
Photodynamic Therapy-Mediated Immune Responses in Three-Dimensional Tumor Models



 and
and 
Abstract
:1. Introduction
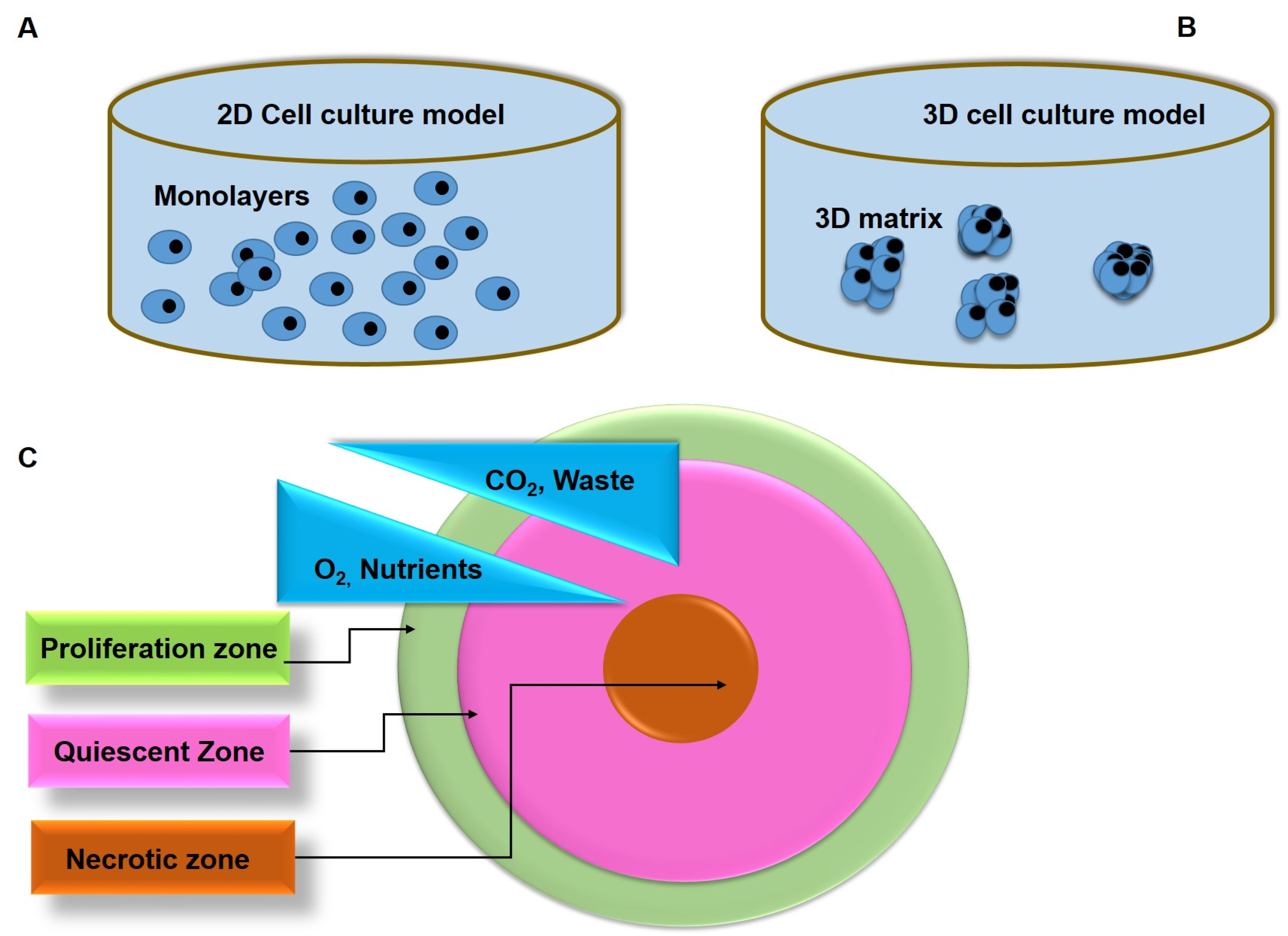
2. Comparison of 2D and 3D Tumor Models
3. Methods for 3D Tumor Generation
3.1. Hanging Drop Method
3.2. Liquid Overlay Method
3.3. Bioreactor-Based 3D Culture Method
3.4. Hydrogels
3.5. Magnetic Levitation Method
4. PDT-Induced Antitumor Immune Responses
4.1. PDT and Innate Immune Response
4.2. PDT and Adaptive Immune Response
5. Experimental Studies on Immune Responses to PDT in Cancer Treatment
6. Clinical Studies on PDT Immune Responses in Cancer Treatment
7. Enhancing PDT-Induced Antitumor Immune Responses
7.1. Intracellular Accumulation of PSs
7.2. ER-Targeted PDT
7.3. Mitochondria Targeted PDT
7.4. Application of Nanotechnology
8. Experimental Models for Evaluation of PDT-Induced Antitumor Immune Responses
8.1. Immunomodulation
8.1.1. Immunostimulants
8.1.2. Blocking Immunocompromising (Cellular) Factors
8.1.3. Recognition of Tumor-Associated Antigens (TAA)
9. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 2D | Two-dimensional |
| 3D | Three-dimensional |
| APCs | Antigen-presenting cells |
| BCC | Basal cell carcinoma |
| BCG | Bacillus Calmette–Geurin |
| CLIPT | Continuous low irradiance PDT (CLIPT) |
| CRT | Calreticulin |
| CTL | Cytotoxic tumor-specific T lymphocytes |
| DAMPs | Damage-associated molecular patterns |
| DCs | Dendritic cells |
| ECM | Extracellular matrix |
| EPR | Enhanced permeability and retention |
| ER | Endoplasmic reticulum |
| ESCC | Esophageal squamous cell carcinoma |
| HMGB1 | High-mobility group |
| Hsp60 | Heat shock-related proteins |
| ICD | Immunogenic cell death |
| IL | Interleukins |
| IFNγ | Interferon-gamma |
| LLC | Lewis lung carcinoma |
| MCTS | Multicellular tumor spheroids |
| MCWE | Mycobacterium cell wall extract |
| MDSCs | Myeloid-derived suppressive cells |
| MHC | Major histocompatibility complex |
| NK | Natural killer cells |
| NP | Nanoparticles |
| PBMCs | Peripheral blood mononuclear |
| PDT | Photodynamic therapy |
| PGE2 | Prostaglandin E2 |
| PRRs | Pattern recognition receptors |
| PS | Photosensitizer |
| ROS | Reactive oxygen species |
| TAA | Tumor-associated antigens |
| TGF-β | Transforming growth factor beta |
| TME | Tumor microenvironment |
| TNF-α | Tumor necrosis factor alpha |
| TPP | Triphenylphosphonium |
| Tregs | Regulatory T cells |
| TRL | Toll-like receptor |
| VIN | Vulval intraepithelial neoplasia |
References
- Kruger, C.A.; Abrahamse, H. Utilisation of Targeted Nanoparticle Photosensitiser Drug Delivery Systems for the Enhancement of Photodynamic Therapy. Molecules 2018, 23, 2628. [Google Scholar] [CrossRef] [Green Version]
- dos Santos, A.F.; de Almeida, D.R.Q.; Terra, L.F.; Baptista, M.S.; Labriola, L. Photodynamic Therapy in Cancer Treatment—An Update Review. J. Cancer Metastatis Treat. 2019, 5, 25. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Wang, B.; Zhao, R.; Zhang, Q.; Kong, X. Multifunctional Nanoparticles as Photosensitizer Delivery Carriers for Enhanced Photodynamic Cancer Therapy. Mater. Sci. Eng. C Mater. Biol. Appl. 2020, 115, 111099. [Google Scholar] [CrossRef]
- Yang, Y.; Hu, Y.; Wang, H. Targeting Antitumor Immune Response for Enhancing the Efficacy of Photodynamic Therapy of Cancer: Recent Advances and Future Perspectives. Oxid. Med. Cell. Longev. 2016, 2016, e5274084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reginato, E.; Wolf, P.; Hamblin, M.R. Immune Response after Photodynamic Therapy Increases Anti-Cancer and Anti-Bacterial Effects. World J. Immunol. 2014, 4, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Beltrán Hernández, I.; Yu, Y.; Ossendorp, F.; Korbelik, M.; Oliveira, S. Preclinical and Clinical Evidence of Immune Responses Triggered in Oncologic Photodynamic Therapy: Clinical Recommendations. J. Clin. Med. 2020, 9, 333. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Dai, Z. Antitumor Immune Responses Induced by Photodynamic and Sonodynamic Therapy: A Narrative Review. J. Bio-X Res. 2021, 4, 77–86. [Google Scholar] [CrossRef]
- Pitt, J.M.; Marabelle, A.; Eggermont, A.; Soria, J.-C.; Kroemer, G.; Zitvogel, L. Targeting the Tumor Microenvironment: Removing Obstruction to Anticancer Immune Responses and Immunotherapy. Ann. Oncol. 2016, 27, 1482–1492. [Google Scholar] [CrossRef]
- Hwang, H.S.; Shin, H.; Han, J.; Na, K. Combination of Photodynamic Therapy (PDT) and Anti-Tumor Immunity in Cancer Therapy. J. Pharm. Investig. 2018, 48, 143–151. [Google Scholar] [CrossRef] [Green Version]
- Pinto, B.; Henriques, A.C.; Silva, P.M.A.; Bousbaa, H. Three-Dimensional Spheroids as In Vitro Preclinical Models for Cancer Research. Pharmaceutics 2020, 12, 1186. [Google Scholar] [CrossRef]
- Poggi, A.; Villa, F.; Fernadez, J.L.C.; Costa, D.; Zocchi, M.R.; Benelli, R. Three-Dimensional Culture Models to Study Innate Anti-Tumor Immune Response: Advantages and Disadvantages. Cancers 2021, 13, 3417. [Google Scholar] [CrossRef] [PubMed]
- Chaicharoenaudomrung, N.; Kunhorm, P.; Noisa, P. Three-Dimensional Cell Culture Systems as an In Vitro Platform for Cancer and Stem Cell Modeling. World J. Stem Cells 2019, 11, 1065–1083. [Google Scholar] [CrossRef]
- Mohammad-Hadi, L.; MacRobert, A.J.; Loizidou, M.; Yaghini, E. Photodynamic Therapy in 3D Cancer Models and the Utilisation of Nanodelivery Systems. Nanoscale 2018, 10, 1570–1581. [Google Scholar] [CrossRef] [Green Version]
- Yoon, P.S.; Del Piccolo, N.; Shirure, V.S.; Peng, Y.; Kirane, A.; Canter, R.J.; Fields, R.C.; George, S.C.; Gholami, S. Advances in Modeling the Immune Microenvironment of Colorectal Cancer. Front. Immunol. 2021, 11, 3876. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Keller, E.T.; Garfield, D.H.; Shen, K.; Wang, J. Stromal Cells in Tumor Microenvironment and Breast Cancer. Cancer Metastasis Rev. 2013, 32, 303–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khanna, S.; Chauhan, A.; Bhatt, A.N.; Dwarakanath, B.S.R. Chapter 13—Multicellular Tumor Spheroids as In Vitro Models for Studying Tumor Responses to Anticancer Therapies. In Animal Biotechnology, 2nd ed.; Verma, A.S., Singh, A., Eds.; Academic Press: Cambridge, MA, USA, 2020; pp. 251–268. ISBN 978-0-12-811710-1. [Google Scholar]
- Lazzari, G.; Couvreur, P.; Mura, S. Multicellular Tumor Spheroids: A Relevant 3D Model for the In Vitro Preclinical Investigation of Polymer Nanomedicines. Polym. Chem. 2017, 8, 4947–4969. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Farach-Carson, M.C.; Jia, X. Three-Dimensional In Vitro Tumor Models for Cancer Research and Drug Evaluation. Biotechnol. Adv. 2014, 32, 1256–1268. [Google Scholar] [CrossRef] [Green Version]
- Nath, S.; Devi, G.R. Three-Dimensional Culture Systems in Cancer Research: Focus on Tumor Spheroid Model. Pharmacol. Ther. 2016, 163, 94–108. [Google Scholar] [CrossRef] [Green Version]
- Gurski, L.A.; Petrelli, N.J.; Jia, X.; Farach-Carson, M.C. 3D Matrices for Anti-Cancer Drug Testing and Development. Oncol. Issues 2010, 25, 20–25. [Google Scholar] [CrossRef]
- Takai, A.; Fako, V.; Dang, H.; Forgues, M.; Yu, Z.; Budhu, A.; Wang, X.W. Three-Dimensional Organotypic Culture Models of Human Hepatocellular Carcinoma. Sci. Rep. 2016, 6, 21174. [Google Scholar] [CrossRef] [Green Version]
- Florczyk, S.J.; Wang, K.; Jana, S.; Wood, D.L.; Sytsma, S.K.; Sham, J.; Kievit, F.M.; Zhang, M. Porous Chitosan-Hyaluronic Acid Scaffolds as a Mimic of Glioblastoma Microenvironment ECM. Biomaterials 2013, 34, 10143–10150. [Google Scholar] [CrossRef] [Green Version]
- Antoni, D.; Burckel, H.; Josset, E.; Noel, G. Three-Dimensional Cell Culture: A Breakthrough in Vivo. Int. J. Mol. Sci. 2015, 16, 5517–5527. [Google Scholar] [CrossRef]
- Breslin, S.; O’Driscoll, L. The Relevance of Using 3D Cell Cultures, in Addition to 2D Monolayer Cultures, When Evaluating Breast Cancer Drug Sensitivity and Resistance. Oncotarget 2016, 7, 45745–45756. [Google Scholar] [CrossRef] [Green Version]
- Krausz, E.; de Hoogt, R.; Gustin, E.; Cornelissen, F.; Grand-Perret, T.; Janssen, L.; Vloemans, N.; Wuyts, D.; Frans, S.; Axel, A.; et al. Translation of a Tumor Microenvironment Mimicking 3D Tumor Growth Co-Culture Assay Platform to High-Content Screening. J. Biomol. Screen. 2013, 18, 54–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bingel, C.; Koeneke, E.; Ridinger, J.; Bittmann, A.; Sill, M.; Peterziel, H.; Wrobel, J.K.; Rettig, I.; Milde, T.; Fernekorn, U.; et al. Three-Dimensional Tumor Cell Growth Stimulates Autophagic Flux and Recapitulates Chemotherapy Resistance. Cell Death Dis. 2017, 8, e3013. [Google Scholar] [CrossRef] [Green Version]
- Ravi, M.; Paramesh, V.; Kaviya, S.R.; Anuradha, E.; Solomon, F.D.P. 3D Cell Culture Systems: Advantages and Applications. J. Cell Physiol. 2015, 230, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Ma, C.; Lan, Q.; Xu, T. 3D Bioprinted Glioma Stem Cells for Brain Tumor Model and Applications of Drug Susceptibility. Biofabrication 2016, 8, 045005. [Google Scholar] [CrossRef] [PubMed]
- Hickman, J.A.; Graeser, R.; de Hoogt, R.; Vidic, S.; Brito, C.; Gutekunst, M.; van der Kuip, H.; Consortium, I.P. Three-Dimensional Models of Cancer for Pharmacology and Cancer Cell Biology: Capturing Tumor Complexity In Vitro/Ex Vivo. Biotechnol. J. 2014, 9, 1115–1128. [Google Scholar] [CrossRef]
- Friedrich, J.; Seidel, C.; Ebner, R.; Kunz-Schughart, L.A. Spheroid-Based Drug Screen: Considerations and Practical Approach. Nat. Protoc. 2009, 4, 309–324. [Google Scholar] [CrossRef]
- Lv, D.; Hu, Z.; Lu, L.; Lu, H.; Xu, X. Three-Dimensional Cell Culture: A Powerful Tool in Tumor Research and Drug Discovery. Oncol. Lett. 2017, 14, 6999–7010. [Google Scholar] [CrossRef] [Green Version]
- Ryu, N.-E.; Lee, S.-H.; Park, H. Spheroid Culture System Methods and Applications for Mesenchymal Stem Cells. Cells 2019, 8, 1620. [Google Scholar] [CrossRef] [Green Version]
- Huang, B.-W.; Gao, J.-Q. Application of 3D Cultured Multicellular Spheroid Tumor Models in Tumor-Targeted Drug Delivery System Research. J. Control Release 2018, 270, 246–259. [Google Scholar] [CrossRef]
- Schneider, L.; Kalt, M.; Larocca, M.; Babu, V.; Spingler, B. Potent PBS/Polysorbate-Soluble Transplatin-Derived Porphyrin-Based Photosensitizers for Photodynamic Therapy. Inorg. Chem. 2021, 60, 9416–9426. [Google Scholar] [CrossRef] [PubMed]
- Karges, J.; Kuang, S.; Maschietto, F.; Blacque, O.; Ciofini, I.; Chao, H.; Gasser, G. Rationally Designed Ruthenium Complexes for 1- and 2-Photon Photodynamic Therapy. Nat. Commun. 2020, 11, 3262. [Google Scholar] [CrossRef]
- Sontheimer-Phelps, A.; Hassell, B.A.; Ingber, D.E. Modelling Cancer in Microfluidic Human Organs-on-Chips. Nat. Rev. Cancer 2019, 19, 65–81. [Google Scholar] [CrossRef] [PubMed]
- Joseph, J.S.; Malindisa, S.T.; Ntwasa, M. Two-Dimensional (2D) and Three-Dimensional (3D) Cell Culturing in Drug Discovery; IntechOpen: London, UK, 2018; ISBN 978-1-78984-867-0. [Google Scholar]
- Li, Y.; Kumacheva, E. Hydrogel Microenvironments for Cancer Spheroid Growth and Drug Screening. Sci. Adv. 2018, eaas8998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nii, T.; Makino, K.; Tabata, Y. Three-Dimensional Culture System of Cancer Cells Combined with Biomaterials for Drug Screening. Cancers 2020, 12, 2754. [Google Scholar] [CrossRef]
- Nii, T.; Makino, K.; Tabata, Y. A Cancer Invasion Model Combined with Cancer-Associated Fibroblasts Aggregates Incorporating Gelatin Hydrogel Microspheres Containing a P53 Inhibitor. Tissue Eng. Part. C Methods 2019, 25, 711–720. [Google Scholar] [CrossRef]
- Anil-Inevi, M.; Yaman, S.; Yildiz, A.A.; Mese, G.; Yalcin-Ozuysal, O.; Tekin, H.C.; Ozcivici, E. Biofabrication of in Situ Self Assembled 3D Cell Cultures in a Weightlessness Environment Generated Using Magnetic Levitation. Sci. Rep. 2018, 8, 7239. [Google Scholar] [CrossRef]
- Kim, J.A.; Choi, J.-H.; Kim, M.; Rhee, W.J.; Son, B.; Jung, H.-K.; Park, T.H. High-Throughput Generation of Spheroids Using Magnetic Nanoparticles for Three-Dimensional Cell Culture. Biomaterials 2013, 34, 8555–8563. [Google Scholar] [CrossRef]
- Lewis, N.S.; Lewis, E.E.; Mullin, M.; Wheadon, H.; Dalby, M.J.; Berry, C.C. Magnetically Levitated Mesenchymal Stem Cell Spheroids Cultured with a Collagen Gel Maintain Phenotype and Quiescence. J. Tissue Eng. 2017, 8, 2041731417704428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mroz, P.; Hashmi, J.T.; Huang, Y.-Y.; Lange, N.; Hamblin, M.R. Stimulation of Anti-Tumor Immunity by Photodynamic Therapy. Expert Rev. Clin. Immunol. 2011, 7, 75–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vatansever, F.; Hamblin, M.R. Photodynamic Therapy and Antitumor Immune Response. Cancer Immunol. 2015, 383–399. [Google Scholar] [CrossRef]
- Wachowska, M.; Muchowicz, A.; Demkow, U. Immunological Aspects of Antitumor Photodynamic Therapy Outcome. Cent. Eur. J. Immunol. 2015, 40, 481–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donohoe, C.; Senge, M.O.; Arnaut, L.G.; Gomes-da-Silva, L.C. Cell Death in Photodynamic Therapy: From Oxidative Stress to Anti-Tumor Immunity. Biochim. Biophys. Acta (BBA) Rev. Cancer 2019, 1872, 188308. [Google Scholar] [CrossRef] [PubMed]
- Korbelik, M.; Zhang, W.; Merchant, S. Involvement of Damage-Associated Molecular Patterns in Tumor Response to Photodynamic Therapy: Surface Expression of Calreticulin and High-Mobility Group Box-1 Release. Cancer Immunol. Immunother. 2011, 60, 1431–1437. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.W.; Snyder, E.; Miller, J.; Carter, S.; Houser, C.; Klampatsa, A.; Albelda, S.M.; Cengel, K.A.; Busch, T.M. Luminol Chemiluminescence Reports Photodynamic Therapy-Generated Neutrophil Activity In Vivo and Serves as a Biomarker of Therapeutic Efficacy. Photochem. Photobiol. 2019, 95, 430–438. [Google Scholar] [CrossRef] [Green Version]
- Trempolec, N.; Doix, B.; Degavre, C.; Brusa, D.; Bouzin, C.; Riant, O.; Feron, O. Photodynamic Therapy-Based Dendritic Cell Vaccination Suited to Treat Peritoneal Mesothelioma. Cancers 2020, 12, 545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doix, B.; Trempolec, N.; Riant, O.; Feron, O. Low Photosensitizer Dose and Early Radiotherapy Enhance Antitumor Immune Response of Photodynamic Therapy-Based Dendritic Cell Vaccination. Front. Oncol. 2019, 9, 811. [Google Scholar] [CrossRef]
- Garg, A.D.; Krysko, D.V.; Verfaillie, T.; Kaczmarek, A.; Ferreira, G.B.; Marysael, T.; Rubio, N.; Firczuk, M.; Mathieu, C.; Roebroek, A.J.M.; et al. A Novel Pathway Combining Calreticulin Exposure and ATP Secretion in Immunogenic Cancer Cell Death. EMBO J. 2012, 31, 1062–1079. [Google Scholar] [CrossRef] [Green Version]
- Garg, A.; Vandenberk, L.; Koks, C.; Verschuere, T.; Boon, L.; Gool, S.; Agostinis, P. Dendritic Cell Vaccines Based on Immunogenic Cell Death Elicit Danger Signals and T Cell–Driven Rejection of High-Grade Glioma. Sci. Transl. Med. 2016, 8, 328ra27. [Google Scholar] [CrossRef]
- Qin, J.; Kunda, N.; Qiao, G.; Calata, J.F.; Pardiwala, K.; Prabhakar, B.S.; Maker, A.V. Colon Cancer Cell Treatment with Rose Bengal Generates a Protective Immune Response via Immunogenic Cell Death. Cell Death Dis. 2017, 8, e2584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, J.; Fan, Z.; Zhou, F.; Wang, X.; Shi, L.; Zhang, H.; Wang, P.; Yang, D.; Zhang, L.; Chen, W.R.; et al. Improvement of DC Vaccine with ALA-PDT Induced Immunogenic Apoptotic Cells for Skin Squamous Cell Carcinoma. Oncotarget 2015, 6, 17135–17146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Ji, J.; Zhang, H.; Fan, Z.; Zhang, L.; Shi, L.; Zhou, F.; Chen, W.R.; Wang, H.; Wang, X. Stimulation of Dendritic Cells by DAMPs in ALA-PDT Treated SCC Tumor Cells. Oncotarget 2015, 6, 44688–44702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Etminan, N.; Peters, C.; Lakbir, D.; Bünemann, E.; Börger, V.; Sabel, M.C.; Hänggi, D.; Steiger, H.-J.; Stummer, W.; Sorg, R.V. Heat-Shock Protein 70-Dependent Dendritic Cell Activation by 5-Aminolevulinic Acid-Mediated Photodynamic Treatment of Human Glioblastoma Spheroids in vitro. Br. J. Cancer 2011, 105, 961–969. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, P.; Wang, X.; Shi, L.; Fan, Z.; Zhang, G.; Yang, D.; Bahavar, C.F.; Zhou, F.; Chen, W.R.; et al. Antitumor Effects of DC Vaccine with ALA-PDT-Induced Immunogenic Apoptotic Cells for Skin Squamous Cell Carcinoma in Mice. Technol. Cancer Res. Treat. 2018, 17, 1533033818785275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lobo, A.C.S.; Gomes-da-Silva, L.C.; Rodrigues-Santos, P.; Cabrita, A.; Santos-Rosa, M.; Arnaut, L.G. Immune Responses after Vascular Photodynamic Therapy with Redaporfin. J. Clin. Med. 2019, 9, 104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turubanova, V.D.; Balalaeva, I.V.; Mishchenko, T.A.; Catanzaro, E.; Alzeibak, R.; Peskova, N.N.; Efimova, I.; Bachert, C.; Mitroshina, E.V.; Krysko, O.; et al. Immunogenic Cell Death Induced by a New Photodynamic Therapy Based on Photosens and Photodithazine. J. Immunother. Cancer 2019, 7, 350. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Liu, L.; Liang, R.; Zhou, H.; Pan, H.; Zhang, S.; Cai, L. Tumor-Targeted Nanoplatform for in Situ Oxygenation-Boosted Immunogenic Phototherapy of Colorectal Cancer. Acta Biomater. 2020, 104, 188–197. [Google Scholar] [CrossRef]
- Ogawa, M.; Tomita, Y.; Nakamura, Y.; Lee, M.-J.; Lee, S.; Tomita, S.; Nagaya, T.; Sato, K.; Yamauchi, T.; Iwai, H.; et al. Immunogenic Cancer Cell Death Selectively Induced by near Infrared Photoimmunotherapy Initiates Host Tumor Immunity. Oncotarget 2017, 8, 10425–10436. [Google Scholar] [CrossRef] [Green Version]
- Liang, R.; Liu, L.; He, H.; Chen, Z.; Han, Z.; Luo, Z.; Wu, Z.; Zheng, M.; Ma, Y.; Cai, L. Oxygen-Boosted Immunogenic Photodynamic Therapy with Gold Nanocages@manganese Dioxide to Inhibit Tumor Growth and Metastases. Biomaterials 2018, 177, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Liu, L.; Liang, R.; Luo, Z.; He, H.; Wu, Z.; Tian, H.; Zheng, M.; Ma, Y.; Cai, L. Bioinspired Hybrid Protein Oxygen Nanocarrier Amplified Photodynamic Therapy for Eliciting Anti-Tumor Immunity and Abscopal Effect. ACS Nano 2018, 12, 8633–8645. [Google Scholar] [CrossRef] [PubMed]
- Silva, P.D.; Bano, S.; Pogue, B.W.; Wang, K.K.; Maytin, E.V.; Hasan, T. Photodynamic Priming with Triple-Receptor Targeted Nanoconjugates That Trigger T Cell-Mediated Immune Responses in a 3D In Vitro Heterocellular Model of Pancreatic Cancer. Nanophotonics 2021, 10, 3199–3214. [Google Scholar] [CrossRef]
- Li, Z.; Wang, C.; Deng, H.; Wu, J.; Huang, H.; Sun, R.; Zhang, H.; Xiong, X.; Feng, M. Robust Photodynamic Therapy Using 5-ALA-Incorporated Nanocomplexes Cures Metastatic Melanoma through Priming of CD4+CD8+ Double Positive T Cells. Adv. Sci. 2019, 6, 1802057. [Google Scholar] [CrossRef] [Green Version]
- Morais, J.A.V.; Almeida, L.R.; Rodrigues, M.C.; Azevedo, R.B.; Muehlmann, L.A. The Induction of Immunogenic Cell Death by Photodynamic Therapy in B16F10 Cells In Vitro Is Effected by the Concentration of the Photosensitizer. Photodiagn. Photodyn. Ther. 2021, 35, 102392. [Google Scholar] [CrossRef]
- Abdel-Hady, E.S.; Martin-Hirsch, P.; Duggan-Keen, M.; Stern, P.L.; Moore, J.V.; Corbitt, G.; Kitchener, H.C.; Hampson, I.N. Immunological and Viral Factors Associated with the Response of Vulval Intraepithelial Neoplasia to Photodynamic Therapy. Cancer Res. 2001, 61, 192–196. [Google Scholar] [PubMed]
- Kabingu, E.; Oseroff, A.R.; Wilding, G.E.; Gollnick, S.O. Enhanced Systemic Immune Reactivity to a Basal Cell Carcinoma Associated Antigen Following Photodynamic Therapy. Clin. Cancer Res. 2009, 15, 4460–4466. [Google Scholar] [CrossRef] [Green Version]
- Thong, P.S.-P.; Ong, K.-W.; Goh, N.S.-G.; Kho, K.-W.; Manivasager, V.; Bhuvaneswari, R.; Olivo, M.; Soo, K.-C. Photodynamic-Therapy-Activated Immune Response against Distant Untreated Tumours in Recurrent Angiosarcoma. Lancet Oncol. 2007, 8, 950–952. [Google Scholar] [CrossRef]
- Morrison, S.A.; Hill, S.L.; Rogers, G.S.; Graham, R.A. Efficacy and Safety of Continuous Low-Irradiance Photodynamic Therapy in the Treatment of Chest Wall Progression of Breast Cancer. J. Surg. Res. 2014, 192, 235–241. [Google Scholar] [CrossRef]
- Reginato, E.; Lindenmann, J.; Langner, C.; Schweintzger, N.; Bambach, I.; Smolle-Jüttner, F.; Wolf, P. Photodynamic Therapy Downregulates the Function of Regulatory T Cells in Patients with Esophageal Squamous Cell Carcinoma. Photochem. Photobiol. Sci. 2014, 13, 1281–1289. [Google Scholar] [CrossRef]
- Gift, M.; Ann, K.; Mfouo Tynga, I.; Abrahamse, H. A Review of Nanoparticle Photosensitizer Drug Delivery Uptake Systems for Photodynamic Treatment of Lung Cancer. Photodiagn. Photodyn. Ther. 2018, 22, 147–154. [Google Scholar] [CrossRef]
- Kwiatkowski, S.; Knap, B.; Przystupski, D.; Saczko, J.; Kędzierska, E.; Knap-Czop, K.; Kotlińska, J.; Michel, O.; Kotowski, K.; Kulbacka, J. Photodynamic Therapy—Mechanisms, Photosensitizers and Combinations. Biomed. Pharmacother. 2018, 106, 1098–1107. [Google Scholar] [CrossRef]
- Yuan, B.; Liu, J.; Guan, R.; Jin, C.; Ji, L.; Chao, H. Endoplasmic Reticulum Targeted Cyclometalated Iridium(III) Complexes as Efficient Photodynamic Therapy Photosensitizers. Dalton Trans. 2019, 48, 6408–6415. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Z.; Dai, J.; Yu, M.; Li, J.; Shen, P.; Hu, R.; Lou, X.; Zhao, Z.; Tang, B.Z. Type I Photosensitizers Based on Phosphindole Oxide for Photodynamic Therapy: Apoptosis and Autophagy Induced by Endoplasmic Reticulum Stress. Chem. Sci. 2020, 11, 3405–3417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- West, A.P.; Shadel, G.S.; Ghosh, S. Mitochondria in Innate Immune Responses. Nat. Rev. Immunol. 2011, 11, 389–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, K.; Kaufman, R.J. From Endoplasmic-Reticulum Stress to the Inflammatory Response. Nature 2008, 454, 455–462. [Google Scholar] [CrossRef] [Green Version]
- Garg, A.D.; Dudek, A.M.; Ferreira, G.B.; Verfaillie, T.; Vandenabeele, P.; Krysko, D.V.; Mathieu, C.; Agostinis, P. ROS-Induced Autophagy in Cancer Cells Assists in Evasion from Determinants of Immunogenic Cell Death. Autophagy 2013, 9, 1292–1307. [Google Scholar] [CrossRef]
- Michaud, M.; Sukkurwala, A.Q.; Di Sano, F.; Zitvogel, L.; Kepp, O.; Kroemer, G. Synthetic Induction of Immunogenic Cell Death by Genetic Stimulation of Endoplasmic Reticulum Stress. OncoImmunology 2014, 3, e28276. [Google Scholar] [CrossRef] [Green Version]
- Garg, A.D.; Agostinis, P. ER Stress, Autophagy and Immunogenic Cell Death in Photodynamic Therapy-Induced Anti-Cancer Immune Responses. Photochem. Photobiol. Sci. 2014, 13, 474–487. [Google Scholar] [CrossRef]
- Agostinis, P.; Berg, K.; Cengel, K.A.; Foster, T.H.; Girotti, A.W.; Gollnick, S.O.; Hahn, S.M.; Hamblin, M.R.; Juzeniene, A.; Kessel, D.; et al. Photodynamic Therapy of Cancer: An Update. CA Cancer J. Clin. 2011, 61, 250–281. [Google Scholar] [CrossRef]
- Md, S.; Haque, S.; Madheswaran, T.; Zeeshan, F.; Meka, V.S.; Radhakrishnan, A.K.; Kesharwani, P. Lipid Based Nanocarriers System for Topical Delivery of Photosensitizers. Drug Discov. Today 2017, 22, 1274–1283. [Google Scholar] [CrossRef]
- Li, W.; Yang, J.; Luo, L.; Jiang, M.; Qin, B.; Yin, H.; Zhu, C.; Yuan, X.; Zhang, J.; Luo, Z.; et al. Targeting Photodynamic and Photothermal Therapy to the Endoplasmic Reticulum Enhances Immunogenic Cancer Cell Death. Nat. Commun. 2019, 10, 3349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noh, I.; Lee, D.; Kim, H.; Jeong, C.-U.; Lee, Y.; Ahn, J.-O.; Hyun, H.; Park, J.-H.; Kim, Y.-C. Enhanced Photodynamic Cancer Treatment by Mitochondria-Targeting and Brominated Near-Infrared Fluorophores. Adv. Sci. 2018, 5, 1700481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Gao, N.; Hu, Y.; Jia, C.; Chou, T.; Du, H.; Wang, H. Gold Nanoparticle-Enhanced Photodynamic Therapy: Effects of Surface Charge and Mitochondrial Targeting. Ther. Deliv. 2015, 6, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zeng, F.; Wu, H.; Yu, C.; Wu, S. Dual-Targeting Nanosystem for Enhancing Photodynamic Therapy Efficiency. ACS Appl. Mater. Interfaces 2015, 7, 9287–9296. [Google Scholar] [CrossRef]
- Soler, D.C.; Ohtola, J.; Sugiyama, H.; Rodriguez, M.E.; Han, L.; Oleinick, N.L.; Lam, M.; Baron, E.D.; Cooper, K.D.; McCormick, T.S. Activated T Cells Exhibit Increased Uptake of Silicon Phthalocyanine Pc 4 and Increased Susceptibility to Pc 4-Photodynamic Therapy-Mediated Cell Death. Photochem. Photobiol. Sci. 2016, 15, 822–831. [Google Scholar] [CrossRef] [Green Version]
- Marrache, S.; Tundup, S.; Harn, D.A.; Dhar, S. Ex Vivo Programming of Dendritic Cells by Mitochondria-Targeted Nanoparticles to Produce Interferon-Gamma for Cancer Immunotherapy. ACS Nano 2013, 7, 7392–7402. [Google Scholar] [CrossRef] [PubMed]
- Senapati, S.; Mahanta, A.K.; Kumar, S.; Maiti, P. Controlled Drug Delivery Vehicles for Cancer Treatment and Their Performance. Signal Transduct. Target. Ther. 2018, 3, 7. [Google Scholar] [CrossRef] [Green Version]
- Tang, C.; Li, H. Application of Nanoparticles in the Early Diagnosis and Treatment of Tumors: Current Status and Progress. Tradit. Med. Res. 2020, 5, 34–43. [Google Scholar] [CrossRef]
- Monge-Fuentes, V.; Muehlmann, L.A.; de Azevedo, R.B. Perspectives on the Application of Nanotechnology in Photodynamic Therapy for the Treatment of Melanoma. Nano Rev. 2014, 5, 24381. [Google Scholar] [CrossRef] [Green Version]
- Nkune, N.; Kruger, C.; Abrahamse, H. Possible Enhancement of Photodynamic Therapy (PDT) Colorectal Cancer Treatment When Combined with Cannabidiol. Anti-Cancer Agents Med. Chem. 2020, 20, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Naidoo, C.; Kruger, C.A.; Abrahamse, H. Photodynamic Therapy for Metastatic Melanoma Treatment: A Review. Technol. Cancer Res. Treat. 2018, 17, 1533033818791795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, E.J.; Choi, D.G.; Shim, M.S. Targeted and Effective Photodynamic Therapy for Cancer Using Functionalized Nanomaterials. Acta Pharm. Sin. B 2016, 6, 297–307. [Google Scholar] [CrossRef] [Green Version]
- Chattopadhyay, S.; Dash, S.K.; Mandal, D.; Das, B.; Tripathy, S.; Dey, A.; Pramanik, P.; Roy, S. Metal Based Nanoparticles as Cancer Antigen Delivery Vehicles for Macrophage Based Antitumor Vaccine. Vaccine 2016, 34, 957–967. [Google Scholar] [CrossRef] [PubMed]
- Boraschi, D.; Italiani, P. From Antigen Delivery System to Adjuvanticy: The Board Application of Nanoparticles in Vaccinology. Vaccines 2015, 3, 930–939. [Google Scholar] [CrossRef] [Green Version]
- Tian, G.; Zhang, X.; Gu, Z.; Zhao, Y. Recent Advances in Upconversion Nanoparticles-Based Multifunctional Nanocomposites for Combined Cancer Therapy. Adv. Mater. 2015, 27, 7692–7712. [Google Scholar] [CrossRef] [PubMed]
- Aldinucci, A.; Turco, A.; Biagioli, T.; Toma, F.M.; Bani, D.; Guasti, D.; Manuelli, C.; Rizzetto, L.; Cavalieri, D.; Massacesi, L.; et al. Carbon Nanotube Scaffolds Instruct Human Dendritic Cells: Modulating Immune Responses by Contacts at the Nanoscale. Nano Lett. 2013, 13, 6098–6105. [Google Scholar] [CrossRef]
- Xiang, J.; Xu, L.; Gong, H.; Zhu, W.; Wang, C.; Xu, J.; Feng, L.; Cheng, L.; Peng, R.; Liu, Z. Antigen-Loaded Upconversion Nanoparticles for Dendritic Cell Stimulation, Tracking, and Vaccination in Dendritic Cell-Based Immunotherapy. ACS Nano 2015, 9, 6401–6411. [Google Scholar] [CrossRef]
- Pathak, R.K.; Kolishetti, N.; Dhar, S. Targeted Nanoparticles in Mitochondrial Medicine. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2015, 7, 315–329. [Google Scholar] [CrossRef] [Green Version]
- Irvine, D.J.; Hanson, M.C.; Rakhra, K.; Tokatlian, T. Synthetic Nanoparticles for Vaccines and Immunotherapy. Chem. Rev. 2015, 115, 11109–11146. [Google Scholar] [CrossRef] [Green Version]
- Lei, Z.; Ren, X.; Wang, S.; Liang, X.; Tang, Y. Immunocompromised and Immunocompetent Mouse Models for Head and Neck Squamous Cell Carcinoma. OncoTargets Ther. 2016, 9, 545–555. [Google Scholar] [CrossRef] [Green Version]
- Holzapfel, B.M.; Wagner, F.; Thibaudeau, L.; Levesque, J.-P.; Hutmacher, D.W. Concise Review: Humanized Models of Tumor Immunology in the 21st Century: Convergence of Cancer Research and Tissue Engineering. Stem Cells 2015, 33, 1696–1704. [Google Scholar] [CrossRef] [PubMed]
- Manoto, S.; Houreld, N.; Abrahamse, H. Resistance of Lung Cancer Cells Grown as Multicellular Tumour Spheroids to Zinc Sulfophthalocyanine Photosensitization. Int. J. Mol. Sci. 2015, 16, 10185–10200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.-I.; Lee, J.; Kwon, J.-L.; Park, H.-B.; Lee, S.-Y.; Kim, J.-Y.; Sung, J.; Kim, J.M.; Song, K.S.; Kim, K.-H. Scaffold-Free Coculture Spheroids of Human Colonic Adenocarcinoma Cells and Normal Colonic Fibroblasts Promote Tumorigenicity in Nude Mice. Transl. Oncol. 2016, 9, 79–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szade, K.; Zukowska, M.; Szade, A.; Collet, G.; Kloska, D.; Kieda, C.; Jozkowicz, A.; Dulak, J. Spheroid-Plug Model as a Tool to Study Tumor Development, Angiogenesis, and Heterogeneity in vivo. Tumor Biol. 2016, 37, 2481–2496. [Google Scholar] [CrossRef] [Green Version]
- Brodin, N.P.; Guha, C.; Tomé, W.A. Photodynamic Therapy and Its Role in Combined Modality Anticancer Treatment. Technol. Cancer Res. Treat. 2015, 14, 355–368. [Google Scholar] [CrossRef]
- Anzengruber, F.; Avci, P.; de Freitas, L.F.; Hamblin, M.R. T-Cell Mediated Anti-Tumor Immunity after Photodynamic Therapy: Why Does It Not Always Work and How Can We Improve It? Photochem. Photobiol. Sci. 2015, 14, 1492–1509. [Google Scholar] [CrossRef] [Green Version]
- Nath, S.; Obaid, G.; Hasan, T. The Course of Immune Stimulation by Photodynamic Therapy: Bridging Fundamentals of Photochemically Induced Immunogenic Cell Death to the Enrichment of T-Cell Repertoire. Photochem. Photobiol. 2019, 95, 1288–1305. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.S.; Mellman, I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Kalathil, S.G.; Thanavala, Y. High Immunosuppressive Burden in Cancer Patients: A Major Hurdle for Cancer Immunotherapy. Cancer Immunol. Immunother. 2016, 65, 813–819. [Google Scholar] [CrossRef] [Green Version]
- Circelli, L.; Tornesello, M.; Buonaguro, F.M.; Buonaguro, L. Use of Adjuvants for Immunotherapy. Hum. Vaccines Immunother. 2017, 13, 1774–1777. [Google Scholar] [CrossRef] [PubMed]
- Korbelik, M.; Sun, J.; Posakony, J.J. Interaction between Photodynamic Therapy and BCG Immunotherapy Responsible for the Reduced Recurrence of Treated Mouse Tumors. Photochem. Photobiol. 2001, 73, 403–409. [Google Scholar] [CrossRef]
- Bae, S.-M.; Kim, Y.-W.; Kwak, S.-Y.; Kim, Y.-W.; Ro, D.-Y.; Shin, J.-C.; Park, C.-H.; Han, S.-J.; Oh, C.-H.; Kim, C.-K.; et al. Photodynamic Therapy-Generated Tumor Cell Lysates with CpG-Oligodeoxynucleotide Enhance Immunotherapy Efficacy in Human Papillomavirus 16 (E6/E7) Immortalized Tumor Cells. Cancer Sci. 2007, 98, 747–752. [Google Scholar] [CrossRef]
- Xia, Y.; Gupta, G.K.; Castano, A.P.; Mroz, P.; Avci, P.; Hamblin, M.R. CpG Oligodeoxynucleotide as Immune Adjuvant Enhances Photodynamic Therapy Response in Murine Metastatic Breast Cancer. J. Biophotonics 2014, 7, 897–905. [Google Scholar] [CrossRef] [PubMed]
- Bhatta, A.K.; Wang, P.; Keyal, U.; Zhao, Z.; Ji, J.; Zhu, L.; Wang, X.; Zhang, G. Therapeutic Effect of Imiquimod Enhanced ALA-PDT on Cutaneous Squamous Cell Carcinoma. Photodiagnosis Photodyn. Ther. 2018, 23, 273–280. [Google Scholar] [CrossRef]
- Korbelik, M.; Sun, J.; Cecic, I.; Serrano, K. Adjuvant Treatment for Complement Activation Increases the Effectiveness of Photodynamic Therapy of Solid Tumors. Photochem. Photobiol. Sci. 2004, 3, 812–816. [Google Scholar] [CrossRef]
- Korbelik, M.; Cecic, I. Enhancement of Tumour Response to Photodynamic Therapy by Adjuvant Mycobacterium Cell-Wall Treatment. J Photochem. Photobiol. B 1998, 44, 151–158. [Google Scholar] [CrossRef]
- Korbelik, M.; Banáth, J.; Zhang, W.; Gallagher, P.; Hode, T.; Lam, S.S.K.; Chen, W.R. N-Dihydrogalactochitosan as Immune and Direct Antitumor Agent Amplifying the Effects of Photodynamic Therapy and Photodynamic Therapy-Generated Vaccines. Int. Immunopharmacol. 2019, 75, 105764. [Google Scholar] [CrossRef]
- Korbelik, M.; Banáth, J.; Saw, K.M.; Zhang, W.; Čiplys, E. Calreticulin as Cancer Treatment Adjuvant: Combination with Photodynamic Therapy and Photodynamic Therapy-Generated Vaccines. Front. Oncol. 2015, 5, 15. [Google Scholar] [CrossRef] [Green Version]
- Kleinovink, J.W.; van Driel, P.B.; Snoeks, T.J.; Prokopi, N.; Fransen, M.F.; Cruz, L.J.; Mezzanotte, L.; Chan, A.; Löwik, C.W.; Ossendorp, F. Combination of Photodynamic Therapy and Specific Immunotherapy Efficiently Eradicates Established Tumors. Clin. Cancer Res. 2016, 22, 1459–1468. [Google Scholar] [CrossRef] [Green Version]
- Seidel, J.A.; Otsuka, A.; Kabashima, K. Anti-PD-1 and Anti-CTLA-4 Therapies in Cancer: Mechanisms of Action, Efficacy, and Limitations. Front. Oncol. 2018, 8, 86. [Google Scholar] [CrossRef]
- Duan, X.; Chan, C.; Guo, N.; Han, W.; Weichselbaum, R.R.; Lin, W. Photodynamic Therapy Mediated by Nontoxic Core–Shell Nanoparticles Synergizes with Immune Checkpoint Blockade to Elicit Antitumor Immunity and Antimetastatic Effect on Breast Cancer. J. Am. Chem. Soc. 2016, 138, 16686–16695. [Google Scholar] [CrossRef] [Green Version]
- Kleinovink, J.W.; Fransen, M.F.; Löwik, C.W.; Ossendorp, F. Photodynamic-Immune Checkpoint Therapy Eradicates Local and Distant Tumors by CD8+ T Cells. Cancer Immunol. Res. 2017, 5, 832–838. [Google Scholar] [CrossRef] [Green Version]
- O’Shaughnessy, M.J.; Murray, K.S.; Rosa, S.P.L.; Budhu, S.; Merghoub, T.; Somma, A.; Monette, S.; Kim, K.; Corradi, R.B.; Scherz, A.; et al. Systemic Antitumor Immunity by PD-1/PD-L1 Inhibition Is Potentiated by Vascular-Targeted Photodynamic Therapy of Primary Tumors. Clin. Cancer Res. 2018, 24, 592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muchowicz, A.; Wachowska, M.; Stachura, J.; Tonecka, K.; Gabrysiak, M.; Wołosz, D.; Pilch, Z.; Kilarski, W.W.; Boon, L.; Klaus, T.J.; et al. Inhibition of Lymphangiogenesis Impairs Antitumour Effects of Photodynamic Therapy and Checkpoint Inhibitors in Mice. Eur. J. Cancer 2017, 83, 19–27. [Google Scholar] [CrossRef]
- Bao, R.; Wang, Y.; Lai, J.; Zhu, H.; Zhao, Y.; Li, S.; Li, N.; Huang, J.; Yang, Z.; Wang, F.; et al. Enhancing Anti-PD-1/PD-L1 Immune Checkpoint Inhibitory Cancer Therapy by CD276-Targeted Photodynamic Ablation of Tumor Cells and Tumor Vasculature. Mol. Pharm. 2019, 16, 339–348. [Google Scholar] [CrossRef]
- Nagaya, T.; Friedman, J.; Maruoka, Y.; Ogata, F.; Okuyama, S.; Clavijo, P.E.; Choyke, P.L.; Allen, C.; Kobayashi, H. Host Immunity Following Near-Infrared Photoimmunotherapy Is Enhanced with PD-1 Checkpoint Blockade to Eradicate Established Antigenic Tumors. Cancer Immunol. Res. 2019, 7, 401–413. [Google Scholar] [CrossRef]
- Reginato, E.; Mroz, P.; Chung, H.; Kawakubo, M.; Wolf, P.; Hamblin, M.R. Photodynamic Therapy plus Regulatory T-Cell Depletion Produces Immunity against a Mouse Tumour That Expresses a Self-Antigen. Br. J. Cancer 2013, 109, 2167–2174. [Google Scholar] [CrossRef] [PubMed]
- Korbelik, M.; Banáth, J.; Zhang, W. Mreg Activity in Tumor Response to Photodynamic Therapy and Photodynamic Therapy-Generated Cancer Vaccines. Cancers 2016, 8, 94. [Google Scholar] [CrossRef] [Green Version]
- Kumai, T.; Oikawa, K.; Aoki, N.; Kimura, S.; Harabuchi, Y.; Celis, E.; Kobayashi, H. Tumor-Derived TGF-β and Prostaglandin E2 Attenuate Anti-Tumor Immune Responses in Head and Neck Squamous Cell Carcinoma Treated with EGFR Inhibitor. J. Transl. Med. 2014, 12, 265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.-C.; Yang, H.; Zhang, R.; Zhao, J.-J.; Hao, D.-J. Tumour-Associated Antigens and Their Anti-Cancer Applications. Eur. J. Cancer Care 2017, 26, e12446. [Google Scholar] [CrossRef] [PubMed]
- Wachowska, M.; Muchowicz, A.; Golab, J. Targeting Epigenetic Processes in Photodynamic Therapy-Induced Anticancer Immunity. Front. Oncol. 2015, 5, 176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wachowska, M.; Gabrysiak, M.; Muchowicz, A.; Bednarek, W.; Barankiewicz, J.; Rygiel, T.; Boon, L.; Mroz, P.; Hamblin, M.R.; Golab, J. 5-Aza-2′-Deoxycytidine Potentiates Antitumour Immune Response Induced by Photodynamic Therapy. Eur. J. Cancer 2014, 50, 1370–1381. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Characteristic | 2D | 3D | Refs. |
|---|---|---|---|
| In vivo-like | Poor resemblance of the 3D architecture of tumor tissue | Mimic the 3D structure of in vivo tumor tissues | [21] |
| Proliferation | Cells grown in monolayers proliferate faster than in 3D tumor models | A relatively slow proliferation rate is similar to that of human tumor cells | [22] |
| Polarity | Partial polarization | A precise portrayal of cell polarization | [23] |
| Morphology | Flat and sheet-like cells with a stretched appearance | Form aggregated cells. | [24] |
| Rigidity | Strong rigid (about 3 × 109 Pascals) | Less rigid (>4000 Pascals) | [25] |
| Cellular interactions | Limited cellular interactions and cellular extracellular matrix | Exhibit cellular interactions and cell-extracellular matrix-like solid tumors | [16] |
| Gene/protein expression | Alterations in gene expression, mRNA splicing, topology, and biochemistry of cells, often show discrepancies in gene/protein levels when compared to in vivo models | Genes and protein expressions in solid tumors pertinently resemble 3D tumor models | [26,27] |
| Response to therapeutics | Monolayer cell cultures are more susceptible to drugs than human tumors | Tumor cells in 3D cultures exhibit drug resistance characteristics similar to those observed in vivo human tumors | [16,26] |
| Culture formation | Takes minutes–hours | Take hours–days | [28] |
| Culture quality | Good performance, reproducible, long-term culture, ease of interpretation, and culture simplicity | Poor performance and reproducibility, difficult interpretation, and cultures | |
| Access to growth factors | Constant exposure of cells to oxygen, nutrients, metabolites, and signaling molecules (as opposed to in vivo) | Limited distribution of oxygen, nutrients, metabolites, and signaling molecules (similar to in vivo) | [16,29] |
| Cost of maintenance | Cost-effective, abundant commercially available tests and media | Costly, laborious, and lack of commercially available tests | [30] |
| Generation | PS | Localization | Cell Line | Tumor Model | Animal Species | Hallmarks of Immunogenic Cell Death (ICD) In Vitro | Hallmarks of ICD In Vivo | Refs. |
|---|---|---|---|---|---|---|---|---|
| 1st | Photofrin | Mitochondria, cellular membrane | Lewis lung carcinoma (LLC) cells | 2D monolayer cell culture and in vivo | C57BL/6 mice | PDT-treated LLC increased the expression of high-mobility group box-1 (HMGB1) protein in macrophages | PDT accelerated the expression of calreticulin (CRT) and (HMGB1) protein in LLC tumors in vivo. | [48] |
| AB12 Mesothelioma | in vivo | Balb/c mice | Localized neutrophil function at 1 h and then drops at 4 h. Increased infiltration of neutrophils at the treated at 24 h | N/A | [49] | |||
| 2nd | OR141 | Endoplasmic reticulum (ER) | AB12 Mesothelioma | 2D monolayer cell culture, in vivo | Balb/c mice | Maturation of DCs (increased levels of CD80, CD86, CD40 and MHC) | PDT-OR141 showed robust CD8+ and CD4+T responses with increased proliferation, cytotoxic reactions and increased production of interferon-gamma (IFNγ). | [50] |
| Mouse SCC7, Human A431 squamous cell carcinoma cells and mouse B16 melanoma cells | 2D monolayer cell culture | N/A | Maturation of DCs (increased expression of MHC-ll+, CD80+ and CD86+) | N/A | [51] | |||
| Hypericin | ER | T25 human bladder carcinoma cells | 2D monolayer cell culture | N/A | Maturation of DCs (increased CD80, CD83, CD86, and MCH ll) and functional stimulation (increased NO and L-1β, absent IL-10) | N/A | [52] | |
| GL261 glioma cells | 2D monolayer cell culture and in vivo | C57BL/6 mice | Maturation of DCs (elevated levels of CD80, CD86, CD40 and MHC I) | PDT stimulated the accumulation of T-lymphocytes (CD3+, CD4+ and CD8+), TH1 cells, CTLs and TH17 cells at the treated sites | [53] | |||
| Rose bengal (RB) | N/A | CT26 colorectal carcinoma cell line | 2D monolayer cell culture and in vivo | Balb/c mice | Upregulation of CRT expression A dose-dependent decrease in ATP Increased extracellular content of HMGB1 Increased expression of HSP90 | PDT-RB stimulated the expression of CRT and HSP90 on tumor cells and the release of HMGB1. | [54] | |
| 5-Aminolevulinic acid (5-ALA) | ER | PECA squamous cell carcinoma cell line | 2D monolayer cell culture and in vivo | SKH-1 mice | Maturation of DCs (upregulation of MHC-II, DC80, and CD86) and increased production of IFN-γ and IL-12 | PDT upregulated expression of CD80, CD86, and MHC-II and induced T cell proliferation | [55] | |
| PECA squamous cell carcinoma cell line | 2D monolayer cell culture and in vivo | SKH-1 mice | PDT improved the expression of CRT, HSP70, and HMGB1 | Simulated phenotypic maturation (increased MHCII, CD80, and CD86) | [56] | |||
| Glioblastoma (GB) cell lines U87 and U251 | 3D tumor spheroids | N/A | Maturation of DCs (increased levels of CD40, CD80, CD83, and CD86) | N/A | [57] | |||
| PECA squamous cell carcinoma | in vivo | SKH-1 mice | N/A | Infiltration of T-lymphocytes (CD4+/CD8+) at 7 days | [58] | |||
| Redaporfin | ER and Golgi apparatus GA | CT26 colorectal carcinoma cell line | in vivo | Balb/c mice | N/A | PDT resulted in a strong neutrophilia (2–24 h), the systemic elevation of IL-6 (24 h), increased number of CD4+ and CD8+ T cells, as well as increased production of IFN-γ or CD69+. | [59] | |
| Photodithazine | ER and Golgi apparatus | GL261 murine glioma, MCA205 murine sarcoma | 2D monolayer cell culture and in vivo | C57BL/6J | Maturation of DCs (increased CD40, CD86, and MHC II) and increase in IL-6 | PDT stimulated the release of calreticulin, HMGB1 and ATP, which activated the production of IL-6. | [60] | |
| 3rd | Core–shell gold nanocage coated with manganese dioxide and hyaluronic acid (AMH) | Hyaluronic acid targets CD44-overexpressed on the plasma membrane of CT26 cancer cells | CT26 colorectal carcinoma cell line | 2D monolayer cell culture | N/A | Maturation of DCs (upregulation of CD83, CD86, MHC II) | N/A | [61] |
| Cetuximab-IR700 | Cetuximab binds to HER1-overexpressed on the plasma membrane of cancer cells | A431 human epidermoid carcinoma | 2D monolayer cell culture and in vivo | Athymic nude mice | Maturation of DCs (increased expression of CD80, CD86, MHC II) and increased production of IL-12 | Increased population of CD86+ DCs, CD11c, CD205, and MHC II positive cells. | [62] | |
| Core–shell gold nanocage@manganese dioxide (AuNC@MnO2, AM) | N/A | 4T1 murine mammary carcinoma | 2D monolayer cell culture and in vivo | Balb/c mice | Maturation of DCs (overexpression of CD83, and CD86) and increased production of IL-12 | PDT resulted in intratumoral increase in CD11c+CD86+ and CD11c+CD83+ DCs, as well as increased NK cells and CD8+ and CD4+ | [63] | |
| Hybrid protein oxygen nanocarrier with chlorin e6 encapsulated (C@HPOC) | N/A | 4T1 murine mammary carcinoma | 2D monolayer cell culture and in vivo | Balb/c mice | Maturation of DCs (increased CD86 and MHC II) | An influx of NK cells, T cells (CD8+ CD4+) at the tumor site, and maturation of DCs. | [64] | |
| Benzoporphyrin Derivative nanoconjugates modified with cetuximab, transferrin and trastuzumab | Cetuximab binds with anti-EGFR mAb, transferrin with glycoprotein and trastuzumab binds with anti-HER-2 mAb | PDAC Pancreatic cancer cells | 3D tumor spheroids | N/A | PDT triggered the expression of heat shock-related proteins (Hsp60, Hsp70), caltreticulin and high-mobility group box 1 in light intensity and time-dependent manner. A similar trend was observed in CD4+ and CD8+ T cells antitumor reactivity by upregulating CD107a and IFN-γ | N/A | [65] | |
| 5-ALAdoamine) dendrimers generation two (PAMAM-G2) | Endo-lysosomes and mitochondria | B16 and A375 metastatic melanoma cells | in vivo | C57BL6J mice | N/A | Prevented tumor metastases. Inhibited tumor-recurrence. Infiltration of CD4+ CD8+ T cells at the tumor region, predominately central memory T cells (CD44high CD62Lhigh). Insignificant change of CD3+ T cells in the spleen. Increased levels of TNF-α and IFN-γ in serum. Maintained immune balance and prolonged recurrence-free survival | [66] | |
| Aluminum-phthalocyanine nanoemulsion (AlPcNE) | N/A | B16F10 cells | in vivo | C57BL/6 mice | N/A | PDT-AlPcNE induced a significant release of HMGB1 and ATP as well as the expression of CRT on the plasma membrane | [67] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nkune, N.W.; Simelane, N.W.N.; Montaseri, H.; Abrahamse, H. Photodynamic Therapy-Mediated Immune Responses in Three-Dimensional Tumor Models. Int. J. Mol. Sci. 2021, 22, 12618. https://doi.org/10.3390/ijms222312618
Nkune NW, Simelane NWN, Montaseri H, Abrahamse H. Photodynamic Therapy-Mediated Immune Responses in Three-Dimensional Tumor Models. International Journal of Molecular Sciences. 2021; 22(23):12618. https://doi.org/10.3390/ijms222312618
Chicago/Turabian StyleNkune, Nkune Williams, Nokuphila Winifred Nompumelelo Simelane, Hanieh Montaseri, and Heidi Abrahamse. 2021. "Photodynamic Therapy-Mediated Immune Responses in Three-Dimensional Tumor Models" International Journal of Molecular Sciences 22, no. 23: 12618. https://doi.org/10.3390/ijms222312618