
DNA Repair in Haploid Context

Abstract
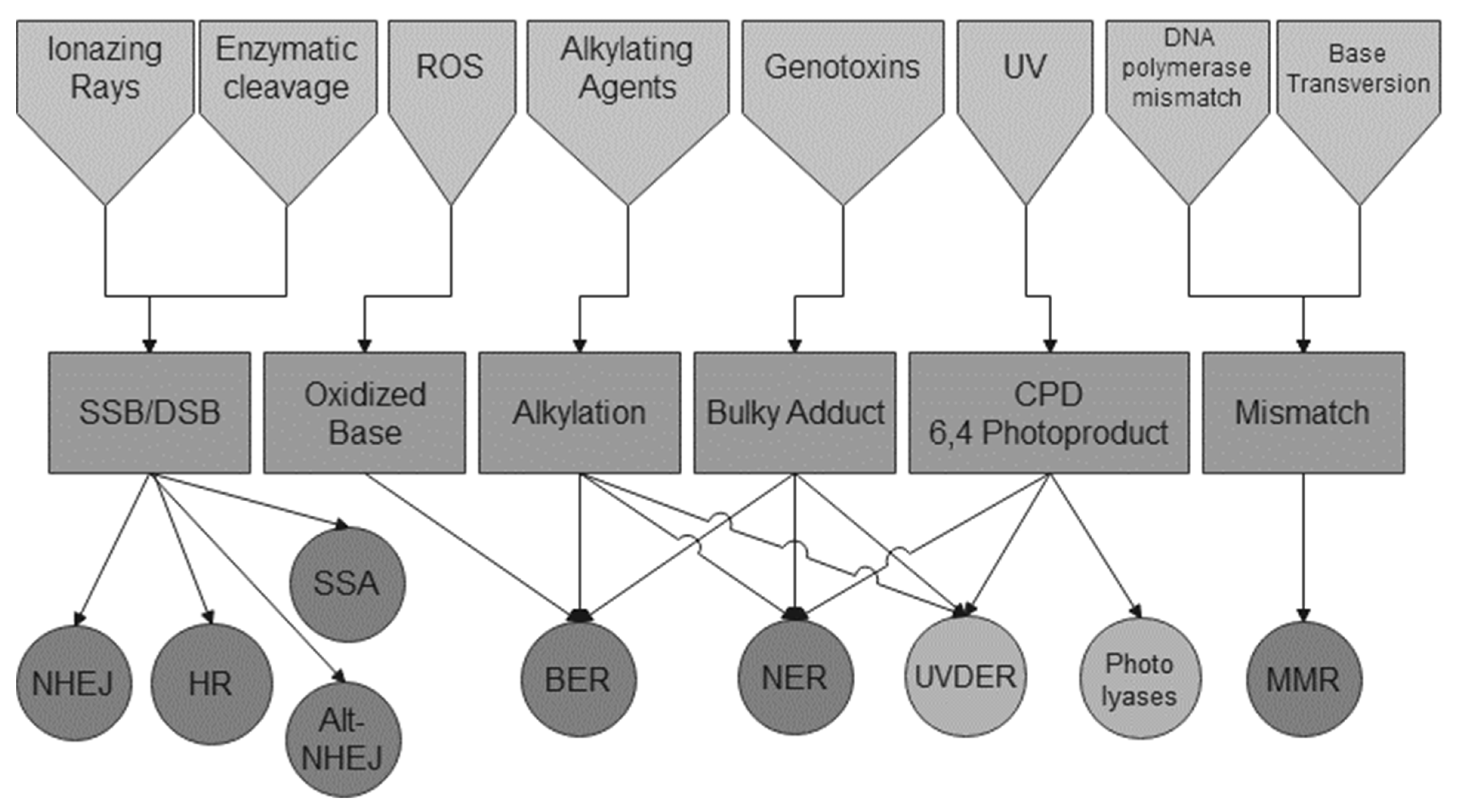
:1. Introduction
2. General Consideration Regarding Ploidy States
3. DNA Repair Systems Comparison between S. cerevisiae and Sc. pombe
3.1. Nucleotide Excision Repair
3.1.1. General Mechanism
3.1.2. NER in Yeast
3.2. Base Excision Repair
3.2.1. General Mechanism
3.2.2. BER in Yeast
3.3. Mismatch Repair
3.3.1. General Mechanism
3.3.2. MMR in Yeast
3.4. Double Strand Breaks (DSBs) Repair Pathways Selection
3.4.1. DSB Regulation in Mammals
3.4.2. DSB Regulation in Yeast
3.5. Homologous Recombination
3.5.1. General Mechanism
3.5.2. HR in Yeast
3.6. Non-Homologous End-Joining
3.6.1. General Mechanism
3.6.2. NHEJ in Yeast
3.7. Single-Strand Annealing
3.7.1. General Mechanism
3.7.2. SSA in Yeast
3.8. Yeast Specific DNA Repair Mechanisms
3.8.1. UV Damage Excision Repair
3.8.2. Photolyases
3.9. Alternative DNA Repair Approaches in Yeast
4. Repair in Human Germ Cells
4.1. Gametes
4.1.1. Spermatozoa
4.1.2. Oocyte
5. Conclusions
Funding
Conflicts of Interest
References
- Gerstein, A.C.; Otto, S.P. Ploidy and the causes of genomic evolution. J. Hered. 2009, 100, 571–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gresham, D.; Desai, M.M.; Tucker, C.M.; Jenq, H.T.; Pai, D.A.; Ward, A.; DeSevo, C.G.; Botstein, D.; Dunham, M.J. The repertoire and dynamics of evolutionary adaptations to controlled nutrient-limited environments in yeast. PLoS Genet. 2008, 4, e1000303. [Google Scholar] [CrossRef] [Green Version]
- Zeyl, C.; Vanderford, T.; Carter, M. An evolutionary advantage of haploidy in large yeast populations. Science 2003, 299, 555–558. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zeidler, A.F.B.; Song, W.; Puccia, C.M.; Malc, E.; Greenwell, P.W.; Mieczkowski, P.A.; Petes, T.D.; Argueso, J.L. Gene copy-number variation in haploid and diploid strains of the yeast saccharomyces cerevisiae. Genetics 2013, 193, 785–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crow, J.F.; Kimura, M. Evolution in sexual and asexual populations. Am. Nat. 1965, 99, 439–450. [Google Scholar] [CrossRef]
- Kondrashov, A.S.; Crow, J.F. Haploidy or Diploidy: Which Is Better? Nature 1991, 351, 314–315. [Google Scholar] [CrossRef]
- Sharp, N.P.; Sandell, L.; James, C.G.; Otto, S.P. The Genome-Wide Rate and Spectrum of Spontaneous Mutations Differ between Haploid and Diploid Yeast. Proc. Natl. Acad. Sci. USA 2018, 115, E5046–E5055. [Google Scholar] [CrossRef] [Green Version]
- Lang, G.I.; Murray, A.W. Mutation rates across budding yeast chromosome VI are correlated with replication timing. Genome Biol. Evol. 2011, 3, 799–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orr, H.A.; Otto, S.P. Does Diploidy Increase the Rate of Adaptation? Genetics 1994, 136, 1475–1480. [Google Scholar] [CrossRef]
- Sia, E.A.; Butler, C.A.; Dominska, M.; Greenwell, P.; Fox, T.D.; Petes, T.D. Analysis of Microsatellite Mutations in the Mitochondrial DNA of Saccharomyces Cerevisiae. Proc. Natl. Acad. Sci. USA 2000, 97, 250–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selmecki, A.M. Polyploidy Can Drive Rapid Adaptation in Yeast. Nature 2015, 519, 349–352. [Google Scholar] [CrossRef]
- Spivak, G. Nucleotide Excision Repair in Humans. DNA Repair 2015, 36, 13–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanawalt, P.C.; Spivak, G. Transcription-Coupled DNA Repair: Two Decades of Progress and Surprises. Nat. Rev. Mol. Cell Biol. 2008, 9, 958–970. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.H.; Xu, L.; Wang, D. Mechanism of Transcription-Coupled DNA Modification Recognition. Cell Biosci. 2017, 7, 9. [Google Scholar] [CrossRef] [Green Version]
- Dawlaty, M.M.; Malureanu, L.; Jeganathan, K.B. Resolution of Sister Centromeres Requires RanBP2-Mediated SUMOylation of Topoisomerase IIalpha. Cell 2008, 133, 103–115. [Google Scholar] [CrossRef] [Green Version]
- Fousteri, M. Cockayne Syndrome A and B Proteins Differentially Regulaterecruitment of Chromatin Remodeling and Repair Factors to Stalled RNApolymerase II in vivo. Mol. Cell 2006, 23, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Belmont, A.S. Mitotic Chromosome Structure and Condensation. Curr. Opin. Cell Biol. 2006, 18, 632–638. [Google Scholar] [CrossRef] [PubMed]
- Okuda, M.; Nakazawa, Y.; Guo, C.; Ogi, T.; Nishimura, Y. Common TFIIH Recruitment Mechanism in Global Genome and Transcription-Coupled Repair Subpathways. Nucleic Acids Res. 2017, 45, 13043–13055. [Google Scholar] [CrossRef]
- Reardon, J.T.; Sancar, A. Recognition and Repair of the Cyclobutane Thymine Dimer, a Major Cause of Skin Cancers, by the Human Excision Nuclease. Genes Dev. 2003, 17, 2539–2551. [Google Scholar] [CrossRef] [Green Version]
- Li, C.-L. Tripartite DNA Lesion Recognition and Verification by XPC, TFIIH, and XPA in Nucleotide Excision Repair. Mol. Cell 2015, 59, 1025–1034. [Google Scholar] [CrossRef] [Green Version]
- Cai, Y.; Patel, D.J.; Broyde, S.; Geacintov, N.E. Base sequence context effects on nucleotide excision repair. J. Nucleic Acids 2010, 2010, 174252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kropachev, K.; Kolbanovskii, M.; Cai, Y.; Rodriguez, F.; Kolbanovskii, A.; Liu, Y.; Zhang, L.; Amin, S.; Patel, D.; Broyde, S.; et al. The Sequence Dependence of Human Nucleotide Excision Repair Efficiencies of Benzo[a]Pyrene-Derived DNA Lesions: Insights into the Structural Factors That Favor Dual Incisions. J. Mol. Biol. 2009, 386, 1193–1203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mocquet, V.; Kropachev, K.; Kolbanovskiy, M.; Kolbanovskiy, A.; Tapias, A.; Cai, Y.; Broyde, S.; Geacintov, N.E.; Egly, J.M. The Human DNA Repair Factor XPC-HR23B Distinguishes Stereoisomeric Benzo[a]Pyrenyl-DNA Lesions. EMBO J. 2007, 26, 2923–2932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mu, H.; Geacintov, N.E.; Min, J.H.; Zhang, Y.; Broyde, S. Nucleotide Excision Repair Lesion-Recognition Protein Rad4 Captures a Pre-Flipped Partner Base in a Benzo[a] Pyrene-Derived DNA Lesion: How Structure Impacts the Binding Pathway. Chem. Res. Toxicol. 2017, 30, 1344–1354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akita, M.; Tak, Y.S.; Shimura, T.; Matsumoto, S.; Okuda-Shimizu, Y.; Shimizu, Y.; Nishi, R.; Saitoh, H.; Iwai, S.; Mori, T.; et al. SUMOylation of Xeroderma Pigmentosum Group C Protein Regulates DNA Damage Recognition during Nucleotide Excision Repair. Sci. Rep. 2015, 5, 10984. [Google Scholar] [CrossRef] [Green Version]
- Cuijk, L.; Vermeulen, W.; Marteijn, J.A. Ubiquitin at work: The ubiquitousregulation of the damage recognition step of NER. Exp. Cell Res. 2014, 329, 101–109. [Google Scholar] [CrossRef]
- Sugasawa, K.; Akagi, J.; Nishi, R.; Iwai, S.; Hanaoka, F. Two-Step Recognition of DNA Damage for Mammalian Nucleotide Excision Repair: Directional Binding of the XPC Complex and DNA Strand Scanning. Mol. Cell 2009, 36, 642–653. [Google Scholar] [CrossRef]
- Singh, A. TFIIH Subunit Alterations Causing Xeroderma Pigmentosumand Trichothiodystrophy Specifically Disturb Several Steps Duringtranscription. Am. J. Hum. Genet. 2015, 96, 194–207. [Google Scholar] [CrossRef] [Green Version]
- Tsodikov, O.V.; Ivanov, D.; Orelli, B.; Staresincic, L.; Shoshani, I.; Oberman, R.; Scharer, O.D.; Wagner, G.; Ellenberger, T. Structural Basis for the Recruitment of ERCC1-XPF to Nucleotide Excision Repair Complexes by XPA. EMBO J. 2007, 26, 4768–4776. [Google Scholar] [CrossRef] [Green Version]
- Sijbers, A.M.; Laat, W.L.; Ariza, R.R.; Biggerstaff, M.; Wei, Y.F.; Moggs, J.G.; Carter, K.C.; Shell, B.K.; Evans, E.; Jong, M.C.; et al. Xeroderma Pigmentosum Group F Caused by a Defect in a Structure- Specific DNA Repair Endonuclease. Cell 1996, 86, 811–822. [Google Scholar] [CrossRef] [Green Version]
- Araujo, S.J.; Nigg, E.A.; Wood, R.D. Strong Functional Interactions of TFIIH with XPC and XPG in Human DNA Nucleotide Excision Repair, without a Preassembled Repairosome. Mol. Cell. Biol. 2001, 21, 2281–2291. [Google Scholar] [CrossRef] [Green Version]
- Ito, S.; Kuraoka, I.; Chymkowitch, P.; Compe, E.; Takedachi, A.; Ishigami, C.; Coin, F.; Egly, J.M.; Tanaka, K. XPG stabilizes TFIIH Allowing Transactivation of Nuclear Receptors: Implications for Cockayne Syndrome in XP-G/CS Patients. Mol. Cell 2007, 26, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Fagbemi, A.F.; Orelli, B.; Schärer, O.D. Regulation of Endonuclease Activity in Human Nucleotide Excision Repair. DNA Repair 2011, 10, 722–729. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Smerdon, M.J. Rpb4 and Rpb9 Mediate Subpathways of Transcription-Coupled DNA Repair in Saccharomyces Cerevisiae. EMBO J. 2002, 21, 5921–5929. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.-K.; Yu, S.-L.; Prakash, L.; Prakash, S. Yeast RAD26, a Homolog of the Human CSB Gene, Functions Independently of Nucleotide Excision Repair and Base Excision Repair in Promoting Transcription through Damaged Bases. Mol. Cell. Biol. 2002, 22, 4383–4389. [Google Scholar] [CrossRef] [Green Version]
- Yasuhira, S.; Yasui, A.; Morimyo, M. Transcription Dependence and the Roles of Two Excision Repair Pathways for UV Damage in Fission Yeast Schizosaccharomyces Pombe. J. Biol. Chem. 1999, 274, 26822–26827. [Google Scholar] [CrossRef] [Green Version]
- Guzder, S.N.; Sung, P.; Prakash, L.; Prakash, S. The DNA-Dependent ATPase Activity of Yeast Nucleotide Excision Repair Factor 4 and Its Role in DNA Damage Recognition. J. Biol. Chem. 1998, 273, 6292–6296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, S.; Smirnova, J.B.; Friedberg, E.C.; Stillman, B.; Akiyama, M. ABF1-binding sites promote efficient global genome nucleotide excision repair. J. Biol. Chem. 2009, 284, 966–973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Laat, W.L.; Jaspers, N.G.; Hoeijmakers, J.H. Molecular mechanism of nucleotide excision repair. Genes Dev. 1999, 13, 768–785. [Google Scholar] [CrossRef] [Green Version]
- Sugasawa, K.; Okamoto, T.; Shimizu, Y.; Masutani, C.; Iwai, S.; Hanaoka, F. A multistep damage recognition mechanism for global genomic nucleotide excision repair. Genes Dev. 2001, 15, 507–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukumoto, Y.; Hiyama, H.; Yokoi, M.; Nakaseko, Y.; Yanagida, M.; Hanaoka, F. Two Budding Yeast RAD4 Homologs in Fission Yeast Play Different Roles in the Repair of UV-Induced DNA Damage. DNA Repair 2002, 1, 833–845. [Google Scholar] [CrossRef]
- Marti, T.M.; Kunz, C.; Fleck, O. Repair of Damaged and Mismatched DNA by the XPC Homologues Rhp41 and Rhp42 of Fission Yeast. Genetics 2003, 164, 457–467. [Google Scholar] [CrossRef] [PubMed]
- Verhage, R.; Zeeman, A.M.; Groot, N.D.; Gleig, F.; Bang, D.D. The RAD7 and RAD16 Genes, Which Are Essential for Pyrimidine Dimer Removal from the Silent Mating Type Loci, Are Also Required for Repair of the Nontranscribed Strand of an Active Gene in Saccharomyces Cerevisiae. Mol. Cell. Biol. 1994, 14, 6135–6142. [Google Scholar] [PubMed]
- Bardwell, A.J.; Bardwell, L.; Tomkinson, A.E.; Friedberg, E.C. Specific Cleavage of Model Recombination and Repair Intermediates by the Yeast Rad1-Rad10 DNA Endonuclease. Science 1994, 265, 2082–2085. [Google Scholar] [CrossRef]
- Carr, A.M.; Schmidt, H.; Kirchoff, S.; Muriel, W.J.; Sheldrick, K.S.; Griffiths, D.J.; Basmacioglu, C.N.; Subramani, S.; Clegg, M.; Nasim, A.; et al. The Rad16 Gene of Schizosaccharomyces Pombe: A Homolog of the RAD1 Gene of Saccharomyces CereÍisiae. Mol. Cell. Biol. 1994, 14, 2029–2040. [Google Scholar]
- McCready, S.J.; Burkhill, H.; Evans, S. The Saccharomyces CereÍisiae RAD2 Gene Complements a Schizosaccharomyces Pombe Repair Mutation. Curr. Genet. 1989, 15, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Beard, W.A.; Horton, J.K.; Prasad, R.; Wilson, S.H. Eukaryotic Base Excision Repair: New Approaches Shine Light on Mechanism. Annu. Rev. Biochem. 2019, 88, 137–162. [Google Scholar] [CrossRef]
- Chastain, P.D.; Nakamura, J.; Rao, S.; Chu, H.; Ibrahim, J.G. Abasic Sites Preferentially Form at Regions Undergoing DNA Replication. FASEB J. 2010, 24, 3674–3680. [Google Scholar]
- Nakamura, J.; Swenberg, J.A. Endogenous Apurinic/Apyrimidinic Sites in Genomic DNA of Mammalian Tissues. Cancer Res. 1999, 59, 2522–2526. [Google Scholar]
- Hegde, M.L.; Hazra, T.K.; Mitra, S. Functions of Disordered Regions in Mammalian Early Base Excision Repair Proteins. Cell. Mol. Life Sci. 2010, 67, 3573–3587. [Google Scholar] [CrossRef] [Green Version]
- Svilar, D.; Goellner, E.M.; Almeida, K.H.; Sobol, R.W. Base Excision Repair and Lesion-Dependent Subpathways for Repair of Oxidative DNA Damage. Antioxid. Redox Signal. 2011, 14, 2491–2507. [Google Scholar] [CrossRef] [Green Version]
- Jacobs, A.L.; Schar, P. DNA glycosylases: In DNA repair and beyond. Chromosoma 2012, 121, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Huffman, J.L.; Sundheim, O.; Tainer, J.A. DNA Base Damage Recognition and Removal: New Twists and Grooves. Mutat. Res. 2005, 577, 55–76. [Google Scholar] [CrossRef] [PubMed]
- Sassa, A.; Beard, W.A.; Prasad, R.; Wilson, S.H. DNA sequence context effects on the glycosylase activity of human 8-oxoguanine DNA glycosylase. J. Biol. Chem. 2012, 287, 36702–36710. [Google Scholar] [CrossRef] [Green Version]
- Wong, I.; Lundquist, A.J.; Bernards, A.S.; Mosbaugh, D.W. Presteady-state analysis of a single catalytic turnover by Escherichia coli uracil-DNA glycosylase reveals a pinch-pull-push mechanism. J. Biol. Chem. 2002, 277, 19424–19432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robertson, A.B.; Klungland, A.; Rognes, T.; Leiros, I. DNA Repair in Mammalian Cells: Base Excision Repair: The Long and Short of It. Cell. Mol. Life Sci. 2009, 66, 981–993. [Google Scholar] [CrossRef] [PubMed]
- Beard, W.A.; Wilson, S.H. Structure and mechanism of DNA polymerase b. Chem. Rev. 2006, 106, 361–382. [Google Scholar] [CrossRef] [PubMed]
- Prasad, R.; Batra, V.K.; Yang, X.-P.; Krahn, J.M.; Pedersen, L.C. Structural Insight into the DNA Polymerase Βdeoxyribose Phosphate Lyase Mechanism. DNA Repair 2005, 4, 1347–1357. [Google Scholar] [CrossRef]
- Fortini, P.; Parlanti, E.; Sidorkina, O.M.; Laval, J.; Dogliotti, E. The Type of DNA Glycosylase Determines the Base Excision Repair Pathway in Mammalian Cells. J. Biol. Chem. 1999, 274, 15230–15236. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Beard, W.A.; Shock, D.D.; Prasad, R.; Hou, E.W.; Wilson, S.H. DNA Polymerase β and Flap Endonuclease 1 Enzymatic Specificities Sustain DNA Synthesis for Long Patch Base Excision Repair. J. Biol. Chem. 2005, 280, 3665–3674. [Google Scholar] [CrossRef] [Green Version]
- Elder, R.T.; Zhu, X.; Priet, S. A Fission Yeast Homologue of the Human Uracil-DNA-Glycosylase and Their Roles in Causing DNA Damage after Overexpression. Biochem. Biophys. Res. Commun. 2003, 306, 693–700. [Google Scholar] [CrossRef]
- Hardeland, U.; Bentele, M.; Jiricny, J.; Schär, P. The Versatile Thymine DNA-glycosylase: A Comparative Characterization of the Human, Drosophila and Fission Yeast Orthologs. Nucleic Acids Res. 2003, 31, 2261–2271. [Google Scholar] [CrossRef]
- Dong, L.; Mi, R.; Glass, R.A.; Barry, J.N.; Cao, W. Repair of Deaminated Base Damage by Schizosaccharomyces Pombe Thymine DNA Glycosylase. DNA Repair 2008, 7, 1962–1972. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Derfler, B.; Samson, L. Saccharomyces Cerevisiae 3-Methyladenine DNA Glycosylase Has Homology to the AlkA Glycosylase of E. Coli and Is Induced in Response to DNA Alkylation Damage. EMBO J. 1990, 9, 4569–4575. [Google Scholar] [CrossRef]
- Berdal, K.G.; Johansen, R.F.; Seeberg, E. Release of Normal Bases from Intact DNA by a Native DNA Repair Enzyme. EMBO J. 1998, 17, 363–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanamitsu, K.; Tanihigashi, H.; Tanita, Y.; Inatani, S.; Ikeda, S. Involvement of 3-Methyladenine DNA Glycosylases Mag1p and Mag2p in Base Excision Repair of Methyl Methanesulfonate-Damaged DNA in the Fission Yeast Schizosaccharomyces Pombe. Genes Genet. Syst. 2007, 82, 489–494. [Google Scholar] [CrossRef] [Green Version]
- Memisoglu, A.; Samson, L. Contribution of Base Excision Repair, Nucleotide Excision Repair, and DNA Recombination to Alkylation Resistance of the Fission Yeast Schizosaccharomyces Pombe. J. Bacteriol. 2000, 182, 2104–2112. [Google Scholar] [CrossRef] [Green Version]
- Alseth, I.; Osman, F.; Korvald, H. Biochemical Characterization and DNA Repair Pathway Interactions of Mag1-Mediated Base Excision Repair in Schizosaccharomyces Pombe. Nucleic Acids Res. 2005, 33, 1123–1131. [Google Scholar] [CrossRef] [Green Version]
- Alseth, I.; Korvald, H.; Osman, F.; Seeberg, E.; Bjørås, M. A General Role of the DNA Glycosylase Nth1 in the Abasic Sites Cleavage Step of Base Excision Repair in Schizosaccharomyces Pombe. Nucleic Acids Res. 2004, 32, 5119–5125. [Google Scholar] [CrossRef] [Green Version]
- Ribar, B.; Izumi, T.; Mitra, S. The Major Role of Human AP-Endonuclease Homolog Apn2 in Repair of Abasic Sites in Schizosaccharomyces Pombe. Nucleic Acids Res. 2004, 32, 115–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugimoto, T.; Igawa, E.; Tanihigashi, H.; Matsubara, M.; Ide, H.; Ikeda, S. Roles of Base Excision Repair Enzymes Nth1p and Apn2p from Schizosaccharomyces Pombe in Processing Alkylation and Oxidative DNA Damage. DNA Repair 2005, 4, 1270–1280. [Google Scholar] [CrossRef] [PubMed]
- Alseth, I.; Eide, L.; Pirovano, M.; Rognes, T.; Seeberg, E. The Saccharomyces Cerevisiae Homologues of Endonuclease III from Escherichia Coli, Ntg1 and Ntg2, Are Both Required for Efficient Repair of Spontaneous and Induced Oxidative DNA Damage in Yeast. Mol. Cell. Biol. 1999, 19, 3779–3787. [Google Scholar] [CrossRef] [Green Version]
- Senturker, S.; Kemp, P.A.V.D.; You, H.J.; Doetsch, P.W.; Dizdaroglu, M. Substrate Specificities of the Ntg1 and Ntg2 Proteins of Saccharomyces Cerevisiae for Oxidized DNA Bases Are Not Identical. Nucleic Acids Res. 1998, 26, 5270–5276. [Google Scholar] [CrossRef] [Green Version]
- Girard, P.M.; Guibourt, N.; Boiteux, S. The Ogg1 Protein of Saccharomyces Cerevisiae: A 7,8-Dihydro-8-Oxoguanine DNA Glycosylase/AP Lyase Whose Lysine 241 Is a Critical Residue for Catalytic Activity. Nucleic Acids Res. 1997, 25, 3204–3211. [Google Scholar] [CrossRef] [Green Version]
- Fleck, O. DNA repair pathway. In The Molecular Biology of Schizosaccharomyces Pombe; Egel, R., Ed.; Springer: Berlin/Heidelberg, Germany, 2004; pp. 101–115. [Google Scholar]
- Karahalil, B.; Roldán-Arjona, T.; Dizdaroglu, M. Substrate Specificity of Schizosaccharomyces Pombe Nth Protein for Products of Oxidative DNA Damage. Biochemistry 1998, 37, 590–595. [Google Scholar] [CrossRef]
- Lu, A.-L.; Fawcett, W.P. Characterization of the Recombinant MutY Homolog, an Adenine DNA Glycosylase, from Yeast Schizosaccharomyces Pombe. J. Biol. Chem. 1998, 273, 25098–25105. [Google Scholar] [CrossRef] [Green Version]
- Doi, T.; Yonekura, S.-I.; Tano, K.; Yasuhira, S.; Yonei, S.; Zhang, Q.-M. The Shizosaccharomyces Pombe Homolog (SpMYH) of the Escherichia Coli MutY Is Required for Removal of Guanine from 8-Oxoguanine/Guanine Mispairs to Prevent G:C to C:G Transversions. J. Radiat. Res. 2005, 46, 205–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, D.-Y.; Lu, A.-L. Interaction of Checkpoint Proteins Hus1/Rad1/Rad9 with DNA Base Excision Repair Enzyme MutY Homolog in Fission Yeast, Schizosaccharomyces Pombe. J. Biol. Chem. 2005, 280, 408–417. [Google Scholar] [CrossRef] [Green Version]
- Jansson, K.; Warringer, J.; Farewell, A. The Tumor Suppressor Homolog in Fission Yeast, Myh1+, Displays a Strong Interaction with the Checkpoint Gene Rad1+. Mutat. Res. 2008, 644, 48–55. [Google Scholar] [CrossRef]
- Popoff, S.C.; Spira, A.I.; Johnson, A.W.; Demple, B. Yeast structural gene (APN1) for the major apurinic endonuclease: Homology to Escherichia coli endonuclease IV. Proc. Natl. Acad. Sci. USA 1990, 87, 4193–4197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acevedo-Torres, K.; Fonseca-Williams, S.; Ayala-Torres, S.; Torres-Ramos, C.A. Requirement of the Saccharomyces Cerevisiae APN1 Gene for the Repair of Mitochondrial DNA Alkylation Damage. Environ. Mol. Mutagen. 2009, 50, 317–327. [Google Scholar] [CrossRef] [Green Version]
- Tanihigashi, H.; Yamada, A.; Igawa, E.; Ikeda, S. The Role of Schizosaccharomyces Pombe DNA Repair Enzymes Apn1p and Uve1p in the Base Excision Repair of Apurinic/Apyrimidinic Sites. Biochem. Biophys. Res. Commun. 2006, 347, 889–894. [Google Scholar] [CrossRef] [PubMed]
- Alleva, J.L.; Zuo, S.; Hurwitz, J.; Doetsch, P.W. In Vitro Reconstitution of the Schizosaccharomyces Pombe Alternative Excision Repair Pathway. Biochemistry 2000, 39, 2659–2666. [Google Scholar] [CrossRef] [PubMed]
- Yasuhira, S.; Yasui, A. Alternative Excision Repair Pathway of UV-Damaged DNA in Schizosaccharomyces Pombe Operates Both in Nucleus and in Mitochondria. J. Biol. Chem. 2000, 275, 11824–11828. [Google Scholar] [CrossRef] [Green Version]
- Kunkel, T.A.; Erie, D.A. Eukaryotic Mismatch Repair in Relation to DNA Replication. Annu. Rev. Genet. 2015, 49, 291–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamers, M.H.; Perrakis, A.; Enzlin, J.H.; Winterwerp, H.H.K.; Wind, N.D. The Crystal Structure of DNA Mismatch Repair Protein MutS Binding to a G:T Mismatch. Nature 2000, 407, 711–717. [Google Scholar] [CrossRef]
- Obmolova, G.; Ban, C.; Hsieh, P.; Yang, W. Crystal Structures of Mismatch Repair Protein MutS and Its Complex with a Substrate DNA. Nature 2000, 407, 703–710. [Google Scholar] [CrossRef] [PubMed]
- Iyer, R.R.; Pluciennik, A.; Burdett, V.; Modrich, P.L. DNA mismatch repair: Functions and mechanisms. Chem. Rev. 2006, 106, 302–323. [Google Scholar] [CrossRef]
- Cerretelli, G.; Ager, A.; Arends, M.J.; Frayling, I.M. Molecular Pathology of Lynch Syndrome. J. Pathol. 2020, 250, 518–531. [Google Scholar] [CrossRef] [Green Version]
- Surtees, J.A.; Argueso, J.L.; Alani, E. Mismatch Repair Proteins: Key Regulators of Genetic Recombination. CGR 2004, 107, 146–159. [Google Scholar] [CrossRef]
- Kawasoe, Y.; Tsurimoto, T.; Nakagawa, T.; Masukata, H.; Takahashi, T.S. MutSalpha Maintains the Mismatch Repair Capability by Inhibiting PCNA Unloading. eLife 2016, 5, e15155. [Google Scholar] [CrossRef]
- Erdeniz, N.; Nguyen, M.; Deschenes, S.M.; Liskay, R.M. Mutations Affecting a Putative MutLa Endonuclease Motif Impact Multiple Mismatch Repair Functions. DNA Repair 2007, 6, 1463–1470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Yuan, F.; Presnell, S.R.; Tian, K.; Gao, Y.; Tomkinson, A.E.; Gu, L.; Li, G.M. Reconstitution of 5’-Directed Human Mismatch Repair in a Purified System. Cell 2005, 122, 693–705. [Google Scholar] [CrossRef] [Green Version]
- Kadyrov, F.A.; Genschel, J.; Fang, Y.; Penland, E.; Edelmann, W.; Modrich, P. A possible mechanism for exonuclease 1-independent eukaryotic mismatch repair. Proc. Natl. Acad. Sci. USA 2009, 106, 8495–8500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrington, J.M.; Kolodner, R.D. Saccharomyces Cerevisiae Msh2-Msh3 Acts in Repair of Base-Base Mispairs. Mol. Cell. Biol. 2007, 27, 6546–6554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Detloff, P.; Sieber, J.; Petes, T.D. Repair of Specific Base Pair Mismatches Formed during Meiotic Recombination in the Yeast Saccharomyces Cerevisiae. Mol. Cell. Biol. 1991, 11, 737–745. [Google Scholar]
- Strand, M.; Earley, M.C.; Crouse, G.F.; Petes, T.D. Mutations in the MSH3 Gene Preferentially Lead to Deletions within Tracts of Simple Repetitive DNA in Saccharomyces Cerevisiae. Proc. Natl. Acad. Sci. USA 1995, 92, 10418–10421. [Google Scholar] [CrossRef] [Green Version]
- Harfe, B.D.; Minesinger, B.K.; Jinks-Robertson, S. Discrete in vivo roles for the MutL homologs Mlh2p and Mlh3p in the removal of frameshift intermediates in budding yeast. Curr. Biol. 2002, 10, 145–148. [Google Scholar] [CrossRef] [Green Version]
- Tornier, C.; Bessone, S.; Varlet, I.; Rudolph, C.; Darmon, M.; Fleck, O. Requirement for Msh6, but Not for Swi4 (Msh3), in Msh2-Dependent Repair of Base-Base Mismatches and Mononucleotide Loops in Schizosaccharomyces Pombe. Genetics 2001, 158, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Saparbaev, M.; Prakash, L.; Prakash, S. Requirement of Mismatch Repair Genes MSH2 and MSH3 in the RAD1-RAD10 Pathway of Mitotic Recombination in Saccharomyces Cerevisiae. Genetics 1996, 142, 727–736. [Google Scholar] [CrossRef]
- Rudolph, C.; Kunz, C.; Parisi, S.; Lehmann, E.; Hartsuiker, E.; Fartmann, B.; Kramer, W.; Kohli, J.; Fleck, O. The Msh2 Gene OfSchizosaccharomyces Pombe Is Involved in Mismatch Repair, Mating-Type Switching, and Meiotic Chromosome Organization. Mol. Cell. Biol. 1999, 19, 241–250. [Google Scholar] [CrossRef] [Green Version]
- Ross-MacDonald, P.; Roeder, G.S. Mutation of a Meiosis-Specific MutS Homolog Decreases Crossing over but Not Mismatch Correction. Cell 1994, 79, 1069–1080. [Google Scholar] [CrossRef]
- Hollingsworth, N.M.; Ponte, L.; Halsey, C. MSH5, a Novel MutS Homolog, Facilitates Meiotic Reciprocal Recombination between Homologs in Saccharomyces Cerevisiae but Not Mismatch Repair. Genes Dev. 1995, 9, 1728–1739. [Google Scholar] [CrossRef] [Green Version]
- Zakharyevich, K.; Tang, S.; Ma, Y.; Hunter, N. Delineation of Joint Molecule Resolution Pathways in Meiosis Identifies a Crossover-Specific Resolvase. Cell 2012, 149, 334–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.F.; Kleckner, N.; Hunter, N. Functional Specificity of MutL Homologs in Yeast: Evidence for Three Mlh1- Based Heterocomplexes with Distinct Roles during Meiosis in Recombination and Mismatch Correction. Proc. Natl. Acad. Sci. USA 1999, 96, 13914–13919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, P.T.; Erdeniz, N.; Symington, L.S.; Liskay, R.M. EXO1—A Multi-Tasking Eukaryotic Nuclease. DNA Repair 2004, 3, 1549–1559. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.E.; Kovvali, G.K.; Prakash, L.; Prakash, S. Requirement of the Yeast RTH1 59 to 39 Exonuclease for the Stability of Simple Repetitive DNA. Science 1995, 269, 238–240. [Google Scholar] [CrossRef]
- Mansour, A.A.; Tornier, C.; Lehmann, E.; Darmon, M.; Fleck, O. Control of GT Repeat Stability in Schizosaccharomyces Pombe by Mismatch Repair Factors. Genetics 2001, 158, 77–85. [Google Scholar] [CrossRef]
- Schär, P.; Kohli, J. Marker Effects of G to C Transversions on Intragenic Recombination and Mismatch Repair in Schizosaccharomyces Pombe. Genetics 1993, 133, 825–835. [Google Scholar] [CrossRef]
- Fleck, O.; Lehmann, E.; Schär, P.; Kohli, J. Involvement of Nucleotide-Excision Repair in Msh2 Pms1-Independent Mismatch Repair. Nat. Genet. 1999, 21, 314–317. [Google Scholar] [CrossRef]
- Hohl, M.; Christensen, O.; Kunz, C.; Naegeli, H.; Fleck, O. Binding and Repair of Mismatched DNA Mediated by Rhp14, the Fission Yeast Homologue of Human XPA. J. Biol. Chem. 2001, 276, 30766–30772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunz, C.; Fleck, O. Role of the DNA Repair Nucleases Rad13, Rad2 and Uve1 of Schizosaccharomyces Pombe in Mismatch Correction11Edited by S. Reed. J. Mol. Biol. 2001, 313, 241–253. [Google Scholar] [CrossRef]
- Coïc, E.; Gluck, L.; Fabre, F. Evidence for Short-Patch Mismatch Repair in Saccharomyces Cerevisiae. EMBO J. 2000, 19, 3408–3417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harfe, B.D.; Jinks-Robertson, S. Dna Mismatch Repair and Genetic Instability. Annu. Rev. Genet. 2000, 34, 359–399. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, A.T. Mismatch Repair, Gene Conversion, and Crossing-over in Two Recombination-Defective Mutants of Drosophila Melanogaster. Proc. Natl. Acad. Sci. USA 1982, 79, 5961–5965. [Google Scholar] [CrossRef] [Green Version]
- Bhui-Kaur, A.; Goodman, M.F.; Tower, J. DNA Mismatch Repair Catalyzed by Extracts of Mitotic, Postmitotic, and Senescent Drosophila Tissues and Involvement of Mei-9 Gene Function for Full Activity. Mol. Cell. Biol. 1998, 18, 1436–1443. [Google Scholar] [CrossRef] [Green Version]
- Petit, C.; Sancar, A. Nucleotide Excision Repair: From E. Coli to Man. Biochimie 1999, 81, 15–25. [Google Scholar] [CrossRef]
- Vilenchik, M.M.; Knudson, A.G. Endogenous DNA Double-Strand Breaks: Production, Fidelity of Repair, and Induction of Cancer. Proc. Natl. Acad. Sci. USA 2003, 100, 12871–12876. [Google Scholar] [CrossRef] [Green Version]
- Cannan, W.J.; Pederson, D.S. Mechanisms and Consequences of Double-Strand DNA Break Formation in Chromatin. J. Cell. Physiol. 2016, 231, 3–14. [Google Scholar] [CrossRef] [Green Version]
- Her, J.; Bunting, S.F. How Cells Ensure Correct Repair of DNA Double-Strand Breaks. J. Biol. Chem. 2018, 293, 10502–10511. [Google Scholar] [CrossRef] [Green Version]
- Karanam, K.; Kafri, R.; Loewer, A.; Lahav, G. Quantitative Live Cell Imaging Reveals a Gradual Shift between DNA Repair Mechanisms and a Maximal Use of HR in Mid S Phase. Mol. Cell. 2012, 47, 320–329. [Google Scholar] [CrossRef] [Green Version]
- Mao, Z.; Bozzella, M.; Seluanov, A.; Gorbunova, V. Comparison of Nonhomologous End Joining and Homologous Recombination in Human Cells. DNA Repair 2008, 7, 1765–1771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pierce, A.J.; Hu, P.; Han, M.; Ellis, N.; Jasin, M. Protein Modulates Homologous Repair of Double-Strand Breaks in Mammalian Cells. Genes Dev. 2001, 15, 3237–3242. [Google Scholar] [CrossRef] [Green Version]
- Ahrabi, S.; Sarkar, S.; Pfister, S.X.; Pirovano, G.; Higgins, G.S.; Porter, A.C.; Humphrey, T.C. A Role for Human Homologous Recombination Factors in Suppressing Microhomology-Mediated End Joining. Nucleic Acids Res. 2016, 44, 5743–5757. [Google Scholar] [CrossRef] [Green Version]
- Bétermier, M.; Bertrand, P.; Lopez, B.S. Is Non-Homologous End-Joining Really an Inherently Error-Prone Process? PLoS Genet. 2014, 10, 1004086. [Google Scholar] [CrossRef] [Green Version]
- Rass, E.; Grabarz, A.; Plo, I.; Gautier, J.; Bertrand, P.; Lopez, B.S. Role of Mre11 in Chromosomal Nonhomologous End Joining in Mammalian Cells. Nat. Struct. Mol. Biol. 2009, 16, 819–824. [Google Scholar] [CrossRef]
- So, A.; Le Guen, T.; Lopez, B.S.; Guirouilh-Barbat, J. Genomic rearrangements induced by unscheduled DNA double strand breaks in somatic mammalian cells. FEBS J. 2017, 284, 2324–2344. [Google Scholar] [CrossRef] [Green Version]
- Escribano-Diaz, C. A Cell Cycle-Dependent Regulatory Circuit Composed of 53BP1 RIF1 and BRCA1 CtIP Controls DNA Repair Pathway Choice. Mol. Cell 2013, 49, 872–883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yun, M.H.; Hiom, K. CtIP-BRCA1 Modulates the Choice of DNA Double-Strand-Break Repair Pathway throughout the Cell Cycle. Nature 2009, 459, 460–463. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.D.; Jasin, M. Sister Chromatid Gene Conversion Is a Prominent Double-Strand Break Repair Pathway in Mammalian Cells. EMBO J. 2000, 19, 3398–3407. [Google Scholar] [CrossRef]
- Howard, S.M.; Yanez, D.A.; Stark, J.M. DNA Damage Response Factors from Diverse Pathways, Including DNA Crosslink Repair, Mediate Alternative End Joining. PLoS Genet. 2015, 11, 1004943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhargava, R.; Onyango, D.O.; Stark, J.M. Regulation of Single Strand Annealing and Its Role in Genome Maintenance. Trends Genet. 2016, 32, 566–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krejci, L.; Altmannova, V.; Spirek, M.; Zhao, X. Homologous Recombination and Its Regulation. Nucleic Acids Res. 2012, 40, 5795–5818. [Google Scholar] [CrossRef]
- Tkach, J.M.; Yimit, A.; Lee, A.Y.; Riffle, M.; Costanzo, M.; Jaschob, D.; Hendry, J.A.; Ou, J.; Moffat, J.; Boone, C.; et al. Dissecting DNA Damage Response Pathways by Analysing Protein Localization and Abundance Changes during DNA Replication Stress. Nat. Cell Biol. 2012, 14, 966–976. [Google Scholar] [CrossRef] [Green Version]
- Aylon, Y.; Kupiec, M. DSB Repair: The Yeast Paradigm. DNA Repair 2004, 3, 797–815. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Topper, L.M.; Wilson, T.E. Recruitment and dissociation of nonhomologous end joining proteins at a DNA double-strand break in Saccharomyces cerevisiae. Genetics 2008, 178, 1237–1249. [Google Scholar] [CrossRef] [Green Version]
- Lobachev, K.; Vitriol, E.; Stemple, J.; Resnick, M.A.; Bloom, K. Chromosome Fragmentation after Induction of a Double-Strand Break Is an Active Process Prevented by the RMX Repair Complex. Curr. Biol. 2004, 14, 2107–2112. [Google Scholar] [CrossRef] [Green Version]
- Mimitou, E.P.; Symington, L.S. Ku Prevents Exo1 and Sgs1 Dependent Resection of DNA Ends in the Absence of a Functional MRX Complex or Sae2. EMBO J. 2010, 29, 3358–3369. [Google Scholar] [CrossRef] [Green Version]
- Clerici, M.; Mantiero, D.; Guerini, I.; Lucchini, G.; Longhese, M.P. The Yku70-Yku80 Complex Contributes to Regulate Double-Strand Break Processing and Checkpoint Activation during the Cell Cycle. EMBO Rep. 2008, 9, 810–818. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Shim, E.Y.; Davis, M.; Lee, S.E. Regulation of Repair Choice: Cdk1 Suppresses Recruitment of End Joining Factors at DNA Breaks. DNA Repair 2009, 8, 1235–1241. [Google Scholar] [CrossRef] [Green Version]
- Manolis, K.G.; Nimmo, E.R.; Hartsuiker, E. Novel Functional Requirements for Non-Homologous DNA Endjoining in Schizosaccharomyces Pombe. EMBO J. 2001, 20, 210–221. [Google Scholar] [CrossRef] [Green Version]
- Muris, D.F.; Vreeken, K.; Schmidt, H. Homologous Recombination in the Fission Yeast Schizosaccharomyces Pombe: Different Requirements for the Rhp51+, Rhp54+ and Rad22+ Genes. Curr. Genet. 1997, 31, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Prudden, J.; Evans, J.S.; Hussey, S.P. Pathway Utilization in Response to a Site-Specific DNA Double-Strand Break in Fission Yeast. EMBO J. 2003, 22, 1419–1430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchetti, M.A.; Kumar, S.; Hartsuiker, E. A Single Unbranched S-Phase DNA Damage and Replication Fork Blockage Checkpoint Pathway. Proc. Natl. Acad. Sci. USA 2002, 99, 7472–7477. [Google Scholar] [CrossRef] [Green Version]
- Chahwan, C.; Nakamura, T.M.; Sivakumar, S.; Russell, P.; Rhind, N. The Fission Yeast Rad32(Mre11)–Rad50–Nbs1 Complex Is Required for the S-Phase DNA Damage Checkpoint. Mol. Cell Biol. 2003, 23, 6564–6573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thacker, J. The RAD51 Gene Family, Genetic Instability and Cancer. Cancer Lett. 2005, 219, 125–135. [Google Scholar] [CrossRef]
- Schmidt, H.; Kapitza-Fecke, P.; Stephen, E.R.; Gutz, H. Some of the Swi Genes of Schizosaccharomyces Pombe Also Have a Function in the Repair of Radiation Damage. Curr. Genet. 1989, 16, 89–94. [Google Scholar] [CrossRef]
- Suto, K.; Nagata, A.; Murakami, H.; Okayama, H. A double-strand break repair component is essential for S phase completion in fission yeast cell cycling. Mol. Biol. Cell 1999, 10, 3331–3343. [Google Scholar] [CrossRef] [Green Version]
- Van den Bosch, M.; Vreeken, K.; Zonneveld, J.B.; Brandsma, J.A.; Lombaerts, M.; Murray, J.M.; Lohman, P.H.; Pastink, A. Characterization of RAD52 Homologs in the Fission Yeast Schizosaccharomyces Pombe. Mutat. Res. 2001, 461, 311–323. [Google Scholar] [CrossRef]
- Nimonkar, A.V.; Genschel, J.; Kinoshita, E.; Polaczek, P.; Campbell, J.L.; Wyman, C.; Modrich, P.; Kowalczykowski, S.C. BLM-DNA2-RPA-MRN and EXO1-BLM-RPA-MRN Constitute Two DNA End Resection Machineries for Human DNA Break Repair. Genes Dev. 2011, 25, 350–362. [Google Scholar] [CrossRef] [Green Version]
- Cox, M.M. Motoring along with the Bacterial RecA Protein. Nat. Rev. Mol. Cell. Biol. 2007, 8, 127–138. [Google Scholar] [CrossRef]
- San Filippo, J.; Sung, P.; Klein, H. Mechanism of Eukaryotic Homologous Recombination. Annu. Rev. Biochem. 2008, 77, 229–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, H.L.; Symington, L.S. Sgs1—The Maestro of Recombination. Cell 2012, 149, 257–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mimitou, E.P.; Symington, L.S. Sae2, Exo1 and Sgs1 Collaborate in DNA Double-Strand Break Processing. Nature 2008, 455, 770–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomita, K.; Matsuura, A.; Caspari, T.; Carr, A.M.; Akamatsu, Y.; Iwasaki, H.; Mizuno, K.; Ohta, K.; Uritani, M.; Ushimaru, T.; et al. Competition between the Rad50 Complex and the Ku Heterodimer Reveals a Role for Exo1 in Processing Double-Strand Breaks but Not Telomeres. Mol. Cell. Biol. 2003, 23, 5186–5197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferretti, L.P.; Lafranchi, L.; Sartori, A.A. Controlling DNA-End Resection: A New Task for CDKs. Front. Genet. 2013, 4, 99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugiyama, T.; Zaitseva, E.M.; Kowalczykowski, S.C. A Single-Stranded DNA-Binding Protein Is Needed for Efficient Presynaptic Complex Formation by the Saccharomyces Cerevisiae Rad51 Protein. J. Biol. Chem. 1997, 272, 7940–7945. [Google Scholar] [CrossRef] [Green Version]
- Parker, A.E.; Clyne, R.K.; Carr, A.M.; Kelly, T.J. The Schizosaccharomyces Pombe Rad11+ Gene Encodes the Large Subunit of Replication Protein A. Mol. Cell. Biol. 1997, 17, 2381–2390. [Google Scholar] [CrossRef] [Green Version]
- Krejci, L.; Song, B.; Bussen, W.; Rothstein, R.; Mortensen, U.H.; Sung, P. Interaction with Rad51 Is Indispensable for Recombination Mediator Function of Rad52. J. Biol. Chem. 2002, 277, 40132–40141. [Google Scholar] [CrossRef] [Green Version]
- Hays, S.L.; Firmenich, A.A.; Berg, P. Complex formation in yeast double-strand break repair: Participation of Rad51, Rad52, Rad55, and Rad57 proteins. Proc. Natl. Acad. Sci. USA 1995, 92, 6925–6929. [Google Scholar] [CrossRef] [Green Version]
- Muris, D.F.; Vreeken, K.; Carr, A.M. Isolation of the Schizosaccharomyces Pombe RAD54 Homologue, Rhp54+, a Gene Involved in the Repair of Radiation Damage and Replication Fidelity. J. Cell Sci. 1996, 109, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Sauvageau, S.; Stasiak, A.Z.; Banville, I. Fission Yeast Rad51 and Dmc1, Two Efficient DNA Recombinases Forming Helical Nucleoprotein Filaments. Mol. Cell Biol. 2005, 25, 4377–4387. [Google Scholar] [CrossRef] [Green Version]
- Aylon, Y.; Liefshitz, B.; Bitan-Banin, G.; Kupiec, M. Molecular Dissection of Mitotic Recombination in the Yeast Saccharomyces Cerevisiae. Mol. Cell Biol. 2003, 23, 1403–1417. [Google Scholar] [CrossRef] [Green Version]
- Bzymek, M.; Thayer, N.H.; Oh, S.D.; Kleckner, N.; Hunter, N. Double Holliday Junctions Are Intermediates of DNA Break Repair. Nature 2010, 464, 937–941. [Google Scholar] [CrossRef] [Green Version]
- Grishchuk, A.L.; Kohli, J. Five RecA-like Proteins of Schizosaccharomyces Pombe Are Involved in Meiotic Recombination. Genetics 2003, 165, 1031–1043. [Google Scholar] [CrossRef]
- Segurado, M.; Gómez, M.; Antequera, F. Increased Recombination Intermediates and Homologous Integration Hot Spots at DNA Replication Origins. Mol. Cell 2002, 10, 907–916. [Google Scholar] [CrossRef]
- Lambert, S.; Watson, A.; Sheedy, D.M.; Martin, B.; Carr, A.M. Gross Chromosomal Rearrangements and Elevated Recombination at an Inducible Site-Specific Replication Fork Barrier. Cell 2005, 121, 689–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nemoz, C.; Ropars, V.; Frit, P.; Gontier, A.; Drevet, P.; Yu, J.; Guerois, R.; Pitois, A.; Comte, A.; Deltiel, C.; et al. XLF and APLF Bind Ku80 at Two Remote Sites to Ensure DNA Repair by Non-Homologous End Joining. Nat. Struct. Mol. Biol. 2018, 25, 971–980. [Google Scholar] [CrossRef] [PubMed]
- Meek, K.; Dang, V.; Lees-Miller, S.P. DNA-PK: The Means to Justify the Ends? Adv. Immunol. 2008, 99, 33–58. [Google Scholar]
- Sibanda, B.L.; Chirgadze, D.Y.; Ascher, D.B.; Blundell, T.L. DNA-PKcs Structure Suggests an Allosteric Mechanism Modulating DNA Double-Strand Break Repair. Science 2017, 355, 520–524. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Pannicke, U.; Schwarz, K.; Lieber, M.R. Hairpin Opening and Overhang Processing by an Artemis/DNA-Dependent Protein Kinase Complex in Nonhomologous End Joining and V(D)J Recombination. Cell 2002, 108, 781–794. [Google Scholar] [CrossRef] [Green Version]
- Hammel, M.; Rey, M.; Yu, Y.; Mani, R.S.; Classen, S.; Liu, M.; Pique, M.E.; Fang, S.; Mahaney, B.L.; Weinfeld, M.; et al. XRCC4 Protein Interactions with XRCC4-like Factor (XLF) Create an Extended Grooved Scaffold for DNA Ligation and Double Strand Break Repair. J. Biol. Chem. 2011, 286, 32638–32650. [Google Scholar] [CrossRef] [Green Version]
- Ochi, T.; Blackford, A.N.; Coates, J.; Jhujh, S.; Mehmood, S.; Tamura, N.; Travers, J.; Wu, Q.; Draviam, V.M.; Robinson, C.V. DNA Repair. PAXX, a Paralog of XRCC4 and XLF, Interacts with Ku to Promote DNA Double-Strand Break Repair. Science 2015, 347, 185–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frank-Vaillant, M.; Marcand, S. NHEJ Regulation by Mating Type Is Exercised through a Novel Protein, Lif2p, Essential to the Ligase IV Pathway. Genes Dev. 2001, 15, 3005–3012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kegel, A.; Sjöstrand, J.O.O.; Åström, S.U. Nej1p, a Cell Type-Specific Regulator of Nonhomologous End Joining in Yeast. Curr. Biol. 2001, 11, 1611–1617. [Google Scholar] [CrossRef] [Green Version]
- Valencia, M.; Bentele, M.; Vaze, M.B.; Herrmann, G.; Kraus, E.; Lee, S.E.; Schär, P.; Haber, J.E. NEJ1 Controls Non-Homologous End Joining in Saccharomyces Cerevisiae. Nature 2001, 414, 666–669. [Google Scholar] [CrossRef] [PubMed]
- Dudášová, Z.; Dudáš, A.; Chovanec, M. Non-Homologous End-Joining Factors of Saccharomyces Cerevisiae. FEMS Microbiol. Rev. 2004, 28, 581–601. [Google Scholar] [CrossRef] [Green Version]
- Daley, J.M.; Niu, H.; Miller, A.S.; Sung, P. Biochemical Mechanism of DSB End Resection and Its Regulation. DNA Repair 2015, 32, 66–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Callebaut, I.; Malivert, L.; Fischer, A. Cernunnos Interacts with the XRCC4–DNA-Ligase IV Complex and Is Homologous to the Yeast Nonhomologous End-Joining Factor Nej1. J. Biol. Chem. 2006, 281, 13857–13860. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Wang, J.; Zhou, G.; Lajeunesse, M.; Le, N.; Stawicki, B.N.; Corcino, Y.L.; Berkner, K.L.; Runge, K.W. Nonhomologous End-Joining with Minimal Sequence Loss Is Promoted by the Mre11-Rad50-Nbs1-Ctp1 Complex in Schizosaccharomyces Pombe. Genetics 2017, 206, 481–496. [Google Scholar] [CrossRef] [Green Version]
- Runge, K.W.; Li, Y. A Curious New Role for MRN in Schizosaccharomyces Pombe Non-Homologous End-Joining. Curr. Genet. 2018, 64, 359–364. [Google Scholar] [CrossRef]
- Paques, F.; Haber, J.E. Multiple Pathways of Recombination Induced by Double-Strand Breaks in Saccharomyces Cerevisiae. Microbiol. Mol. Biol. Rev. 1999, 63, 349–404. [Google Scholar] [CrossRef] [Green Version]
- Zhao, F.; Kim, W.; Kloeber, J.A.; Lou, Z. DNA End Resection and Its Role in DNA Replication and DSB Repair Choice in Mammalian Cells. Exp. Mol. Med. 2020, 52, 1705–1714. [Google Scholar] [CrossRef]
- Ma, C.J.; Gibb, B.; Kwon, Y.; Sung, P.; Greene, E.C. Protein Dynamics of Human RPA and RAD51 on SsDNA during Assembly and Disassembly of the RAD51 Filament. Nucleic Acids Res. 2017, 45, 749–761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motycka, T.A.; Bessho, T.; Post, S.M.; Sung, P.; Tomkinson, A.E. Physical and Functional Interaction between the XPF/ERCC1 Endonuclease and HRad52. J. Biol. Chem. 2004, 279, 13634–13639. [Google Scholar] [CrossRef] [Green Version]
- Osman, F.; Fortunato, E.A.; Subramani, S. Double-Strand Break-Induced Mitotic Intrachromosomal Recombination in the Fission Yeast Schizosaccharomyces Pombe. Genetics 1996, 142, 341–357. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, A.; Ogawa, H.; Matsuda, Y.; Ushio, N.; Ikeo, K.; Ogawa, T. Cloning of Human, Mouse and Fission Yeast Recombination Genes Homologous to RAD51 and RecA. Nat. Genet. 1993, 4, 239–243. [Google Scholar] [CrossRef]
- Moore, C.W. Responses of Radiation-Sensitive Mutants of Saccharomyces Cerevisiae to Lethal Effects of Bleomycin. Mutat. Res. 1978, 51, 165–180. [Google Scholar] [CrossRef]
- Fabre, F.; Moustacchi, E. Removal of Pyrimidine Dimers in Cells of Schizosaccharomyces Pombe Mutated in Different Repair Pathways. Biochim. Biophys. Acta (BBA) Nucleic Acids Protein Synth. 1973, 312, 617–625. [Google Scholar] [CrossRef]
- Avery, A.M.; Kaur, B.; Taylor, J.-S.; Mello, J.A.; Essigmann, J.M.; Doetsch, P.W. Substrate Specificity of Ultraviolet DNA Endonuclease (UVDE/Uve1p) from Schizosaccharomyces Pombe. Nucleic Acids Res. 1999, 27, 2256–2264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasui, A.; McCready, S.J. Alternative Repair Pathways for UV-Induced DNA Damage. BioEssays 1998, 20, 291–297. [Google Scholar] [CrossRef]
- Kanno, S.; Shigenori, I.; Takao, M.; Yasui, A. Repair of Apurinic/Apyrimidinic Sites by UV Damage Endonuclease; a Repair Protein for UV and Oxidative Damage. Nucleic Acids Res. 1999, 27, 3096–3103. [Google Scholar] [CrossRef] [Green Version]
- Schupbach, M. The Isolation and Genetic Characterization of UV-Sensitive Mutants of Schizosaccharomyces Pombe. Mutat. Res. 1971, 11, 361–371. [Google Scholar]
- Nasim, A.; Smith, B.P. Dark Repair Inhibitors and Pathways for Repair of Radiation Damage in Schizosaccharomyces Pombe. Molec. Gen. Genet. 1974, 132, 13–22. [Google Scholar] [CrossRef]
- Lehmann, A.R.; Walicka, M.; Griffiths, D.J.; Murray, J.M.; Watts, F.Z.; McCready, S.; Carr, A.M. The Rad18 Gene of Schizosaccharomyces Pombe Defines a New Subgroup of the SMC Superfamily Involved in DNA Repair. Mol. Cell. Biol. 1995, 15, 7067–7080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szanski, P.; Smith, G.R. A Role for Exonuclease I from S. Pombe in Mutation Avoidance and Mismatch Correction. Science 1995, 267, 1166–1169. [Google Scholar]
- Resnick, M.A. A Photoreactivationless Mutant of Saccharomyces Cerevisiae. Photochem. Photobiol. 1969, 9, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Schild, D.; Johnston, J.; Chang, C.; Mortimer, R.K. Cloning and Mapping of Saccharomyces Cerevisiae Photoreactivation Gene PHR1. Mol. Cell. Biol. 1984, 4, 1864–1870. [Google Scholar]
- Sebastian, J.; Kraus, B.; Sancar, G.B. Expression of the Yeast PHR1 Gene Is Induced by DNA-Damaging Agents. Mol. Cell. Biol. 1990, 10, 4630–4637. [Google Scholar] [PubMed] [Green Version]
- Sancar, A. Structure and Function of Photolyase and in Vivo Enzymology: 50th Anniversary. J. Biol. Chem. 2008, 283, 32153–32157. [Google Scholar] [CrossRef] [Green Version]
- Sassanfar, M.; Samson, L. Identification and preliminary characterization of an O6-methylguanine DNA repair methyltransferase in the yeast Saccharomyces cerevisiae. J. Biol. Chem. 1990, 265, 20–25. [Google Scholar] [CrossRef]
- Xiao, W.; Derfler, B.; Chen, J.; Samson, L. Primary Sequence and Biological Functions of a Saccharomyces Cerevisiae O6-Methylguanine/O4-Methylthymine DNA Repair Methyltransferase Gene. EMBO J. 1991, 10, 2179–2186. [Google Scholar] [CrossRef] [PubMed]
- Costa, Y.; Speed, R.; Öllinger, R.; Alsheimer, M.; Semple, C.A.; Gautier, P.; Maratou, K.; Novak, I.; Höög, C.; Benavente, R.; et al. Two Novel Proteins Recruited by Synaptonemal Complex Protein 1 (SYCP1) Are at the Centre of Meiosis. J. Cell Sci. 2005, 118, 2755–2762. [Google Scholar] [CrossRef] [Green Version]
- Marcon, L.; Boissonneault, G. Transient DNA Strand Breaks During Mouse and Human Spermiogenesis:New Insights in Stage Specificity and Link to Chromatin Remodeling. Biol. Reprod. 2004, 70, 910–918. [Google Scholar] [CrossRef]
- Sakkas, D.; Seli, E.; Manicardi, G.C.; Nijs, M.; Ombelet, W.; Bizzaro, D. The Presence of Abnormal Spermatozoa in the Ejaculate: Did Apoptosis Fail? Hum. Fertil. 2004, 7, 99–103. [Google Scholar] [CrossRef]
- Agarwal, A.; Saleh, R.A.; Bedaiwy, M.A. Role of Reactive Oxygen Species in the Pathophysiology of Human Reproduction. Fertil. Steril. 2003, 79, 829–843. [Google Scholar] [CrossRef] [Green Version]
- Gosalvez, J.; Alvarez, J.G.; Fernandez, J.L.; Gosalbez, A.; Gonzalez-Marin, C.; Lopez-Fernandez, C. Assessment of Single and Double-Stranded DNA Damage Following Short- and Long-Term Incubation Post-Thawing of Cryopreserved Human Spermatozoa. Biol. Reprod. 2011, 85, 501. [Google Scholar] [CrossRef]
- Torregrosa, N.; Domínguez-Fandos, D.; Camejo, M.I.; Shirley, C.R.; Meistrich, M.L.; Ballescà, J.L.; Oliva, R. Protamine 2 Precursors, Protamine 1/Protamine 2 Ratio, DNA Integrity and Other Sperm Parameters in Infertile Patients. Hum. Reprod. 2006, 21, 2084–2089. [Google Scholar] [CrossRef]
- Akematsu, T.; Fukuda, Y.; Garg, J.; Fillingham, J.S.; Pearlman, R.E.; Loidl, J. Post-Meiotic DNA Double-Strand Breaks Occur in Tetrahymena, and Require Topoisomerase II and Spo11. eLife 2017, 6, e26176. [Google Scholar] [CrossRef] [PubMed]
- Aoki, V.W.; Moskovtsev, S.I.; Willis, J.; Liu, L.; Mullen, J.B.M.; Carrell, D.T. DNA Integrity Is Compromised in Protamine-Deficient Human Sperm. J. Androl. 2005, 26, 741–748. [Google Scholar] [CrossRef] [PubMed]
- Carrell, D.T.; Emery, B.R.; Hammoud, S. Altered Protamine Expression and Diminished Spermatogenesis: What is the link? Hum. Reprod. Update 2007, 13, 313–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Mateo, S.; Gázquez, C.; Guimerà, M.; Balasch, J.; Meistrich, M.L.; Ballescà, J.L.; Oliva, R. Protamine 2 Precursors (Pre-P2), Protamine 1 to Protamine 2 Ratio (P1/P2), and Assisted Reproduction Outcome. Fertil. Steril. 2009, 91, 715–722. [Google Scholar] [CrossRef]
- Derijck, A.; van der Heijden, G.; Giele, M.; Philippens, M.; de Boer, P. DNA Double-Strand Break Repair in Parental Chromatin of Mouse Zygotes, the First Cell Cycle as an Origin of de Novo Mutation. Hum. Mol. Genet. 2008, 17, 1922–1937. [Google Scholar] [CrossRef] [Green Version]
- Aitken, R.J.; Iuliis, G.N.D.; McLachlan, R.I. Biological and Clinical Significance of DNA Damage in the Male Germ Line. Int. J. Androl. 2009, 32, 46–56. [Google Scholar] [CrossRef]
- Leduc, F.; Maquennehan, V.; Nkoma, G.B.; Boissonneault, G. DNA Damage Response During Chromatin Remodeling in Elongating Spermatids of Mice. Biol. Reprod. 2008, 78, 324–332. [Google Scholar] [CrossRef]
- Sun, G.; Guzman, E.; Balasanyan, V.; Conner, C.M.; Wong, K.; Zhou, H.R.; Kosik, K.S.; Montell, D.J. A Molecular Signature for Anastasis, Recovery from the Brink of Apoptotic Cell Death. J. Cell. Biol. 2017, 216, 3355–3368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaha, C.; Tripathi, R.; Mishra, D.P. Male Germ Cell Apoptosis: Regulation and Biology. Philos. Trans. R. Soc. B Biol. Sci. 2010, 365, 1501–1515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aitken, R.J.; Smith, T.B.; Jobling, M.S.; Baker, M.A.; De Iuliis, G.N. Oxidative Stress and Male Reproductive Health. Asian J. 2014, 16, 31–38. [Google Scholar] [CrossRef]
- Kothari, S.; Thompson, A.; Agarwal, A.; du Plessis, S.S. Free Radicals: Their Beneficial and Detrimental Effects on Sperm Function. Indian J. Exp. Biol. 2010, 48, 425–435. [Google Scholar]
- Kemal Duru, N.; Morshedi, M.; Oehninger, S. Effects of Hydrogen Peroxide on DNA and Plasma Membrane Integrity of Human Spermatozoa. Fertil. Steril. 2000, 74, 1200–1207. [Google Scholar] [CrossRef]
- Burden, H.P.; Holmes, C.H.; Persad, R.; Whittington, K. Prostasomes—Their Effects on Human Male Reproduction and Fertility. Hum. Reprod. Update 2006, 12, 283–292. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, A.; Durairajanayagam, D.; du Plessis, S.S. Utility of Antioxidants during Assisted Reproductive Techniques: An Evidence Based Review. Reprod. Biol. Endocrinol. 2014, 12, 112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, G.; Spivak, G.; Mitchell, D.L.; Mori, T.; McCarrey, J.R.; McMahan, C.A.; Walter, R.B.; Hanawalt, P.C.; Walter, C.A. Nucleotide Excision Repair Activity Varies Among Murine Spermatogenic Cell Types1. Biol. Reprod. 2005, 73, 123–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goukassian, D.; Gad, F.; Yaar, M.; Eller, M.S.; Nehal, U.S.; Gilchrest, B.A. Mechanisms and Implications of the Age-Associated Decrease in DNA Repair Capacity. FASEB J. 2000, 14, 1325–1334. [Google Scholar] [CrossRef]
- Jansen, J.; Olsen, A.K.; Wiger, R.; Naegeli, H.; de Boer, P.; van der Hoeven, F.; Holme, J.A.; Brunborg, G.; Mullenders, L. Nucleotide Excision Repair in Rat Male Germ Cells: Low Level of Repair in Intact Cells Contrasts with High Dual Incision Activity in Vitro. Nucleic Acids Res. 2001, 29, 1791–1800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sassone-Corsi, P. Unique Chromatin Remodeling and Transcriptional Regulation in Spermatogenesis. Science 2002, 296, 2176–2178. [Google Scholar] [CrossRef]
- Ahmed, E.A.; Scherthan, H.; De Rooij, D.G. DNA Double Strand Break Response and Limited Repair Capacity in Mouse Elongated Spermatids. Int. J. Mol. Sci. 2015, 16, 29923–29935. [Google Scholar] [CrossRef] [Green Version]
- Zenzes, M.T. Smoking and Reproduction: Gene Damage to Human Gametes and Embryos. Hum. Reprod. Update 2000, 6, 122–131. [Google Scholar] [CrossRef]
- Stringer, J.M.; Winship, A.; Liew, S.H.; Hutt, K. The Capacity of Oocytes for DNA Repair. Cell. Mol. Life Sci. 2018, 75, 2777–2792. [Google Scholar] [CrossRef]
- Menezo, Y.J.; Russo, G.; Tosti, E.; Mouatassim, S.E.; Benkhalifa, M. Expression Profile of Genes Coding for DNA Repair in Human Oocytes Using Pangenomic Microarrays, with a Special Focus on ROS Linked Decays. J. Assist. Reprod. Genet. 2007, 24, 513–520. [Google Scholar] [CrossRef] [Green Version]
- Jaroudi, S.; Kakourou, G.; Cawood, S.; Doshi, A.; Ranieri, D.M.; Serhal, P.; Harper, J.C.; SenGupta, S.B. Expression Profiling of DNA Repair Genes in Human Oocytes and Blastocysts Using Microarrays. Hum. Reprod. 2009, 24, 2649–2655. [Google Scholar] [CrossRef] [Green Version]
- Zeng, F.; Baldwin, D.A.; Schultz, R.M. Transcript Profiling during Preimplantation Mouse Development. Dev. Biol. 2004, 272, 483–496. [Google Scholar] [CrossRef] [Green Version]
- Zheng, P.; Schramm, R.D.; Latham, K.E. Developmental Regulation and In Vitro Culture Effects on Expression of DNA Repair and Cell Cycle Checkpoint Control Genes in Rhesus Monkey Oocytes and Embryos1. Biol. Reprod. 2005, 72, 1359–1369. [Google Scholar] [CrossRef] [Green Version]
- Martin, J.H.; Bromfield, E.G.; Aitken, R.J.; Lord, T.; Nixon, B. Double Strand Break DNA Repair Occurs via Non-Homologous End-Joining in Mouse MII Oocytes. Sci. Rep. 2018, 8, 9685. [Google Scholar] [CrossRef]
- Hamatani, T.; Falco, G.; Carter, M.G.; Akutsu, H.; Stagg, C.A.; Sharov, A.A.; Dudekula, D.B.; VanBuren, V.; Ko, M.S.H. Age-Associated Alteration of Gene Expression Patterns in Mouse Oocytes. Hum. Mol. Genet. 2004, 13, 2263–2278. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, Y.; Tobari, I.; Yamagiwa, J.; Utsugi, T.; Okamoto, M.; Nakai, S. Dose-Response Relationship of γ-Ray-Induced Reciprocal Translocations at Low Doses in Spermatogonia of the Crab-Eating Monkey (Macaca Fascicularis). Mutat. Res./Fundam. Mol. Mech. Mutagenesis 1985, 151, 121–127. [Google Scholar] [CrossRef]
- Agarwal, A.; Aziz, N.; Rizk, B. Studies on Women’s Health; Humana Press: Totowa, NJ, USA, 2013. [Google Scholar]
- Richards, J.S.; Russell, D.L.; Ochsner, S.; Espey, L.L. Ovulation: New Dimensions and New Regulators of the Inflammatory-Like Response. Annu. Rev. Physiol. 2002, 64, 69–92. [Google Scholar] [CrossRef] [PubMed]
- Revelli, A.; Piane, L.D.; Casano, S.; Molinari, E.; Massobrio, M.; Rinaudo, P. Follicular Fluid Content and Oocyte Quality: From Single Biochemical Markers to Metabolomics. Reprod. Biol. Endocrinol. 2009, 7, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krisher, R.L. In Vivo and In Vitro Environmental Effects on Mammalian Oocyte Quality. Annu. Rev. Anim. Biosci. 2013, 1, 393–417. [Google Scholar] [CrossRef] [PubMed]
- Du Plessis, S.S.; Makker, K.; Desai, N.R.; Agarwal, A. Impact of Oxidative Stress on IVF. Expert Rev. Obstet. Gynecol. 2008, 3, 539–554. [Google Scholar] [CrossRef]
- Borini, A.; Tarozzi, N.; Bizzaro, D.; Bonu, M.A.; Fava, L.; Flamigni, C.; Coticchio, G. Sperm DNA Fragmentation: Paternal Effect on Early Post-Implantation Embryo Development in ART. Hum. Reprod. 2006, 21, 2876–2881. [Google Scholar] [CrossRef] [PubMed]
- Braude, P.; Bolton, V.; Moore, S. Human Gene Expression First Occurs between the Four- and Eight-Cell Stages of Preimplantation Development. Nature 1988, 332, 459–461. [Google Scholar] [CrossRef] [PubMed]
- Ahmadi, A.; Ng, S. Fertilizing Ability of DNA-damaged Spermatozoa. J. Exp. Zool. 1999, 284, 696–704. [Google Scholar] [CrossRef]
- Genescà, A.; Caballín, M.R.; Miró, R.; Benet, J.; Germà, J.R.; Egozcue, J. Repair of Human Sperm Chromosome Aberrations in the Hamster Egg. Hum. Genet. 1992, 89, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Gawecka, J.E.; Marh, J.; Ortega, M.; Yamauchi, Y.; Ward, M.A.; Ward, W.S. Mouse Zygotes Respond to Severe Sperm DNA Damage by Delaying Paternal DNA Replication and Embryonic Development. PLoS ONE 2013, 8, e56385. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| H. sapiens | S. cerevisiae | S. pombe | Functions | |
|---|---|---|---|---|
| TC NER | RNA pol II | RNA pol II | RNA pol II | mRNA synthesis |
| CSB | Rad 26 | Rhp 26 | Detection of RNA pol II arrest | |
| - | Rpd 9 | - | Part of RNA pol II, detection of arrest | |
| CSA | Rad 28 | Ckn1 | TC-NER protein hub | |
| UVSSA-USP7 | - | - | Ubiquitine ligase | |
| GG NER | XPC | Rad4 | rhp41, rhp42 | Scan to detect damages |
| Rad23b | Rad23 | Rhp 23 | XPC partner | |
| TFIIH | TFIIH | TFIIH | Transcription factor, check for damage | |
| XPB | Rad 25 | Ptr 8 | Core units of TFIIH (helicases) | |
| XPD | Rad 3 | Rad 15 | ||
| XPA | Rad 14 | Rph 14 | TFIIH helicases stimulator | |
| ERCCI-XPF | Rad 10 | Swi 10 | Endonuclease complex, cleaves 5′ to damage | |
| XPF | Rad 1 | Rad 16 | ||
| XPG | Rad 2 | Rad 13 | Endonuclease, cleaves 3′ to damage | |
| BER | UNG | Ung 1 | Ung 1 | Uracil DNA Glycosylase (UDG) |
| TDG | - | Thp 1 | UDG, T:G, U:G mismatches, 5-fluorouracil, 3,N4-ethanolcytosine, 5-hydroxyuracil, Xanthine, Oxanine, Hypoxanthine DNA glycosylase | |
| - | Mag 1 | Mag 1 | Alkylation DNA Glycosylase | |
| - | Mag 2 | Mag 2 | ||
| OGG 1 | Ogg 1 | - | 8-oxoG, fapy-G, 7,8-dihydro8oxoG DNA Glycosylase | |
| NTHL 1 | Ntg1/Ntg2 | Nth 1 | 8-oxoG, 8-hydroxycytosine, thimidineglycol, 8-hydroxyuracil DNA Glycosylase, AP site β lyase | |
| MUTYH | - | Myh 1 | Glycosylase of adenine mismatches | |
| APE1 | Apn 2 | Apn 2 | AP endonucleases | |
| - | Apn 1 | Apn 1 | ||
| FEN 1 | Rad 27 | Rad 2 | Structure specific endonuclease, cleaves at ssDNA-dsDNA transition | |
| MMR | MutS α | MutS α | MutS α | Mismatch detection |
| MutS β | MutS β | MutS β | Long insertion deletion loops detection, involved in recombination | |
| MutS γ | MutS γ | MutS γ | Holliday Junction resolvase | |
| MutL α | MutL α | MutL α | MutSα and MutSβ interactor, weakly endonuclease | |
| MutL β | MutL β | - | ||
| MutL γ | MutL γ | - | MutSγ interactor | |
| EXO 1 | EXO 1 | EXO 1 | Exonuclease | |
| PCNA | PCNA | PCNA | DNA polymerase helicase | |
| UVDER | - | - | UVE 1 | UV damage endonucleases |
| - | - | UVDE | ||
| Photolyase | - | Phr1 | - | CPD specific photolyase |
| - | Mgt1 | - | O6-MeG, O4-MeT methyl transferase | |
| HR | Mre11/Rad50/Nbs1 | Mre11/Rad50/Xrs1 | Mre11/Rad50/Nbs1 | MRN/X complex, DNA ends detection, resection initiation |
| CtIP | Sae2 | Ctp 1 | Endonuclease | |
| RPA | RPA | Rad 11 | Single strand DNA binding protein | |
| Rad 52 | Rad 52 | Rad 22 | Displace RPA to form Rad51 filament | |
| Rad51 | Rad 51 | Rhp 51 | Globular protein forming filaments for strand invasion | |
| NHEJ | Ku 70/80 | YKu 70/80 | PKu 70/80 | DNA ends detection and protection |
| DNA-Pkc | - | - | Protein kinase | |
| Artemis | - | - | Endonuclease, DNA ends processing | |
| XRCC4 | Nej 1 | - | DNA ends bridging by filaments formation | |
| XLF | Lif 1 | Xlfl | ||
| DNA ligase IV | Dnl 4 | Lig 4 | DNA ends ligation |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mourrain, L.; Boissonneault, G. DNA Repair in Haploid Context. Int. J. Mol. Sci. 2021, 22, 12418. https://doi.org/10.3390/ijms222212418
Mourrain L, Boissonneault G. DNA Repair in Haploid Context. International Journal of Molecular Sciences. 2021; 22(22):12418. https://doi.org/10.3390/ijms222212418
Chicago/Turabian StyleMourrain, Loïs, and Guylain Boissonneault. 2021. "DNA Repair in Haploid Context" International Journal of Molecular Sciences 22, no. 22: 12418. https://doi.org/10.3390/ijms222212418