HeLa cells. Cell Signal. 2014, 26, 784–796. [Google Scholar] [CrossRef]
- Arsalane, K.; Dubois, C.M.; Muanza, T.; Begin, R.; Boudreau, F.; Asselin, C.; Cantin, A.M. Transforming growth factor-beta(1) is a potent inhibitor of glutathione synthesis in the lung epithelial cell line A549: Transcriptional effect on the GSH rate-limiting enzyme gamma-glutamylcysteine synthetase. Am. J. Resp. Cell. Mol. 1997, 17, 599–607. [Google Scholar] [CrossRef]
- Zhu, G.H.; Huang, C.; Feng, Z.Z.; Lv, X.H.; Qiu, Z.J. Hypoxia-Induced Snail Expression Through Transcriptional Regulation by HIF-1 alpha in Pancreatic Cancer Cells. Digest. Dis. Sci. 2013, 58, 3503–3515. [Google Scholar] [CrossRef]
- Souto, E.B.; Severino, P.; Basso, R.; Santana, M.H. Encapsulation of antioxidants in gastrointestinal-resistant nanoparticulate carriers. Methods Mol. Biol. 2013, 1028, 37–46. [Google Scholar] [PubMed]
- Barcellos-Hoff, M.H.; Dix, T.A. Redox-mediated activation of latent transforming growth factor-beta 1. Mol. Endocrinol. 1996, 10, 1077–1083. [Google Scholar] [PubMed] [Green Version]
- Leonarduzzi, G.; Scavazza, A.; Biasi, F.; Chiarpotto, E.; Camandola, S.; Vogel, S.; Dargel, R.; Poli, G. The lipid peroxidation end product 4-hydroxy-2,3-nonenal up-regulates transforming growth factor beta1 expression in the macrophage lineage: A link between oxidative injury and fibrosclerosis. FASEB J. 1997, 11, 851–857. [Google Scholar] [CrossRef] [PubMed]
- Cucoranu, I.; Clempus, R.; Dikalova, A.; Phelan, P.J.; Ariyan, S.; Dikalov, S.; Sorescu, D. NAD(P)H oxidase 4 mediates transforming growth factor-beta1-induced differentiation of cardiac fibroblasts into myofibroblasts. Circ. Res. 2005, 97, 900–907. [Google Scholar] [CrossRef] [Green Version]
- Carnesecchi, S.; Deffert, C.; Donati, Y.; Basset, O.; Hinz, B.; Preynat-Seauve, O.; Guichard, C.; Arbiser, J.L.; Banfi, B.; Pache, J.C.; et al. A key role for NOX4 in epithelial cell death during development of lung fibrosis. Antioxid. Redox. Signal. 2011, 15, 607–619. [Google Scholar] [CrossRef] [Green Version]
- Amara, N.; Goven, D.; Prost, F.; Muloway, R.; Crestani, B.; Boczkowski, J. NOX4/NADPH oxidase expression is increased in pulmonary fibroblasts from patients with idiopathic pulmonary fibrosis and mediates TGF beta 1-induced fibroblast differentiation into myofibroblasts. Thorax 2010, 65, 733–738. [Google Scholar] [CrossRef] [Green Version]
- Wrighton, K.H.; Lin, X.; Feng, X.H. Phospho-control of TGF-beta superfamily signaling. Cell Res. 2009, 19, 8–20. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.S.; Song, M.C.; Kwak, I.H.; Park, T.J.; Lim, I.K. Constitutive induction of p-Erk1/2 accompanied by reduced activities of protein phosphatases 1 and 2A and MKP3 due to reactive oxygen species during cellular senescence. J. Biol. Chem. 2003, 278, 37497–37510. [Google Scholar] [CrossRef] [Green Version]
- Jiang, F.; Zhang, Y.; Dusting, G.J. NADPH oxidase-mediated redox signaling: Roles in cellular stress response, stress tolerance, and tissue repair. Pharmacol. Rev. 2011, 63, 218–242. [Google Scholar] [CrossRef] [Green Version]
- Moustakas, A.; Heldin, C.H. Non-Smad TGF-beta signals. J. Cell Sci. 2005, 118 Pt 16, 3573–3584. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chung, J.; Huda, M.N.; Shin, Y.; Han, S.; Akter, S.; Kang, I.; Ha, J.; Choe, W.; Choi, T.G.; Kim, S.S. Correlation between Oxidative Stress and Transforming Growth Factor-Beta in Cancers. Int. J. Mol. Sci. 2021, 22, 13181. https://doi.org/10.3390/ijms222413181
Chung J, Huda MN, Shin Y, Han S, Akter S, Kang I, Ha J, Choe W, Choi TG, Kim SS. Correlation between Oxidative Stress and Transforming Growth Factor-Beta in Cancers. International Journal of Molecular Sciences. 2021; 22(24):13181. https://doi.org/10.3390/ijms222413181
Chicago/Turabian StyleChung, Jinwook, Md Nazmul Huda, Yoonhwa Shin, Sunhee Han, Salima Akter, Insug Kang, Joohun Ha, Wonchae Choe, Tae Gyu Choi, and Sung Soo Kim. 2021. "Correlation between Oxidative Stress and Transforming Growth Factor-Beta in Cancers" International Journal of Molecular Sciences 22, no. 24: 13181. https://doi.org/10.3390/ijms222413181