
Keratins as an Inflammation Trigger Point in Epidermolysis Bullosa Simplex

 , and
, and
Abstract
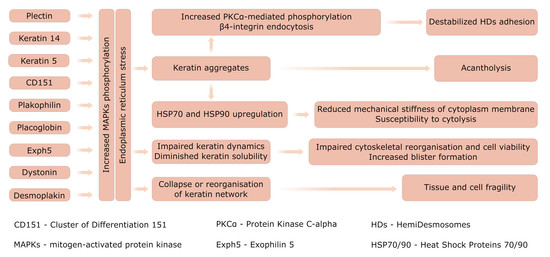
:
{kind=link}
{kind=link}
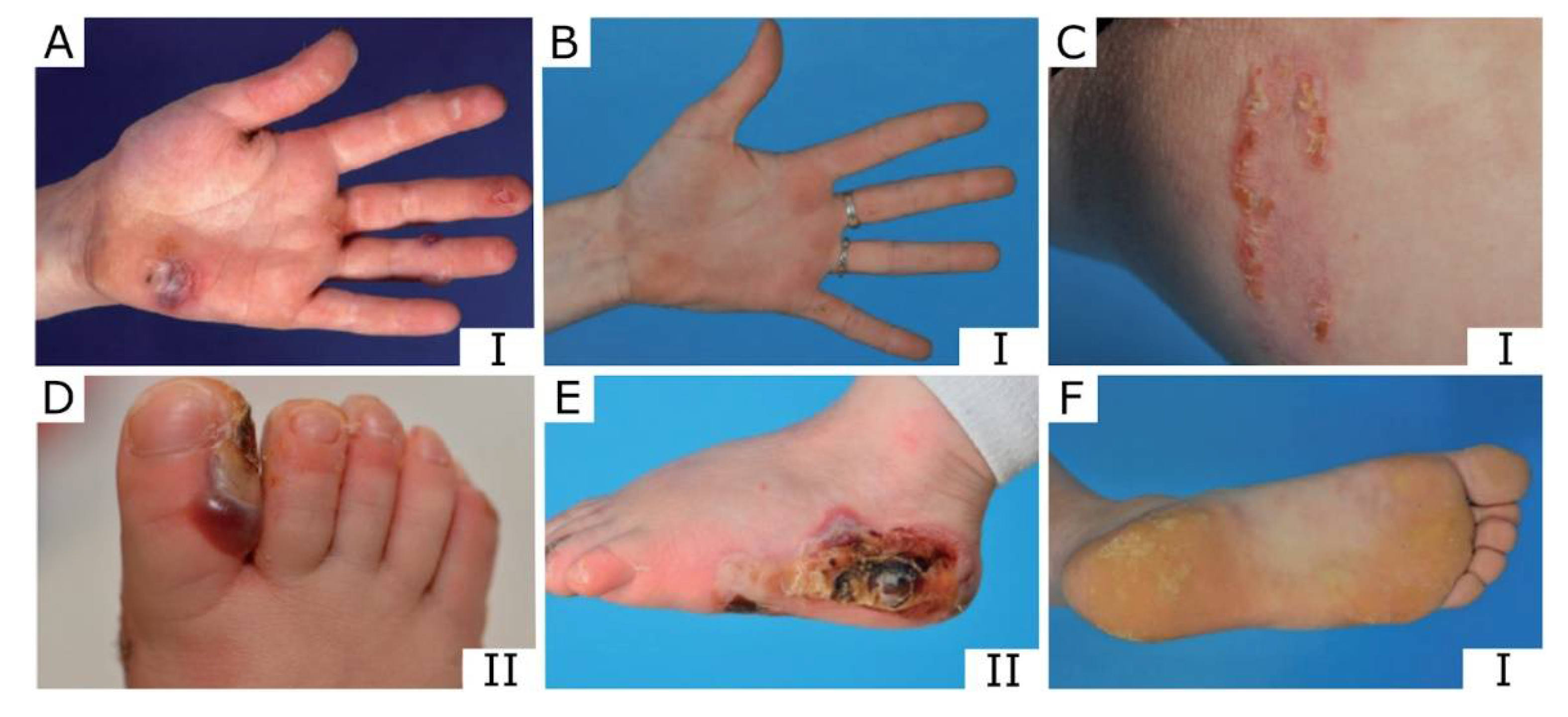{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Several Aspects of Epidermal Keratinocyte Growth, Proliferation, and Response to Inflammatory Stimuli under Normal Healthy Conditions
3. Epidemiology
4. Brief Phenotypic and Genotypic Characteristics
4.1. The Basic Pathology of Most EBS Subtypes
4.2. List of Genes Involved in EBS and EBS Subtypes
4.3. EBS Subtypes
- Localized EBS is characterized by skin blistering that develops any time between childhood and adulthood and is usually limited to the hands and feet. Later in life, the hand palms, and feet skin may thicken and harden (hyperkeratosis).
- Intermediate EBS is associated with widespread blistering that can be present from birth or develop in early infancy. The blistering tends to be more severe than that in localized EBS but milder than that in severe EBS.
- Severe EBS is the most severe type of EB simplex, in which extensive blistering can occur anywhere on the body, including the inside of the mouth cavity. Blistering tends to be present from birth and may improve with age, but older individuals can also be affected by hyperkeratosis. The severity and extent of the blistering vary greatly, and it can be fatal in infancy in very severe cases.
- EBS with mottled pigmentation is the fourth type of EB simplex, in which skin fragility is present at birth and, over time, brown pigmentation interspersed with spots develops on the body. The pigmentation can reduce and disappear in adult life.
- Severe EBS with pyloric atresia (EBS-PA);
- Muscular dystrophy EBS with PLEC deficiency (EBS-MD);
- EBS with migratory circinate erythema (EBS-MCE);
- Intermediate EBS with cardiomyopathy (EBS-CM);
- EBS with nephrotic syndrome due to CD151 mutations.
4.4. Mutations as the Causes of Pathology
5. K5/K14 Heterohybrid Structure and Function
6. Turnover of Keratins in Healthy and EBS Cell Conditions
6.1. Dynamics of the Keratins
6.2. Posttranslational Modifications (PTMs) of Keratins
6.3. Phosphorylation-Dependent Aggregation in EBS Cells
7. Stress-Mediated Cellular Responses
7.1. Mechanical Properties of EBS Mutated Keratins
7.2. ER Stress and the Ubiquitin–Proteasome System of Degradation
7.3. ER Stress and UPR in Inflammatory Cascades in EBS
8. Molecular Pathways Orchestrated in the EBS-Specific Profile
9. Inflammation Initiation and Progression
9.1. Sterile Inflammation (SI)
9.2. Chemokines
9.3. Immune Cells
10. Disease Modeling
11. Abnormal Cellular Structures and Function
12. Microbial Infections
13. WH and Carcinogenesis in EBS
14. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wang, F.; Zieman, A.; Coulombe, P.A. Skin Keratins. Methods Enzymol. 2016, 568, 303–350. [Google Scholar] [CrossRef] [Green Version]
- Fine, J.-D. Inherited Epidermolysis Bullosa: Past, Present, and Future. Ann. N. Y. Acad. Sci. 2010, 1194, 213–222. [Google Scholar] [CrossRef]
- Morley, S.M.; D’Alessandro, M.; Sexton, C.; Rugg, E.L.; Navsaria, H.; Shemanko, C.S.; Huber, M.; Hohl, D.; Heagerty, A.I.; Leigh, I.M.; et al. Generation and Characterization of Epidermolysis Bullosa Simplex Cell Lines: Scratch Assays Show Faster Migration with Disruptive Keratin Mutations. Br. J. Dermatol. 2003, 149, 46–58. [Google Scholar] [CrossRef] [PubMed]
- Cao, T.; Longley, M.A.; Wang, X.J.; Roop, D.R. An Inducible Mouse Model for Epidermolysis Bullosa Simplex: Implications for Gene Therapy. J. Cell Biol. 2001, 152, 651–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, H.; Chen, J.; Planko, L.; Zigrino, P.; Klein-Hitpass, L.; Magin, T.M. Induction of Inflammatory Cytokines by a Keratin Mutation and Their Repression by a Small Molecule in a Mouse Model for EBS. J. Investig. Dermatol. 2007, 127, 2781–2789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peters, B.; Kirfel, J.; Büssow, H.; Vidal, M.; Magin, T.M. Complete Cytolysis and Neonatal Lethality in Keratin 5 Knockout Mice Reveal Its Fundamental Role in Skin Integrity and in Epidermolysis Bullosa Simplex. Mol. Biol. Cell 2001, 12, 1775–1789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoneda, K.; Furukawa, T.; Zheng, Y.-J.; Momoi, T.; Izawa, I.; Inagaki, M.; Manabe, M.; Inagaki, N. An Autocrine/Paracrine Loop Linking Keratin 14 Aggregates to Tumor Necrosis Factor α-Mediated Cytotoxicity in a Keratinocyte Model of Epidermolysis Bullosa Simplex. J. Biol. Chem. 2004, 279, 7296–7303. [Google Scholar] [CrossRef] [Green Version]
- Coulombe, P.A.; Lee, C.-H. Defining Keratin Protein Function in Skin Epithelia: Epidermolysis Bullosa Simplex and Its Aftermath. J. Investig. Dermatol. 2012, 132, 763–775. [Google Scholar] [CrossRef] [Green Version]
- Ujiie, I.; Fujita, Y.; Nakayama, C.; Matsumura, W.; Suzuki, S.; Shinkuma, S.; Nomura, T.; Abe, R.; Shimizu, H. Altered Balance of Epidermis-Related Chemokines in Epidermolysis Bullosa. J. Dermatol. Sci. 2017, 86, 37–45. [Google Scholar] [CrossRef] [Green Version]
- Bardhan, A.; Bruckner-Tuderman, L.; Chapple, I.L.C.; Fine, J.-D.; Harper, N.; Has, C.; Magin, T.M.; Marinkovich, M.P.; Marshall, J.F.; McGrath, J.A.; et al. Epidermolysis Bullosa. Nat. Rev. Dis. Primer 2020, 6, 78. [Google Scholar] [CrossRef]
- Nguyen, A.V.; Soulika, A.M. The Dynamics of the Skin’s Immune System. Int. J. Mol. Sci. 2019, 20, 1811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, S.J. Cell Death and Inflammation: The Case for IL-1 Family Cytokines as the Canonical DAMPs of the Immune System. FEBS J. 2016, 283, 2599–2615. [Google Scholar] [CrossRef] [PubMed]
- Bchetnia, M.; Tremblay, M.-L.; Leclerc, G.; Dupérée, A.; Powell, J.; McCuaig, C.; Morin, C.; Legendre-Guillemin, V.; Laprise, C. Expression Signature of Epidermolysis Bullosa Simplex. Hum. Genet. 2012, 131, 393–406. [Google Scholar] [CrossRef]
- Herzog, J.; Rid, R.; Wagner, M.; Hundsberger, H.; Eger, A.; Bauer, J.; Önder, K. Whole-Transcriptome Gene Expression Profiling in an Epidermolysis Bullosa Simplex Dowling-Meara Model Keratinocyte Cell Line Uncovered Novel, Potential Therapeutic Targets and Affected Pathways. BMC Res. Notes 2015, 8, 785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandling-Bennett, H.A.; Morel, K.D. Common Wound Colonizers in Patients with Epidermolysis Bullosa. Pediatr. Dermatol. 2010, 27, 25–28. [Google Scholar] [CrossRef]
- Roberts, N.; Horsley, V. Developing Stratified Epithelia: Lessons from the Epidermis and Thymus. Wiley Interdiscip. Rev. Dev. Biol. 2014, 3, 389–402. [Google Scholar] [CrossRef] [Green Version]
- Chessa, C.; Bodet, C.; Jousselin, C.; Wehbe, M.; Lévêque, N.; Garcia, M. Antiviral and Immunomodulatory Properties of Antimicrobial Peptides Produced by Human Keratinocytes. Front. Microbiol. 2020, 11, 1155. [Google Scholar] [CrossRef]
- Fischer, C.L.; Wertz, P.W. Effects of Endogenous Lipids on the Skin Microbiome. In Skin Microbiome Handbook; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2020; pp. 217–235. ISBN 978-1-119-59305-8. [Google Scholar]
- Talagas, M.; Lebonvallet, N.; Leschiera, R.; Sinquin, G.; Elies, P.; Haftek, M.; Pennec, J.-P.; Ressnikoff, D.; La Padula, V.; Le Garrec, R.; et al. Keratinocytes Communicate with Sensory Neurons via Synaptic-like Contacts. Ann. Neurol. 2020, 88, 1205–1219. [Google Scholar] [CrossRef] [PubMed]
- Vidal Yucha, S.E.; Tamamoto, K.A.; Kaplan, D.L. The Importance of the Neuro-Immuno-Cutaneous System on Human Skin Equivalent Design. Cell Prolif. 2019, 52, e12677. [Google Scholar] [CrossRef] [Green Version]
- Seeger, M.A.; Paller, A.S. The Roles of Growth Factors in Keratinocyte Migration. Adv. Wound Care 2015, 4, 213–224. [Google Scholar] [CrossRef] [Green Version]
- Ashrafi, M.; Baguneid, M.; Bayat, A. The Role of Neuromediators and Innervation in Cutaneous Wound Healing. Acta Derm. Venereol. 2016, 96, 587–594. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Tsoi, L.C.; Billi, A.C.; Ward, N.L.; Harms, P.W.; Zeng, C.; Maverakis, E.; Kahlenberg, J.M.; Gudjonsson, J.E. Cytokinocytes: The Diverse Contribution of Keratinocytes to Immune Responses in Skin. JCI Insight 2020, 5, 5. [Google Scholar] [CrossRef] [PubMed]
- Klicznik, M.M.; Szenes-Nagy, A.B.; Campbell, D.J.; Gratz, I.K. Taking the Lead–How Keratinocytes Orchestrate Skin T Cell Immunity. Immunol. Lett. 2018, 200, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Blais, M.; Mottier, L.; Germain, M.-A.; Bellenfant, S.; Cadau, S.; Berthod, F. Sensory Neurons Accelerate Skin Reepithelialization via Substance P in an Innervated Tissue-Engineered Wound Healing Model. Tissue Eng. Part A 2014, 20, 2180–2188. [Google Scholar] [CrossRef] [Green Version]
- Roggenkamp, D.; Köpnick, S.; Stäb, F.; Wenck, H.; Schmelz, M.; Neufang, G. Epidermal Nerve Fibers Modulate Keratinocyte Growth via Neuropeptide Signaling in an Innervated Skin Model. J. Investig. Dermatol. 2013, 133, 1620–1628. [Google Scholar] [CrossRef] [Green Version]
- Pondeljak, N.; Lugović-Mihić, L. Stress-Induced Interaction of Skin Immune Cells, Hormones, and Neurotransmitters. Clin. Ther. 2020, 42, 757–770. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.A.; Edwards, T.N.; Liu, A.W.; Hirai, T.; Jones, M.R.; Wu, J.; Li, Y.; Zhang, S.; Ho, J.; Davis, B.M.; et al. Cutaneous TRPV1+ Neurons Trigger Protective Innate Type 17 Anticipatory Immunity. Cell 2019, 178, 919–932.e14. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Guo, S.; Ranzer, M.J.; DiPietro, L.A. Toll-like Receptor 4 Has an Essential Role in Early Skin Wound Healing. J. Investig. Dermatol. 2013, 133, 258–267. [Google Scholar] [CrossRef] [Green Version]
- Miller, L.S.; Modlin, R.L. Toll-like Receptors in the Skin. Semin. Immunopathol. 2007, 29, 15–26. [Google Scholar] [CrossRef]
- Lebre, M.C.; van der Aar, A.M.G.; van Baarsen, L.; van Capel, T.M.M.; Schuitemaker, J.H.N.; Kapsenberg, M.L.; de Jong, E.C. Human Keratinocytes Express Functional Toll-like Receptor 3, 4, 5, and 9. J. Investig. Dermatol. 2007, 127, 331–341. [Google Scholar] [CrossRef] [Green Version]
- Cañedo-Dorantes, L.; Cañedo-Ayala, M. Skin Acute Wound Healing: A Comprehensive Review. Int. J. Inflamm. 2019, 2019, e3706315. [Google Scholar] [CrossRef] [PubMed]
- Romagnani, P.; Lasagni, L.; Annunziato, F.; Serio, M.; Romagnani, S. CXC Chemokines: The Regulatory Link between Inflammation and Angiogenesis. Trends Immunol. 2004, 25, 201–209. [Google Scholar] [CrossRef]
- Ridiandries, A.; Tan, J.T.M.; Bursill, C.A. The Role of Chemokines in Wound Healing. Int. J. Mol. Sci. 2018, 19, 3217. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, Y. The Role of Chemokines in Neutrophil Biology. Front. Biosci. J. Virtual Libr. 2008, 13, 2400–2407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sprecher, E. Epidermolysis Bullosa Simplex. Dermatol. Clin. 2010, 28, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Feinstein, J.A.; Jambal, P.; Peoples, K.; Lucky, A.W.; Khuu, P.; Tang, J.Y.; Lara-Corrales, I.; Pope, E.; Wiss, K.; Hook, K.P.; et al. Assessment of the Timing of Milestone Clinical Events in Patients With Epidermolysis Bullosa From North America. JAMA Dermatol. 2019, 155, 196–203. [Google Scholar] [CrossRef]
- Fine, J.-D. Epidemiology of Inherited Epidermolysis Bullosa Based on Incidence and Prevalence Estimates From the National Epidermolysis Bullosa Registry. JAMA Dermatol. 2016, 152, 1231–1238. [Google Scholar] [CrossRef] [Green Version]
- Baardman, R.; Yenamandra, V.K.; Duipmans, J.C.; Pasmooij, A.M.G.; Jonkman, M.F.; Akker, P.C.; van den Bolling, M.C. Novel Insights into the Epidemiology of Epidermolysis Bullosa (EB) from the Dutch EB Registry: EB More Common than Previously Assumed? J. Eur. Acad. Dermatol. Venereol. 2021, 35, 995–1006. [Google Scholar] [CrossRef]
- Laly, A.C.; Sliogeryte, K.; Pundel, O.J.; Ross, R.; Keeling, M.C.; Avisetti, D.; Waseem, A.; Gavara, N.; Connelly, J.T. The Keratin Network of Intermediate Filaments Regulates Keratinocyte Rigidity Sensing and Nuclear Mechanotransduction. Sci. Adv. 2021, 7, eabd6187. [Google Scholar] [CrossRef]
- Prodinger, C.; Reichelt, J.; Bauer, J.W.; Laimer, M. Epidermolysis Bullosa: Advances in Research and Treatment. Exp. Dermatol. 2019, 28, 1176–1189. [Google Scholar] [CrossRef] [Green Version]
- Gostyńska, K.B.; Bremer, J.; van Dijk-Bos, K.K.; Sinke, R.; Pasmooij, A.M.G.; Jonkman, M.F. In-Frame Exon Skipping in KRT5 Due to Novel Intronic Deletion Causes Epidermolysis Bullosa Simplex, Generalized Severe. Acta Derm. Venereol. 2017, 97, 105–107. [Google Scholar] [CrossRef] [Green Version]
- Horn, H.M.; Tidman, M.J. The Clinical Spectrum of Epidermolysis Bullosa Simplex. Br. J. Dermatol. 2000, 142, 468–472. [Google Scholar] [CrossRef]
- Has, C.; Bauer, J.W.; Bodemer, C.; Bolling, M.C.; Bruckner-Tuderman, L.; Diem, A.; Fine, J.-D.; Heagerty, A.; Hovnanian, A.; Marinkovich, M.P.; et al. Consensus Reclassification of Inherited Epidermolysis Bullosa and Other Disorders with Skin Fragility. Br. J. Dermatol. 2020, 183, 614–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, F.J.D. The Molecular Genetics of Keratin Disorders. Am. J. Clin. Dermatol. 2003, 4, 347–364. [Google Scholar] [CrossRef] [PubMed]
- Human Intermediate Filament Database. Available online: http://www.interfil.org/diseasesTypeInII.php#1 (accessed on 3 September 2021).
- Sathishkumar, D.; Orrin, E.; Terron-Kwiatkowski, A.; Browne, F.; Martinez, A.E.; Mellerio, J.E.; Ogboli, M.; Hoey, S.; Ozoemena, L.; Liu, L.; et al. The p.Glu477Lys Mutation in Keratin 5 Is Strongly Associated with Mortality in Generalized Severe Epidermolysis Bullosa Simplex. J. Investig. Dermatol. 2016, 136, 719–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bchetnia, M.; Lacroix, J.; Farez, T.; Larouche, M.; Powell, J.; McCuaig, C.; Dupéré, A.; Morin, C.; Legendre-Guillemin, V.; Laprise, C. Reduction in Keratin Aggregates in Epidermolysis Bullosa Simplex Keratinocytes after Pretreatment with Trimethylamine N-Oxide. Exp. Dermatol. 2016, 25, 229–230. [Google Scholar] [CrossRef] [Green Version]
- Spörrer, M.; Prochnicki, A.; Tölle, R.C.; Nyström, A.; Esser, P.R.; Homberg, M.; Athanasiou, I.; Zingkou, E.; Schilling, A.; Gerum, R.; et al. Treatment of Keratinocytes with 4-Phenylbutyrate in Epidermolysis Bullosa: Lessons for Therapies in Keratin Disorders. EBioMedicine 2019, 44, 502–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasukawa, K.; Sawamura, D.; McMillan, J.R.; Nakamura, H.; Shimizu, H. Dominant and Recessive Compound Heterozygous Mutations in Epidermolysis Bullosa Simplex Demonstrate the Role of the Stutter Region in Keratin Intermediate Filament Assembly. J. Biol. Chem. 2002, 277, 23670–23674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batta, K.; Rugg, E.L.; Wilson, N.J.; West, N.; Goodyear, H.; Lane, E.B.; Gratian, M.; Dopping-Hepenstal, P.; Moss, C.; Eady, R.A.J. A Keratin 14 ‘Knockout’ Mutation in Recessive Epidermolysis Bullosa Simplex Resulting in Less Severe Disease. Br. J. Dermatol. 2000, 143, 621–627. [Google Scholar] [CrossRef]
- Vahidnezhad, H.; Youssefian, L.; Daneshpazhooh, M.; Mahmoudi, H.; Kariminejad, A.; Fischer, J.; Christiansen, J.; Schneider, H.; Guy, A.; Liu, L.; et al. Biallelic KRT5 Mutations in Autosomal Recessive Epidermolysis Bullosa Simplex, Including a Complete Human Keratin 5 “Knock-Out”. Matrix Biol. 2019, 83, 48–59. [Google Scholar] [CrossRef]
- Bchetnia, M.; Allard, J.-P.; Boucher-Lafleur, A.-M.; Marino, T.C.; Dupéré, A.; Powell, J.; McCuaig, C.; Bernier, M.-È.; Laprise, C. Severe Epidermolysis Bullosa Simplex Phenotype Caused by Codominant Mutations p.Ile377Thr in Keratin 14 and p.Gly138Glu in Keratin 5. Exp. Dermatol. 2020, 29, 961–969. [Google Scholar] [CrossRef]
- Lugassy, J.; Itin, P.; Ishida-Yamamoto, A.; Holland, K.; Huson, S.; Geiger, D.; Hennies, H.C.; Indelman, M.; Bercovich, D.; Uitto, J.; et al. Naegeli-Franceschetti-Jadassohn Syndrome and Dermatopathia Pigmentosa Reticularis: Two Allelic Ectodermal Dysplasias Caused by Dominant Mutations in KRT14. Am. J. Hum. Genet. 2006, 79, 724–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ralser, D.J.; Kumar, S.; Borisov, O.; Sarig, O.; Richard, G.; Wolf, S.; Krawitz, P.M.; Sprecher, E.; Kreiß, M.; Betz, R.C. Identification of a Founder Mutation in KRT14 Associated with Naegeli–Franceschetti–Jadassohn Syndrome. Br. J. Dermatol. 2020, 183, 756–757. [Google Scholar] [CrossRef] [PubMed]
- Shanker, V.; Gupta, M. Dermatopathia Pigmentosa Reticularis: A Rare Reticulate Pigmentary Disorder. Indian Dermatol. Online J. 2013, 4, 40–42. [Google Scholar] [CrossRef]
- Malchin, N.; Sarig, O.; Grafi-Cohen, M.; Geller, S.; Goldberg, I.; Shani, A.; Gat, A.; Sprecher, E.; Mashiah, J. A Novel Homozygous Deletion in EXPH5 Causes a Skin Fragility Phenotype. Clin. Exp. Dermatol. 2016, 41, 915–918. [Google Scholar] [CrossRef]
- Lee, J.Y.W.; Liu, L.; Hsu, C.-K.; Aristodemou, S.; Ozoemena, L.; Ogboli, M.; Moss, C.; Martinez, A.E.; Mellerio, J.E.; McGrath, J.A. Mutations in KLHL24 Add to the Molecular Heterogeneity of Epidermolysis Bullosa Simplex. J. Investig. Dermatol. 2017, 137, 1378–1380. [Google Scholar] [CrossRef] [Green Version]
- Lin, Z.; Li, S.; Feng, C.; Yang, S.; Wang, H.; Ma, D.; Zhang, J.; Gou, M.; Bu, D.; Zhang, T.; et al. Stabilizing Mutations of KLHL24 Ubiquitin Ligase Cause Loss of Keratin 14 and Human Skin Fragility. Nat. Genet. 2016, 48, 1508–1516. [Google Scholar] [CrossRef]
- Vermeer, M.C.; Bolling, M.C.; Bliley, J.M.; Arevalo Gomez, K.F.; Pavez-Giani, M.G.; Kramer, D.; Romero-Herrera, P.H.; Westenbrink, B.D.; Diercks, G.F.; van den Berg, M.P.; et al. Gain-of-Function Mutation in Ubiquitin-Ligase KLHL24 Causes Desmin Degradation and Dilatation in HiPSC-Derived Engineered Heart Tissues. J. Clin. Investig. 2021, 131, 140615. [Google Scholar] [CrossRef]
- Liu, L.; Dopping-Hepenstal, P.J.; Lovell, P.A.; Michael, M.; Horn, H.; Fong, K.; Lai-Cheong, J.E.; Mellerio, J.E.; Parsons, M.; McGrath, J.A. Autosomal Recessive Epidermolysis Bullosa Simplex Due to Loss of BPAG1-e Expression. J. Investig. Dermatol. 2012, 132, 742–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turcan, I.; Pasmooij, A.M.G.; Van den Akker, P.C.; Lemmink, H.; Sinke, R.J.; Jonkman, M.F. Association of Epidermolysis Bullosa Simplex With Mottled Pigmentation and EXPH5 Mutations. JAMA Dermatol. 2016, 152, 1137–1141. [Google Scholar] [CrossRef]
- Fujiwara, S.; Deguchi, S.; Magin, T.M. Disease-Associated Keratin Mutations Reduce Traction Forces and Compromise Adhesion and Collective Migration. J. Cell Sci. 2020, 133, jcs243956. [Google Scholar] [CrossRef]
- Wang, W.; Zuidema, A.; te Molder, L.; Nahidiazar, L.; Hoekman, L.; Schmidt, T.; Coppola, S.; Sonnenberg, A. Hemidesmosomes Modulate Force Generation via Focal Adhesions. J. Cell Biol. 2020, 219, 219. [Google Scholar] [CrossRef] [PubMed]
- Wilhelmsen, K.; Litjens, S.H.M.; Kuikman, I.; Tshimbalanga, N.; Janssen, H.; van den Bout, I.; Raymond, K.; Sonnenberg, A. Nesprin-3, a Novel Outer Nuclear Membrane Protein, Associates with the Cytoskeletal Linker Protein Plectin. J. Cell Biol. 2005, 171, 799–810. [Google Scholar] [CrossRef] [PubMed]
- Natsuga, K. Plectin-Related Skin Diseases. J. Dermatol. Sci. 2015, 77, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Koss-Harnes, D.; Høyheim, B.; Anton-Lamprecht, I.; Gjesti, A.; Jørgensen, R.S.; Jahnsen, F.L.; Olaisen, B.; Wiche, G.; Gedde-Dahl, T. A Site-Specific Plectin Mutation Causes Dominant Epidermolysis Bullosa Simplex Ogna: Two Identical De Novo Mutations. J. Investig. Dermatol. 2002, 118, 87–93. [Google Scholar] [CrossRef] [Green Version]
- Osmanagic-Myers, S.; Gregor, M.; Walko, G.; Burgstaller, G.; Reipert, S.; Wiche, G. Plectin-Controlled Keratin Cytoarchitecture Affects MAP Kinases Involved in Cellular Stress Response and Migration. J. Cell Biol. 2006, 174, 557–568. [Google Scholar] [CrossRef] [Green Version]
- Szeverenyi, I.; Cassidy, A.J.; Chung, C.W.; Lee, B.T.K.; Common, J.E.A.; Ogg, S.C.; Chen, H.; Sim, S.Y.; Goh, W.L.P.; Ng, K.W.; et al. The Human Intermediate Filament Database: Comprehensive Information on a Gene Family Involved in Many Human Diseases. Hum. Mutat. 2008, 29, 351–360. [Google Scholar] [CrossRef]
- Lee, C.-H.; Kim, M.-S.; Chung, B.M.; Leahy, D.J.; Coulombe, P.A. Structural Basis for Heteromeric Assembly and Perinuclear Organization of Keratin Filaments. Nat. Struct. Mol. Biol. 2012, 19, 707–715. [Google Scholar] [CrossRef] [Green Version]
- Strelkov, S.V.; Herrmann, H.; Geisler, N.; Wedig, T.; Zimbelmann, R.; Aebi, U.; Burkhard, P. Conserved Segments 1A and 2B of the Intermediate Filament Dimer: Their Atomic Structures and Role in Filament Assembly. EMBO J. 2002, 21, 1255–1266. [Google Scholar] [CrossRef] [Green Version]
- Feng, X.; Coulombe, P.A. A Role for Disulfide Bonding in Keratin Intermediate Filament Organization and Dynamics in Skin Keratinocytes. J. Cell Biol. 2015, 209, 59–72. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.-H.; Kim, M.-S.; Li, S.; Leahy, D.J.; Coulombe, P.A. Structure-Function Analyses of a Keratin Heterotypic Complex Identify Specific Keratin Regions Involved in Intermediate Filament Assembly. Structure 2020, 28, 355–362.e4. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.C.; Bryan, J.T.; Morasso, M.I.; Jang, S.-I.; Lee, J.-H.; Yang, J.-M.; Marekov, L.N.; Parry, D.A.D.; Steinert, P.M. Coiled-Coil Trigger Motifs in the 1B and 2B Rod Domain Segments Are Required for the Stability of Keratin Intermediate Filaments. Mol. Biol. Cell 2000, 11, 3539–3558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moll, R.; Dhouailly, D.; Sun, T.T. Expression of Keratin 5 as a Distinctive Feature of Epithelial and Biphasic Mesotheliomas. An Immunohistochemical Study Using Monoclonal Antibody AE14. Virchows Arch. B Cell Pathol. Incl. Mol. Pathol. 1989, 58, 129–145. [Google Scholar] [CrossRef] [PubMed]
- Achtstätter, T.; Moll, R.; Moore, B.; Franke, W.W. Cytokeratin Polypeptide Patterns of Different Epithelia of the Human Male Urogenital Tract: Immunofluorescence and Gel Electrophoretic Studies. J. Histochem. Cytochem. Off. J. Histochem. Soc. 1985, 33, 415–426. [Google Scholar] [CrossRef]
- Lomada, D.; Liu, B.; Coghlan, L.; Hu, Y.; Richie, E.R. Thymus Medulla Formation and Central Tolerance Are Restored in IKKalpha-/- Mice That Express an IKKalpha Transgene in Keratin 5+ Thymic Epithelial Cells. J. Immunol. Baltim. Md 1950 2007, 178, 829–837. [Google Scholar] [CrossRef] [Green Version]
- Byrne, C.; Fuchs, E. Probing Keratinocyte and Differentiation Specificity of the Human K5 Promoter in Vitro and in Transgenic Mice. Mol. Cell. Biol. 1993, 13, 3176–3190. [Google Scholar] [CrossRef]
- Abashev, T.M.; Metzler, M.A.; Wright, D.M.; Sandell, L.L. Retinoic Acid Signaling Regulates Krt5 and Krt14 Independently of Stem Cell Markers in Submandibular Salivary Gland Epithelium. Dev. Dyn. Off. Publ. Am. Assoc. Anat. 2017, 246, 135–147. [Google Scholar] [CrossRef] [Green Version]
- Colopy, S.A.; Bjorling, D.E.; Mulligan, W.A.; Bushman, W. A Population of Progenitor Cells in the Basal and Intermediate Layers of the Murine Bladder Urothelium Contributes to Urothelial Development and Regeneration. Dev. Dyn. Off. Publ. Am. Assoc. Anat. 2014, 243, 988–998. [Google Scholar] [CrossRef] [Green Version]
- Becker, M.T.; Shores, C.G.; Yu, K.K.; Yarbrough, W.G. Molecular Assay to Detect Metastatic Head and Neck Squamous Cell Carcinoma. Arch. Otolaryngol. Head Neck Surg. 2004, 130, 21–27. [Google Scholar] [CrossRef] [Green Version]
- Alam, H.; Sehgal, L.; Kundu, S.T.; Dalal, S.N.; Vaidya, M.M. Novel Function of Keratins 5 and 14 in Proliferation and Differentiation of Stratified Epithelial Cells. Mol. Biol. Cell 2011, 22, 4068–4078. [Google Scholar] [CrossRef]
- Kölsch, A.; Windoffer, R.; Leube, R.E. Actin-Dependent Dynamics of Keratin Filament Precursors. Cell Motil. Cytoskeleton 2009, 66, 976–985. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, N.; Windoffer, R.; Magin, T.M.; Leube, R.E. Dissection of Keratin Network Formation, Turnover and Reorganization in Living Murine Embryos. Sci. Rep. 2015, 5, 9007. [Google Scholar] [CrossRef] [Green Version]
- Windoffer, R.; Wöll, S.; Strnad, P.; Leube, R.E. Identification of Novel Principles of Keratin Filament Network Turnover in Living Cells. Mol. Biol. Cell 2004, 15, 2436–2448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Windoffer, R.; Leube, R.E. Imaging of Keratin Dynamics during the Cell Cycle and in Response to Phosphatase Inhibition. In Methods in Cell Biology; Intermediate Filament Cytoskeleton; Academic Press: Cambridge, MA, USA, 2004; Volume 78, pp. 321–352. [Google Scholar]
- Pan, X.; Hobbs, R.P.; Coulombe, P.A. The Expanding Significance of Keratin Intermediate Filaments in Normal and Diseased Epithelia. Curr. Opin. Cell Biol. 2013, 25, 47–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wöll, S.; Windoffer, R.; Leube, R.E. P38 MAPK-Dependent Shaping of the Keratin Cytoskeleton in Cultured Cells. J. Cell Biol. 2007, 177, 795–807. [Google Scholar] [CrossRef] [PubMed]
- Dmello, C.; Srivastava, S.S.; Tiwari, R.; Chaudhari, P.R.; Sawant, S.; Vaidya, M.M. Multifaceted Role of Keratins in Epithelial Cell Differentiation and Transformation. J. Biosci. 2019, 44, 33. [Google Scholar] [CrossRef]
- Snider, N.T.; Park, H.; Omary, M.B. A Conserved Rod Domain Phosphotyrosine That Is Targeted by the Phosphatase PTP1B Promotes Keratin 8 Protein Insolubility and Filament Organization. J. Biol. Chem. 2013, 288, 31329–31337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, X.; Kane, L.A.; Van Eyk, J.E.; Coulombe, P.A. Type I Keratin 17 Protein Is Phosphorylated on Serine 44 by P90 Ribosomal Protein S6 Kinase 1 (RSK1) in a Growth- and Stress-Dependent Fashion. J. Biol. Chem. 2011, 286, 42403–42413. [Google Scholar] [CrossRef] [Green Version]
- Ku, N.-O.; Omary, M.B. Keratins Turn over by Ubiquitination in a Phosphorylation-Modulated Fashion. J. Cell Biol. 2000, 149, 547–552. [Google Scholar] [CrossRef]
- Homberg, M.; Ramms, L.; Schwarz, N.; Dreissen, G.; Leube, R.E.; Merkel, R.; Hoffmann, B.; Magin, T.M. Distinct Impact of Two Keratin Mutations Causing Epidermolysis Bullosa Simplex on Keratinocyte Adhesion and Stiffness. J. Investig. Dermatol. 2015, 135, 2437–2445. [Google Scholar] [CrossRef] [Green Version]
- Seltmann, K.; Cheng, F.; Wiche, G.; Eriksson, J.E.; Magin, T.M. Keratins Stabilize Hemidesmosomes through Regulation of Β4-Integrin Turnover. J. Investig. Dermatol. 2015, 135, 1609–1620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chamcheu, J.C.; Navsaria, H.; Pihl-Lundin, I.; Liovic, M.; Vahlquist, A.; Törmä, H. Chemical Chaperones Protect Epidermolysis Bullosa Simplex Keratinocytes from Heat Stress-Induced Keratin Aggregation: Involvement of Heat Shock Proteins and MAP Kinases. J. Investig. Dermatol. 2011, 131, 1684–1691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gujrati, M.; Mittal, R.; Ekal, L.; Mishra, R.K. SUMOylation of Periplakin Is Critical for Efficient Reorganization of Keratin Filament Network. Mol. Biol. Cell 2019, 30, 357–369. [Google Scholar] [CrossRef]
- Ku, N.-O.; Toivola, D.M.; Strnad, P.; Omary, M.B. Cytoskeletal Keratin Glycosylation Protects Epithelial Tissue from Injury. Nat. Cell Biol. 2010, 12, 876–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snider, N.T.; Weerasinghe, S.V.W.; Iñiguez-Lluhí, J.A.; Herrmann, H.; Omary, M.B. Keratin Hypersumoylation Alters Filament Dynamics and Is a Marker for Human Liver Disease and Keratin Mutation. J. Biol. Chem. 2011, 286, 2273–2284. [Google Scholar] [CrossRef] [Green Version]
- Snider, N.T.; Omary, M.B. Post-Translational Modifications of Intermediate Filament Proteins: Mechanisms and Functions. Nat. Rev. Mol. Cell Biol. 2014, 15, 163–177. [Google Scholar] [CrossRef] [Green Version]
- Homberg, M.; Tellkamp, F.; Omary, B.; Niessen, C.M.; Magin, T. Cooperation of Keratin Mutations and Posttranslational Modifications. J. Investig. Dermatol 2017, 137, S223. [Google Scholar] [CrossRef]
- Sawant, M.S.; Leube, R.E. Consequences of Keratin Phosphorylation for Cytoskeletal Organization and Epithelial Functions. Int. Rev. Cell Mol. Biol. 2017, 330, 171–225. [Google Scholar] [CrossRef]
- Beriault, D.R.; Haddad, O.; McCuaig, J.V.; Robinson, Z.J.; Russell, D.; Lane, E.B.; Fudge, D.S. The Mechanical Behavior of Mutant K14-R125P Keratin Bundles and Networks in NEB-1 Keratinocytes. PLoS ONE 2012, 7, e31320. [Google Scholar] [CrossRef]
- D’Alessandro, M.; Russell, D.; Morley, S.M.; Davies, A.M.; Lane, E.B. Keratin Mutations of Epidermolysis Bullosa Simplex Alter the Kinetics of Stress Response to Osmotic Shock. J. Cell Sci. 2002, 115, 4341–4351. [Google Scholar] [CrossRef] [Green Version]
- Sawant, M.; Schwarz, N.; Windoffer, R.; Magin, T.M.; Krieger, J.; Mücke, N.; Obara, B.; Jankowski, V.; Jankowski, J.; Wally, V.; et al. Threonine 150 Phosphorylation of Keratin 5 Is Linked to Epidermolysis Bullosa Simplex and Regulates Filament Assembly and Cell Viability. J. Investig. Dermatol. 2018, 138, 627–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, L.; Yamada, S.; Wirtz, D.; Coulombe, P.A. A “hot-Spot” Mutation Alters the Mechanical Properties of Keratin Filament Networks. Nat. Cell Biol. 2001, 3, 503–506. [Google Scholar] [CrossRef] [PubMed]
- Russell, D.; Andrews, P.D.; James, J.; Lane, E.B. Mechanical Stress Induces Profound Remodelling of Keratin Filaments and Cell Junctions in Epidermolysis Bullosa Simplex Keratinocytes. J. Cell Sci. 2004, 117, 5233–5243. [Google Scholar] [CrossRef] [Green Version]
- Kippenberger, S.; Hofmann, M.; Zöller, N.; Thaçi, D.; Müller, J.; Kaufmann, R.; Bernd, A. Ligation of Β4 Integrins Activates PKB/Akt and ERK1/2 by Distinct Pathways—Relevance of the Keratin Filament. Biochim. Biophys. Acta BBA-Mol. Cell Res. 2010, 1803, 940–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kölsch, A.; Windoffer, R.; Würflinger, T.; Aach, T.; Leube, R.E. The Keratin-Filament Cycle of Assembly and Disassembly. J. Cell Sci. 2010, 123, 2266–2272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Büchau, F.; Munz, C.; Has, C.; Lehmann, R.; Magin, T.M. KLHL16 Degrades Epidermal Keratins. J. Investig. Dermatol. 2018, 138, 1871–1873. [Google Scholar] [CrossRef] [Green Version]
- Beilin, A.; Gurskaya, N.; Vorotelyak, E. Establishing a Model of Epidermolysis Bullosa Simplex via CRISPR/Cas9 Editing in HaCaT Cells. FEBS Open Bio 2019, 9, 65–431. [Google Scholar] [CrossRef]
- Sørensen, C.B.; Andresen, B.S.; Jensen, U.B.; Jensen, T.G.; Jensen, P.K.A.; Gregersen, N.; Bolund, L. Functional Testing of Keratin 14 Mutant Proteins Associated with the Three Major Subtypes of Epidermolysis Bullosa Simplex. Exp. Dermatol. 2003, 12, 472–479. [Google Scholar] [CrossRef]
- Lehmann, S.M.; Leube, R.E.; Windoffer, R. Growth, Lifetime, Directional Movement and Myosin-Dependent Motility of Mutant Keratin Granules in Cultured Cells. Sci. Rep. 2021, 11, 2379. [Google Scholar] [CrossRef]
- Werner, N.S.; Windoffer, R.; Strnad, P.; Grund, C.; Leube, R.E.; Magin, T.M. Epidermolysis Bullosa Simplex-Type Mutations Alter the Dynamics of the Keratin Cytoskeleton and Reveal a Contribution of Actin to the Transport of Keratin Subunits. Mol. Biol. Cell 2004, 15, 990–1002. [Google Scholar] [CrossRef] [Green Version]
- Sugiura, K. Unfolded Protein Response in Keratinocytes: Impact on Normal and Abnormal Keratinization. J. Dermatol. Sci. 2013, 69, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Löffek, S.; Wöll, S.; Höhfeld, J.; Leube, R.E.; Has, C.; Bruckner-Tuderman, L.; Magin, T.M. The Ubiquitin Ligase CHIP/STUB1 Targets Mutant Keratins for Degradation. Hum. Mutat. 2010, 31, 466–476. [Google Scholar] [CrossRef] [PubMed]
- El-Hawary, M.S.; Abdel-Halim, M.R.E.; Sayed, S.S.; Abdelkader, H.A. Apocytolysis, a Proposed Mechanism of Blister Formation in Epidermolysis Bullosa Simplex. Arch. Dermatol. Res. 2015, 4, 371–377. [Google Scholar] [CrossRef] [PubMed]
- Chamcheu, J.C.; Wood, G.S.; Siddiqui, I.A.; Syed, D.N.; Adhami, V.M.; Teng, J.M.; Mukhtar, H. Progress towards Genetic and Pharmacological Therapies for Keratin Genodermatoses: Current Perspective and Future Promise. Exp. Dermatol. 2012, 21, 481–489. [Google Scholar] [CrossRef]
- Freedberg, I.M.; Tomic-Canic, M.; Komine, M.; Blumenberg, M. Keratins and the Keratinocyte Activation Cycle. J. Investig. Dermatol. 2001, 116, 633–640. [Google Scholar] [CrossRef] [Green Version]
- Lippens, S.; Denecker, G.; Ovaere, P.; Vandenabeele, P.; Declercq, W. Death Penalty for Keratinocytes: Apoptosis versus Cornification. Cell Death Differ. 2005, 12, 1497–1508. [Google Scholar] [CrossRef]
- Wesemann, D.R.; Qin, H.; Kokorina, N.; Benveniste, E.N. TRADD Interacts with STAT1-Alpha and Influences Interferon-Gamma Signaling. Nat. Immunol. 2004, 5, 199–207. [Google Scholar] [CrossRef]
- Lugassy, J.; McGrath, J.A.; Itin, P.; Shemer, R.; Verbov, J.; Murphy, H.R.; Ishida-Yamamoto, A.; Digiovanna, J.J.; Bercovich, D.; Karin, N.; et al. KRT14 Haploinsufficiency Results in Increased Susceptibility of Keratinocytes to TNF-Alpha-Induced Apoptosis and Causes Naegeli-Franceschetti-Jadassohn Syndrome. J. Investig. Dermatol. 2008, 128, 1517–1524. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Cheng, X.; Merched-Sauvage, M.; Caulin, C.; Roop, D.R.; Koch, P.J. An Unexpected Role for Keratin 10 End Domains in Susceptibility to Skin Cancer. J. Cell Sci. 2006, 119, 5067–5076. [Google Scholar] [CrossRef] [Green Version]
- Stegh, A.H.; Herrmann, H.; Lampel, S.; Weisenberger, D.; Andrä, K.; Seper, M.; Wiche, G.; Krammer, P.H.; Peter, M.E. Identification of the Cytolinker Plectin as a Major Early In Vivo Substrate for Caspase 8 during CD95- and Tumor Necrosis Factor Receptor-Mediated Apoptosis. Mol. Cell. Biol. 2000, 20, 5665–5679. [Google Scholar] [CrossRef] [Green Version]
- Ackerl, R.; Walko, G.; Fuchs, P.; Fischer, I.; Schmuth, M.; Wiche, G. Conditional Targeting of Plectin in Prenatal and Adult Mouse Stratified Epithelia Causes Keratinocyte Fragility and Lesional Epidermal Barrier Defects. J. Cell Sci. 2007, 120, 2435–2443. [Google Scholar] [CrossRef] [Green Version]
- Alexeev, V.; Salas-Alanis, J.C.; Palisson, F.; Mukhtarzada, L.; Fortuna, G.; Uitto, J.; South, A.; Igoucheva, O. Pro-Inflammatory Chemokines and Cytokines Dominate the Blister Fluid Molecular Signature in Patients with Epidermolysis Bullosa and Affect Leukocyte and Stem Cell Migration. J. Investig. Dermatol. 2017, 137, 2298–2308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roth, W.; Reuter, U.; Wohlenberg, C.; Bruckner-Tuderman, L.; Magin, T.M. Cytokines as Genetic Modifiers in K5−/− Mice and in Human Epidermolysis Bullosa Simplex. Hum. Mutat. 2009, 30, 832–841. [Google Scholar] [CrossRef] [PubMed]
- Wally, V.; Lettner, T.; Peking, P.; Peckl-Schmid, D.; Murauer, E.M.; Hainzl, S.; Hintner, H.; Bauer, J.W. The Pathogenetic Role of IL-1β in Severe Epidermolysis Bullosa Simplex. J. Investig. Dermatol. 2013, 133, 1901–1903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Moreno, M.; Davis, M.A.; Wong, E.; Pasolli, H.A.; Reynolds, A.B.; Fuchs, E. P120-Catenin Mediates Inflammatory Responses in the Skin. Cell 2006, 124, 631–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russell, D.; Ross, H.; Lane, E.B. ERK Involvement in Resistance to Apoptosis in Keratinocytes with Mutant Keratin. J. Investig. Dermatol. 2010, 130, 671–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, V.; Behr, M.; Kiritsi, D.; Scheffschick, A.; Grahnert, A.; Homberg, M.; Schwieger-Briel, A.; Jakob, T.; Bruckner-Tuderman, L.; Magin, T.M. Keratin-Dependent Thymic Stromal Lymphopoietin Expression Suggests a Link between Skin Blistering and Atopic Disease. J. Allergy Clin. Immunol. 2016, 138, 1461–1464.e6. [Google Scholar] [CrossRef] [Green Version]
- Castela, E.; Tulic, M.K.; Rozières, A.; Bourrat, E.; Nicolas, J.-F.; Kanitakis, J.; Vabres, P.; Bessis, D.; Mazereeuw, J.; Morice-Picard, F.; et al. Epidermolysis Bullosa Simplex Generalized Severe Induces a T Helper 17 Response and Is Improved by Apremilast Treatment. Br. J. Dermatol. 2019, 180, 357–364. [Google Scholar] [CrossRef]
- Castiglia, D.; El Hachem, M.; Diociaiuti, A.; Carbone, T.; De Luca, N.; Pascucci, M.; Zambruno, G.; Cavani, A. T-Lymphocytes Are Directly Involved in the Clinical Expression of Migratory Circinate Erythema in Epidermolysis Bullosa Simplex Patients. Acta Derm. Venereol. 2014, 94, 307–311. [Google Scholar] [CrossRef]
- Kippenberger, S.; Loitsch, S.; Müller, J.; Guschel, M.; Ramirez-Bosca, A.; Kaufmann, R.; Bernd, A. Melanocytes Respond to Mechanical Stretch by Activation of Mitogen-Activated Protein Kinases (MAPK). Pigment Cell Res. 2000, 13, 278–280. [Google Scholar] [CrossRef] [PubMed]
- Yano, S.; Komine, M.; Fujimoto, M.; Okochi, H.; Tamaki, K. Activation of Akt by Mechanical Stretching in Human Epidermal Keratinocytes. Exp. Dermatol. 2006, 15, 356–361. [Google Scholar] [CrossRef] [PubMed]
- Parcellier, A.; Tintignac, L.A.; Zhuravleva, E.; Hemmings, B.A. PKB and the Mitochondria: AKTing on Apoptosis. Cell. Signal. 2008, 20, 21–30. [Google Scholar] [CrossRef]
- Pang, Y.; Zheng, B.; Fan, L.-W.; Rhodes, P.G.; Cai, Z. IGF-1 Protects Oligodendrocyte Progenitors against TNFalpha-Induced Damage by Activation of PI3K/Akt and Interruption of the Mitochondrial Apoptotic Pathway. Glia 2007, 55, 1099–1107. [Google Scholar] [CrossRef] [PubMed]
- Zupancic, T.; Sersa, G.; Törmä, H.; Lane, E.B.; Herrmann, H.; Komel, R.; Liovic, M. Keratin Gene Mutations Influence the Keratinocyte Response to DNA Damage and Cytokine Induced Apoptosis. Arch. Dermatol. Res. 2017, 309, 587–593. [Google Scholar] [CrossRef]
- Schumann, H.; Roth, W.; Has, C.; Volz, A.; Erfurt-Berge, C.; Magin, T.M.; Bruckner-Tuderman, L. Verrucous Carcinoma in Epidermolysis Bullosa Simplex Is Possibly Associated with a Novel Mutation in the Keratin 5 Gene. Br. J. Dermatol. 2012, 167, 929–936. [Google Scholar] [CrossRef]
- Liovic, M.; Lee, B.; Tomic-Canic, M.; D’Alessandro, M.; Bolshakov, V.N.; Lane, E.B. Dual-Specificity Phosphatases in the Hypo-Osmotic Stress Response of Keratin-Defective Epithelial Cell Lines. Exp. Cell Res. 2008, 314, 2066–2075. [Google Scholar] [CrossRef]
- Wagner, M.; Trost, A.; Hintner, H.; Bauer, J.W.; Onder, K. Imbalance of Intermediate Filament Component Keratin 14 Contributes to Increased Stress Signalling in Epidermolysis Bullosa Simplex. Exp. Dermatol. 2013, 22, 292–294. [Google Scholar] [CrossRef]
- Wagner, M.; Hintner, H.; Bauer, J.W.; Onder, K. Gene Expression Analysis of an Epidermolysis Bullosa Simplex Dowling-Meara Cell Line by Subtractive Hybridization: Recapitulation of Cellular Differentiation, Migration and Wound Healing. Exp. Dermatol. 2012, 21, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Zhang, S.; Wang, G. Keratin 17 in Disease Pathogenesis: From Cancer to Dermatoses. J. Pathol. 2019, 247, 158–165. [Google Scholar] [CrossRef]
- Mariani, R.A.; Paranjpe, S.; Dobrowolski, R.; Weber, G.F. 14-3-3 Targets Keratin Intermediate Filaments to Mechanically Sensitive Cell–Cell Contacts. Mol. Biol. Cell 2020, 31, 930–943. [Google Scholar] [CrossRef]
- Guo, Y.; Redmond, C.J.; Leacock, K.A.; Brovkina, M.V.; Ji, S.; Jaskula-Ranga, V.; Coulombe, P.A. Keratin 14-Dependent Disulfides Regulate Epidermal Homeostasis and Barrier Function via 14-3-3σ and YAP1. eLife 2020, 9, e53165. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.; Chen, L.; Liu, Y.-F.; Gao, T.-W.; Wang, G.; Fan, X.-L.; Fan, J.-Y.; Fan, P.-S.; Li, C.-Y.; Liu, B.; et al. Altered Keratin 17 Peptide Ligands Inhibit in Vitro Proliferation of Keratinocytes and T Cells Isolated from Patients with Psoriasis. J. Am. Acad. Dermatol. 2006, 54, 992–1002. [Google Scholar] [CrossRef]
- Yang, L.; Fan, X.; Cui, T.; Dang, E.; Wang, G. Nrf2 Promotes Keratinocyte Proliferation in Psoriasis through Up-Regulation of Keratin 6, Keratin 16, and Keratin 17. J. Investig. Dermatol. 2017, 137, 2168–2176. [Google Scholar] [CrossRef] [Green Version]
- Pourreyron, C.; Reilly, L.; Proby, C.; Panteleyev, A.; Fleming, C.; McLean, K.; South, A.P.; Foerster, J. Wnt5a Is Strongly Expressed at the Leading Edge in Non-Melanoma Skin Cancer, Forming Active Gradients, While Canonical Wnt Signalling Is Repressed. PLoS ONE 2012, 7, e31827. [Google Scholar] [CrossRef] [Green Version]
- Kurbet, A.S.; Hegde, S.; Bhattacharjee, O.; Marepally, S.; Vemula, P.K.; Raghavan, S. Sterile Inflammation Enhances ECM Degradation in Integrin Β1 KO Embryonic Skin. Cell Rep. 2016, 16, 3334–3347. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.Y.; Nuñez, G. Sterile Inflammation: Sensing and Reacting to Damage. Nat. Rev. Immunol. 2010, 10, 826–837. [Google Scholar] [CrossRef] [Green Version]
- Bianchi, M.E. DAMPs, PAMPs and Alarmins: All We Need to Know about Danger. J. Leukoc. Biol. 2007, 81, 1–5. [Google Scholar] [CrossRef]
- Pfisterer, K.; Shaw, L.E.; Symmank, D.; Weninger, W. The Extracellular Matrix in Skin Inflammation and Infection. Front. Cell Dev. Biol. 2021, 9, 682414. [Google Scholar] [CrossRef]
- Hulina, A.; Grdić Rajković, M.; Jakšić Despot, D.; Jelić, D.; Dojder, A.; Čepelak, I.; Rumora, L. Extracellular Hsp70 Induces Inflammation and Modulates LPS/LTA-Stimulated Inflammatory Response in THP-1 Cells. Cell Stress Chaperones 2018, 23, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Morizane, S.; Kajita, A.; Mizuno, K.; Takiguchi, T.; Iwatsuki, K. Toll-like Receptor Signalling Induces the Expression of Serum Amyloid A in Epidermal Keratinocytes and Dermal Fibroblasts. Clin. Exp. Dermatol. 2019, 44, 40–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eigenbrod, T.; Park, J.-H.; Harder, J.; Iwakura, Y.; Núñez, G. Critical Role for Mesothelial Cells in Necrosis-Induced Inflammation through the Recognition of IL-1α Released from Dying Cells. J. Immunol. 2008, 181, 8194–8198. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.-J.; Kono, H.; Golenbock, D.; Reed, G.; Akira, S.; Rock, K.L. Identification of a Key Pathway Required for the Sterile Inflammatory Response Triggered by Dying Cells. Nat. Med. 2007, 13, 851–856. [Google Scholar] [CrossRef]
- Karppinen, S.-M.; Heljasvaara, R.; Gullberg, D.; Tasanen, K.; Pihlajaniemi, T. Toward Understanding Scarless Skin Wound Healing and Pathological Scarring. F1000Research 2019, 8, 787. [Google Scholar] [CrossRef] [Green Version]
- Annicchiarico, G.; Morgese, M.G.; Esposito, S.; Lopalco, G.; Lattarulo, M.; Tampoia, M.; Bonamonte, D.; Brunetti, L.; Vitale, A.; Lapadula, G.; et al. Proinflammatory Cytokines and Antiskin Autoantibodies in Patients with Inherited Epidermolysis Bullosa. Medicine 2015, 94, e1528. [Google Scholar] [CrossRef]
- Schiraldi, M.; Raucci, A.; Muñoz, L.M.; Livoti, E.; Celona, B.; Venereau, E.; Apuzzo, T.; De Marchis, F.; Pedotti, M.; Bachi, A.; et al. HMGB1 Promotes Recruitment of Inflammatory Cells to Damaged Tissues by Forming a Complex with CXCL12 and Signaling via CXCR4. J. Exp. Med. 2012, 209, 551–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huitema, L.; Phillips, T.; Alexeev, V.; Igoucheva, O. Immunological Mechanisms Underlying Progression of Chronic Wounds in Recessive Dystrophic Epidermolysis Bullosa. Exp. Dermatol. 2021, 30, 1724–1733. [Google Scholar] [CrossRef] [PubMed]
- Mellerio, J.E. Potential Therapeutic Targeting of Inflammation in Epidermolysis Bullosa Simplex. Br. J. Dermatol. 2019, 180, 258–260. [Google Scholar] [CrossRef] [PubMed]
- Bobr, A.; Igyarto, B.Z.; Haley, K.M.; Li, M.O.; Flavell, R.A.; Kaplan, D.H. Autocrine/Paracrine TGF-Β1 Inhibits Langerhans Cell Migration. Proc. Natl. Acad. Sci. USA 2012, 109, 10492–10497. [Google Scholar] [CrossRef] [Green Version]
- Diaz, L.E.A.; Debes, G.F. Integrin A4β1-Dependent Regulatory B-Cell Migration into the Skin Limits Cutaneous Inflammation. J. Immunol. 2021, 206, 11.06. [Google Scholar]
- Debes, G.F.; McGettigan, S.E. Skin-Associated B Cells in Health and Inflammation. J. Immunol. 2019, 202, 1659–1666. [Google Scholar] [CrossRef] [Green Version]
- Rittié, L. Cellular Mechanisms of Skin Repair in Humans and Other Mammals. J. Cell Commun. Signal. 2016, 10, 103–120. [Google Scholar] [CrossRef] [Green Version]
- Lloyd, C.; Yu, Q.C.; Cheng, J.; Turksen, K.; Degenstein, L.; Hutton, E.; Fuchs, E. The Basal Keratin Network of Stratified Squamous Epithelia: Defining K15 Function in the Absence of K14. J. Cell Biol. 1995, 129, 1329–1344. [Google Scholar] [CrossRef] [Green Version]
- Troy, T.-C.; Turksen, K. In vitro characteristics of early epidermal progenitors isolated from keratin 14 (K14)-deficient mice: Insights into the role of keratin 17 in mouse keratinocytes. J. Cell. Physiol. 1999, 180, 409–421. [Google Scholar] [CrossRef]
- Vijayaraj, P.; Kröger, C.; Reuter, U.; Windoffer, R.; Leube, R.E.; Magin, T.M. Keratins Regulate Protein Biosynthesis through Localization of GLUT1 and -3 Upstream of AMP Kinase and Raptor. J. Cell Biol. 2009, 187, 175–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winter, L.; Kuznetsov, A.V.; Grimm, M.; Zeöld, A.; Fischer, I.; Wiche, G. Plectin Isoform P1b and P1d Deficiencies Differentially Affect Mitochondrial Morphology and Function in Skeletal Muscle. Hum. Mol. Genet. 2015, 24, 4530–4544. [Google Scholar] [CrossRef] [Green Version]
- Walko, G.; Vukasinovic, N.; Gross, K.; Fischer, I.; Sibitz, S.; Fuchs, P.; Reipert, S.; Jungwirth, U.; Berger, W.; Salzer, U.; et al. Targeted Proteolysis of Plectin Isoform 1a Accounts for Hemidesmosome Dysfunction in Mice Mimicking the Dominant Skin Blistering Disease EBS-Ogna. PLoS Genet. 2011, 7, e1002396. [Google Scholar] [CrossRef] [Green Version]
- Johnson, A.L.; Peterson, S.M.; Terry, M.M.L.; Ferguson, B.; Colgin, L.M.; Lewis, A.D. Spontaneous KRT5 Gene Mutation in Rhesus Macaques (Macaca Mulatta): A Novel Nonhuman Primate Model of Epidermolysis Bullosa Simplex. Vet. Pathol. 2020, 57, 344–348. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Choi, H.Y.; So, J.-H.; Kim, C.-H.; Ho, S.-Y.; Frank, M.; Li, Q.; Uitto, J. Zebrafish Type XVII Collagen: Gene Structures, Expression Profiles, and Morpholino “Knock-down” Phenotypes. Matrix Biol. J. Int. Soc. Matrix Biol. 2010, 29, 629–637. [Google Scholar] [CrossRef]
- Li, Q.; Uitto, J. Zebrafish as a Model System to Study Skin Biology and Pathology. J. Investig. Dermatol. 2014, 134, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Bohnekamp, J.; Cryderman, D.E.; Paululat, A.; Baccam, G.C.; Wallrath, L.L.; Magin, T.M. A Drosophila Model of Epidermolysis Bullosa Simplex. J. Investig. Dermatol. 2015, 135, 2031–2039. [Google Scholar] [CrossRef] [Green Version]
- Abaci, H.; Guo, Z.; Doucet, Y.; Jacków, J.; Christiano, A. Next Generation Human Skin Constructs as Advanced Tools for Drug Development. Exp. Biol. Med. 2017, 242, 1657–1668. [Google Scholar] [CrossRef]
- Chidgey, M.; Dawson, C. Desmosomes: A Role in Cancer? Br. J. Cancer 2007, 96, 1783–1787. [Google Scholar] [CrossRef] [PubMed]
- Lorch, J.H.; Klessner, J.; Park, J.K.; Getsios, S.; Wu, Y.L.; Stack, M.S.; Green, K.J. Epidermal Growth Factor Receptor Inhibition Promotes Desmosome Assembly and Strengthens Intercellular Adhesion in Squamous Cell Carcinoma Cells. J. Biol. Chem. 2004, 279, 37191–37200. [Google Scholar] [CrossRef] [Green Version]
- Guo, L.; Degenstein, L.; Dowling, J.; Yu, Q.C.; Wollmann, R.; Perman, B.; Fuchs, E. Gene Targeting of BPAG1: Abnormalities in Mechanical Strength and Cell Migration in Stratified Epithelia and Neurologic Degeneration. Cell 1995, 81, 233–243. [Google Scholar] [CrossRef] [Green Version]
- Vetter, A.; Jahn, K.; Bouameur, J.-E.; Kiritsi, D.; Magin, T.M. Epidermolysis Bullosa Simplex Keratinocytes Show Disturbed Mitochondrial Positioning and Activity. J. Investig. Dermatol. 2020, 140, 1438–1442.e5. [Google Scholar] [CrossRef] [PubMed]
- Hamanaka, R.B.; Glasauer, A.; Hoover, P.; Yang, S.; Blatt, H.; Mullen, A.R.; Getsios, S.; Gottardi, C.J.; DeBerardinis, R.J.; Lavker, R.M.; et al. Mitochondrial Reactive Oxygen Species Promote Epidermal Differentiation and Hair Follicle Development. Sci. Signal. 2013, 6, ra8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, V.; Bouameur, J.-E.; Bär, J.; Rice, R.H.; Hornig-Do, H.-T.; Roop, D.R.; Schwarz, N.; Brodesser, S.; Thiering, S.; Leube, R.E.; et al. A Keratin Scaffold Regulates Epidermal Barrier Formation, Mitochondrial Lipid Composition, and Activity. J. Cell Biol. 2015, 211, 1057–1075. [Google Scholar] [CrossRef]
- Glàsz-Bóna, A.; Medvecz, M.; Virágh, Z.; Hatvani, Z.; Blazsek, A.; Kárpáti, S. Epidermolysis Bullosa Simplex with Mottled Pigmentation-Mutation Analysis Proved the Diagnosis in a Four-Generation Pedigree. Eur. J. Dermatol. EJD 2010, 20, 698–700. [Google Scholar] [CrossRef]
- Betz, R.C.; Planko, L.; Eigelshoven, S.; Hanneken, S.; Pasternack, S.M.; Bussow, H.; Van Den Bogaert, K.; Wenzel, J.; Braun-Falco, M.; Rutten, A.; et al. Loss-of-Function Mutations in the Keratin 5 Gene Lead to Dowling-Degos Disease. Am. J. Hum. Genet. 2006, 78, 510–519. [Google Scholar] [CrossRef] [Green Version]
- Planko, L.; Böhse, K.; Höhfeld, J.; Betz, R.C.; Hanneken, S.; Eigelshoven, S.; Kruse, R.; Nöthen, M.M.; Magin, T.M. Identification of a Keratin-Associated Protein with a Putative Role in Vesicle Transport. Eur. J. Cell Biol. 2007, 86, 827–839. [Google Scholar] [CrossRef]
- Cario, M.; Pain, C.; Kaulanjan-Checkmodine, P.; Masia, D.; Delia, G.; Casoli, V.; Costet, P.; Goussot, J.-F.; Guyonnet-Duperat, V.; Bibeyran, A.; et al. Epidermal Keratin 5 Expression and Distribution Is under Dermal Influence. Pigment Cell Melanoma Res. 2020, 33, 435–445. [Google Scholar] [CrossRef]
- Rubin, A.I.; Garzon, M.C.; Morel, K.D. Herpetic Infection in Epidermolysis Bullosa. Pediatr. Dermatol. 2006, 23, 355–357. [Google Scholar] [CrossRef]
- Levin, L.E.; Shayegan, L.H.; Lucky, A.W.; Hook, K.P.; Bruckner, A.L.; Feinstein, J.A.; Whittier, S.; Lauren, C.T.; Pope, E.; Lara-Corrales, I.; et al. Characterization of Wound Microbes in Epidermolysis Bullosa: Results from the Epidermolysis Bullosa Clinical Characterization and Outcomes Database. Pediatr. Dermatol. 2021, 38, 119–124. [Google Scholar] [CrossRef] [PubMed]
- van der Kooi-Pol, M.M.; Sadaghian Sadabad, M.; Duipmans, J.C.; Sabat, A.J.; Stobernack, T.; Omansen, T.F.; Westerhout-Pluister, G.N.; Jonkman, M.F.; Harmsen, H.J.M.; van Dijl, J.M. Topography of Distinct Staphylococcus aureus Types in Chronic Wounds of Patients with Epidermolysis Bullosa. PLoS ONE 2013, 8, e67272. [Google Scholar] [CrossRef] [Green Version]
- Mellerio, J.E. Infection and Colonization in Epidermolysis Bullosa. Dermatol. Clin. 2010, 28, 267–269. [Google Scholar] [CrossRef] [PubMed]
- Hoste, E.; Arwert, E.N.; Lal, R.; South, A.P.; Salas-Alanis, J.C.; Murrell, D.F.; Donati, G.; Watt, F.M. Innate Sensing of Microbial Products Promotes Wound-Induced Skin Cancer. Nat. Commun. 2015, 6, 5932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guttmann-Gruber, C.; Tockner, B.; Scharler, C.; Hüttner, C.; Common, J.E.; Tay, A.S.L.; Denil, S.L.I.J.; Klausegger, A.; Trost, A.; Breitenbach, J.; et al. Low-Dose Calcipotriol Can Elicit Wound Closure, Anti-Microbial, and Anti-Neoplastic Effects in Epidermolysis Bullosa Keratinocytes. Sci. Rep. 2018, 8, 13430. [Google Scholar] [CrossRef]
- Lee, J.T.Y.; Wang, G.; Tam, Y.T.; Tam, C. Membrane-Active Epithelial Keratin 6A Fragments (KAMPs) Are Unique Human Antimicrobial Peptides with a Non-Aβ Structure. Front. Microbiol. 2016, 7, 1799. [Google Scholar] [CrossRef] [Green Version]
- Chan, J.K.L.; Yuen, D.; Too, P.H.-M.; Sun, Y.; Willard, B.; Man, D.; Tam, C. Keratin 6a Reorganization for Ubiquitin–Proteasomal Processing Is a Direct Antimicrobial Response. J. Cell Biol. 2018, 217, 731–744. [Google Scholar] [CrossRef] [Green Version]
- Geisler, F.; Leube, R.E. Epithelial Intermediate Filaments: Guardians against Microbial Infection? Cells 2016, 5, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luan, R.; Liang, Z.; Zhang, Q.; Sun, L.; Zhao, Y. Molecular Regulatory Networks of Thymic Epithelial Cell Differentiation. Differ. Res. Biol. Divers. 2019, 107, 42–49. [Google Scholar] [CrossRef]
- Odaka, C.; Loranger, A.; Takizawa, K.; Ouellet, M.; Tremblay, M.J.; Murata, S.; Inoko, A.; Inagaki, M.; Marceau, N. Keratin 8 Is Required for the Maintenance of Architectural Structure in Thymus Epithelium. PLoS ONE 2013, 8, e75101. [Google Scholar] [CrossRef] [Green Version]
- Farley, A.M.; Morris, L.X.; Vroegindeweij, E.; Depreter, M.L.G.; Vaidya, H.; Stenhouse, F.H.; Tomlinson, S.R.; Anderson, R.A.; Cupedo, T.; Cornelissen, J.J.; et al. Dynamics of Thymus Organogenesis and Colonization in Early Human Development. Dev. Camb. Engl. 2013, 140, 2015–2026. [Google Scholar] [CrossRef] [Green Version]
- Tominaga, M.; Tengara, S.; Kamo, A.; Ogawa, H.; Takamori, K. Matrix Metalloproteinase-8 Is Involved in Dermal Nerve Growth: Implications for Possible Application to Pruritus from in Vitro Models. J. Investig. Dermatol. 2011, 131, 2105–2112. [Google Scholar] [CrossRef] [Green Version]
- Lincoln, V.; Chao, L.; Woodley, D.T.; Murrell, D.; Kim, M.; O’Toole, E.A.; Ly, A.; Cogan, J.; Mosallaei, D.; Wysong, A.; et al. Over-Expression of Stromal Periostin Correlates with Poor Prognosis of Cutaneous Squamous Cell Carcinomas. Exp. Dermatol. 2021, 30, 698–704. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.K.; Wheeler, J.J.; Pitake, S.; Ding, H.; Jiang, C.; Fukuyama, T.; Paps, J.S.; Ralph, P.; Coyne, J.; Parkington, M.; et al. Periostin Activation of Integrin Receptors on Sensory Neurons Induces Allergic Itch. Cell Rep. 2020, 31, 107472. [Google Scholar] [CrossRef] [PubMed]
- Corren, J.; Ziegler, S.F. TSLP: From Allergy to Cancer. Nat. Immunol. 2019, 20, 1603–1609. [Google Scholar] [CrossRef]
- Guillot-Delost, M.; Guilleré, L.; Berger, F.; Ventre, A.; Michea, P.; Sirven, P.; Pattarini, L.; Scholer-Dahirel, A.; Kebir, F.-Z.; Huerre, M.; et al. Ligand–Receptor Dissociated Expression Explains High TSLP without Prognostic Impact in Human Primary Head and Neck Squamous Cell Carcinoma. OncoImmunology 2016, 5, e1179414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.-M.; Lin, L.-W.; Chen, Y.-W.; Ye, Y.-L. The Expression and Prognostic Impact of Proinflammatory Cytokines and Their Associations with Carcinogens in Oropharyngeal Squamous Cell Carcinoma. Cancer Immunol. Immunother. 2020, 69, 549–558. [Google Scholar] [CrossRef] [PubMed]
- DePianto, D.; Kerns, M.L.; Dlugosz, A.A.; Coulombe, P.A. Keratin 17 Promotes Epithelial Proliferation and Tumor Growth by Polarizing the Immune Response in Skin. Nat. Genet. 2010, 42, 910–914. [Google Scholar] [CrossRef] [Green Version]
- Stacey, S.N.; Sulem, P.; Masson, G.; Gudjonsson, S.A.; Thorleifsson, G.; Jakobsdottir, M.; Sigurdsson, A.; Gudbjartsson, D.F.; Sigurgeirsson, B.; Benediktsdottir, K.R.; et al. New Common Variants Affecting Susceptibility to Basal Cell Carcinoma. Nat. Genet. 2009, 41, 909–914. [Google Scholar] [CrossRef]
- Karantza, V. Keratins in Health and Cancer: More than Mere Epithelial Cell Markers. Oncogene 2011, 30, 127–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanley, C.J.; Henriet, E.; Sirka, O.K.; Thomas, G.J.; Ewald, A.J. Tumor-Resident Stromal Cells Promote Breast Cancer Invasion through Regulation of the Basal Phenotype. Mol. Cancer Res. MCR 2020, 18, 1615–1622. [Google Scholar] [CrossRef] [PubMed]
- McGinn, O.; Ward, A.V.; Fettig, L.M.; Riley, D.; Ivie, J.; Paul, K.V.; Kabos, P.; Finlay-Schultz, J.; Sartorius, C.A. Cytokeratin 5 Alters β-Catenin Dynamics in Breast Cancer Cells. Oncogene 2020, 39, 2478–2492. [Google Scholar] [CrossRef] [PubMed]
- Irvine, A.D.; Rugg, E.L.; Lane, E.B.; Hoare, S.; Peret, C.; Hughes, A.E.; Heagerty, A.H. Molecular Confirmation of the Unique Phenotype of Epidermolysis Bullosa Simplex with Mottled Pigmentation. Br. J. Dermatol. 2001, 144, 40–45. [Google Scholar] [CrossRef] [Green Version]
- Hovnanian, A.; Pollack, E.; Hilal, L.; Rochat, A.; Prost, C.; Barrandon, Y.; Goossens, M. A Missense Mutation in the Rod Domain of Keratin 14 Associated with Recessive Epidermolysis Bullosa Simplex. Nat. Genet. 1993, 3, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Rugg, E.L.; McLean, W.H.; Lane, E.B.; Pitera, R.; McMillan, J.R.; Dopping-Hepenstal, P.J.; Navsaria, H.A.; Leigh, I.M.; Eady, R.A. A Functional “Knockout” of Human Keratin 14. Genes Dev. 1994, 8, 2563–2573. [Google Scholar] [CrossRef] [Green Version]
- McGrath, J.A. Recently Identified Forms of Epidermolysis Bullosa. Ann. Dermatol. 2015, 27, 658–666. [Google Scholar] [CrossRef] [Green Version]
- Tu, W.; Chen, P.; Hou, P.; Huang, H.; Wang, J.; Chao, S.; Lee, J.; McGrath, J.; Natsuga, K.; Hsu, C. Plectin Missense Mutation p.Leu319Pro in the Pathogenesis of Autosomal Recessive Epidermolysis Bullosa Simplex. Acta Derm. Venereol. 2020, 100, adv00242. [Google Scholar] [CrossRef] [PubMed]
- Walter, M.C.; Reilich, P.; Krause, S.; Hiebeler, M.; Gehling, S.; Goebel, H.H.; Schoser, B.; Abicht, A. Congenital Myopathy and Epidermolysis Bullosa Due to PLEC Variant. Neuromuscul. Disord. 2021, in press. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.-H.; Kim, S.-C.; Ichiki, Y.; Park, J.; Nagai, M.; Kitajima, Y. A Usual Frameshift and Delayed Termination Codon Mutation in Keratin 5 Causes a Novel Type of Epidermolysis Bullosa Simplex with Migratory Circinate Erythema. J. Investig. Dermatol. 2003, 121, 482–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagao-Watanabe, M.; Fukao, T.; Matsui, E.; Kaneko, H.; Inoue, R.; Kawamoto, N.; Kasahara, K.; Nagai, M.; Ichiki, Y.; Kitajima, Y.; et al. Identification of Somatic and Germline Mosaicism for a Keratin 5 Mutation in Epidermolysis Bullosa Simplex in a Family of Which the Proband Was Previously Regarded as a Sporadic Case. Clin. Genet. 2004, 66, 236–238. [Google Scholar] [CrossRef]
- McGrath, J.A.; Hoeger, P.H.; Christiano, A.M.; McMillan, J.R.; Mellerio, J.E.; Ashton, G.H.; Dopping-Hepenstal, P.J.; Lake, B.D.; Leigh, I.M.; Harper, J.I.; et al. Skin Fragility and Hypohidrotic Ectodermal Dysplasia Resulting from Ablation of Plakophilin 1. Br. J. Dermatol. 1999, 140, 297–307. [Google Scholar] [CrossRef] [PubMed]
- Groves, R.W.; Liu, L.; Dopping-Hepenstal, P.J.; Markus, H.S.; Lovell, P.A.; Ozoemena, L.; Lai-Cheong, J.E.; Gawler, J.; Owaribe, K.; Hashimoto, T.; et al. A Homozygous Nonsense Mutation within the Dystonin Gene Coding for the Coiled-Coil Domain of the Epithelial Isoform of BPAG1 Underlies a New Subtype of Autosomal Recessive Epidermolysis Bullosa Simplex. J. Investig. Dermatol. 2010, 130, 1551–1557. [Google Scholar] [CrossRef] [Green Version]
- Jonkman, M.F.; Pas, H.H.; Nijenhuis, M.; Kloosterhuis, G.; Steege, G. Deletion of a Cytoplasmic Domain of Integrin Beta4 Causes Epidermolysis Bullosa Simplex. J. Investig. Dermatol. 2002, 119, 1275–1281. [Google Scholar] [CrossRef] [Green Version]
- Chung, H.J.; Uitto, J. Epidermolysis Bullosa with Pyloric Atresia. Dermatol. Clin. 2010, 28, 43–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.; Maier, K.; Leppert, J.; Hausser, I.; Schwieger-Briel, A.; Weibel, L.; Theiler, M.; Kiritsi, D.; Busch, H.; Boerries, M.; et al. Monoallelic Mutations in the Translation Initiation Codon of KLHL24 Cause Skin Fragility. Am. J. Hum. Genet. 2016, 99, 1395–1404. [Google Scholar] [CrossRef] [Green Version]
- Vahidnezhad, H.; Youssefian, L.; Saeidian, A.H.; Mahmoudi, H.; Touati, A.; Abiri, M.; Kajbafzadeh, A.-M.; Aristodemou, S.; Liu, L.; McGrath, J.A.; et al. Recessive Mutation in Tetraspanin CD151 Causes Kindler Syndrome-like Epidermolysis Bullosa with Multi-Systemic Manifestations Including Nephropathy. Matrix Biol. 2018, 66, 22–33. [Google Scholar] [CrossRef] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Evtushenko, N.A.; Beilin, A.K.; Kosykh, A.V.; Vorotelyak, E.A.; Gurskaya, N.G. Keratins as an Inflammation Trigger Point in Epidermolysis Bullosa Simplex. Int. J. Mol. Sci. 2021, 22, 12446. https://doi.org/10.3390/ijms222212446
Evtushenko NA, Beilin AK, Kosykh AV, Vorotelyak EA, Gurskaya NG. Keratins as an Inflammation Trigger Point in Epidermolysis Bullosa Simplex. International Journal of Molecular Sciences. 2021; 22(22):12446. https://doi.org/10.3390/ijms222212446
Chicago/Turabian StyleEvtushenko, Nadezhda A., Arkadii K. Beilin, Anastasiya V. Kosykh, Ekaterina A. Vorotelyak, and Nadya G. Gurskaya. 2021. "Keratins as an Inflammation Trigger Point in Epidermolysis Bullosa Simplex" International Journal of Molecular Sciences 22, no. 22: 12446. https://doi.org/10.3390/ijms222212446