
Evaluation of the Binding Mechanism of Human Defensin 5 in a Bacterial Membrane: A Simulation Study


Abstract
:1. Introduction
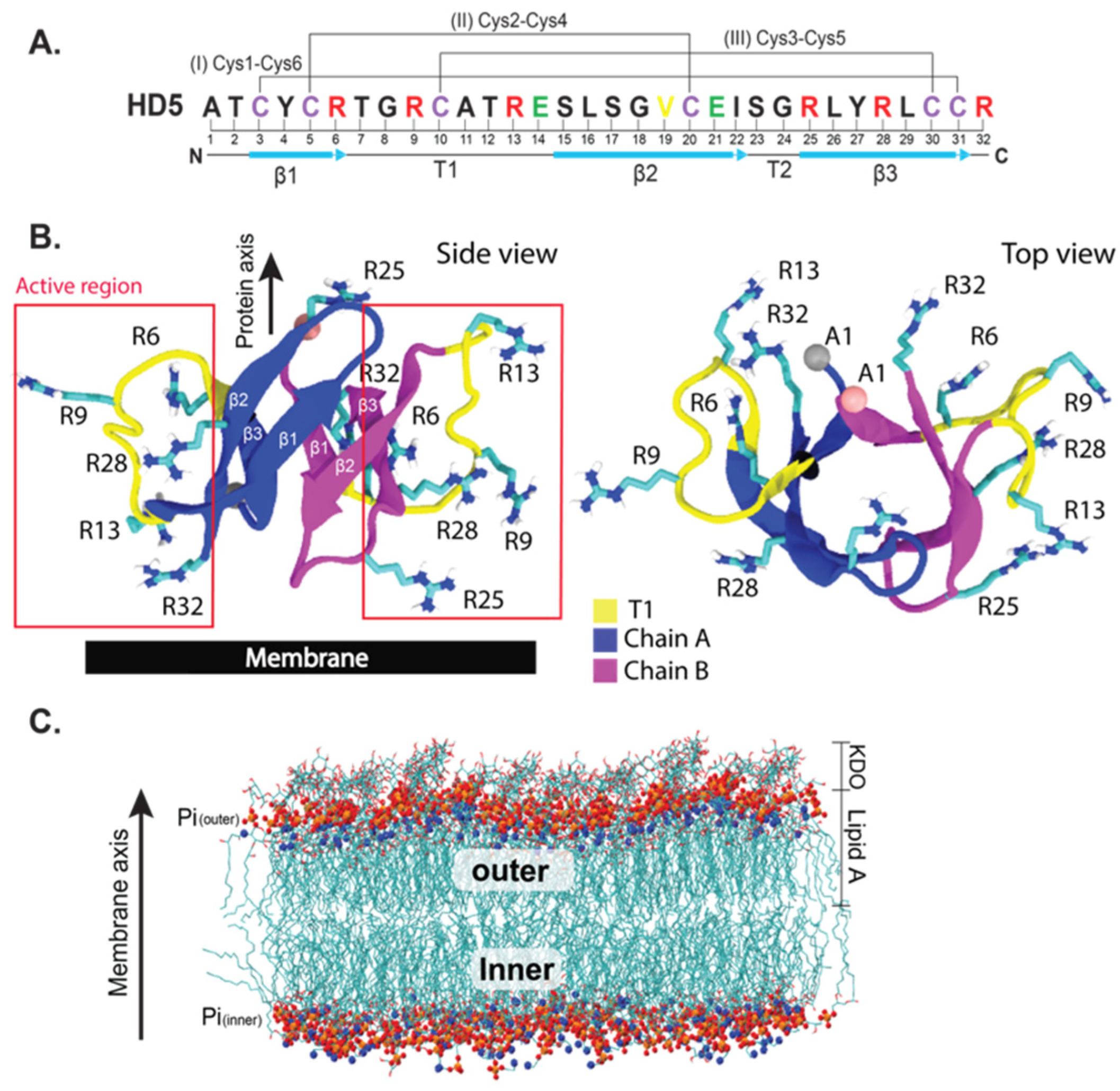
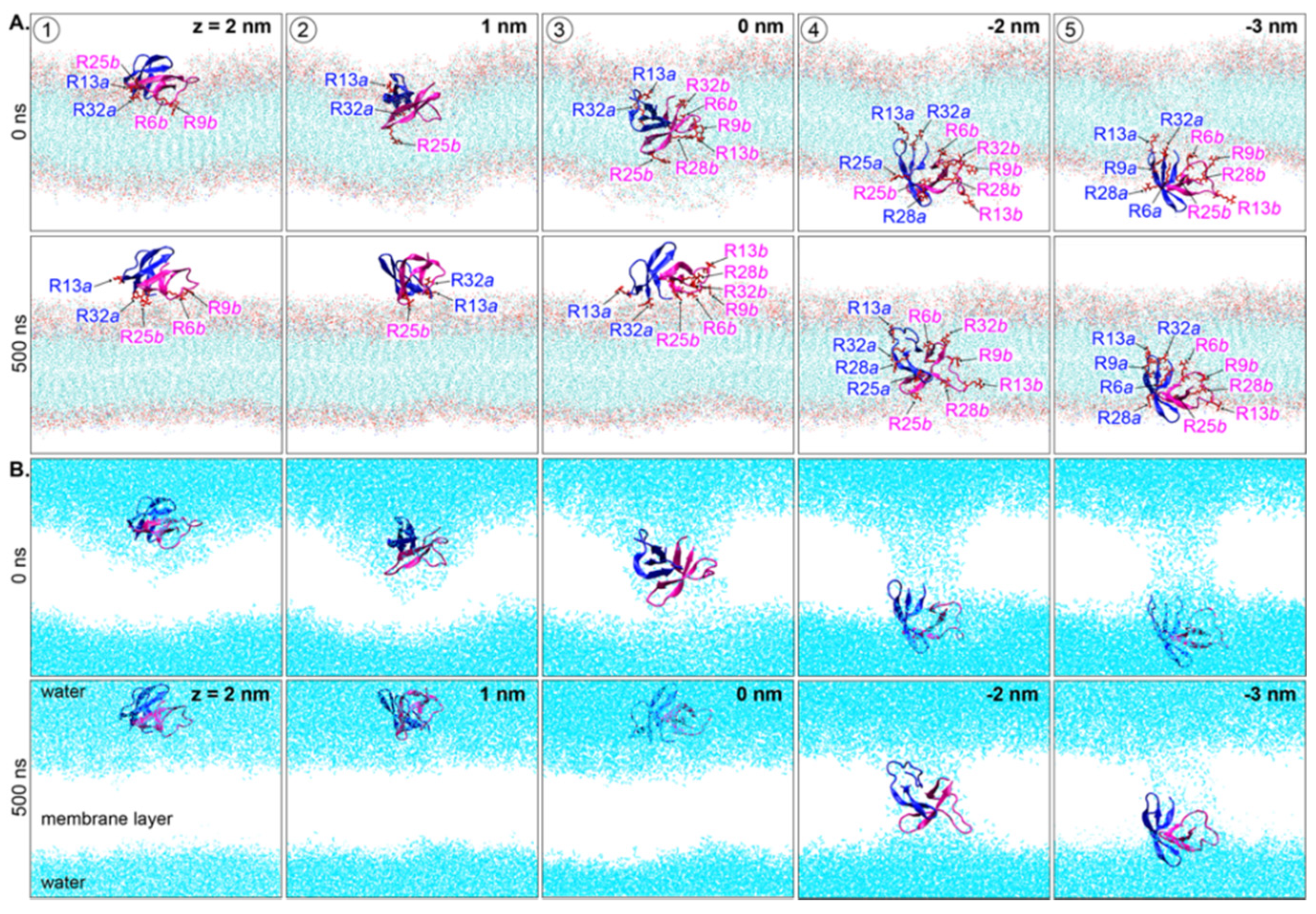
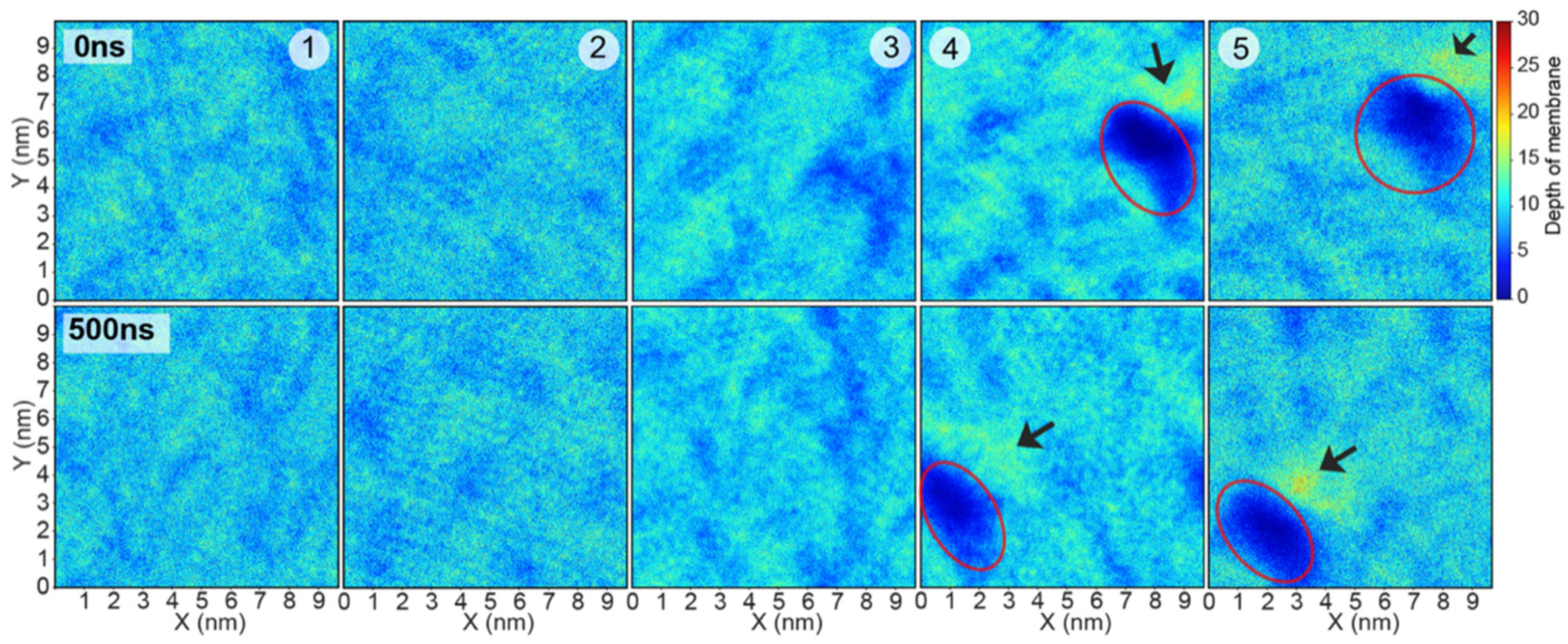
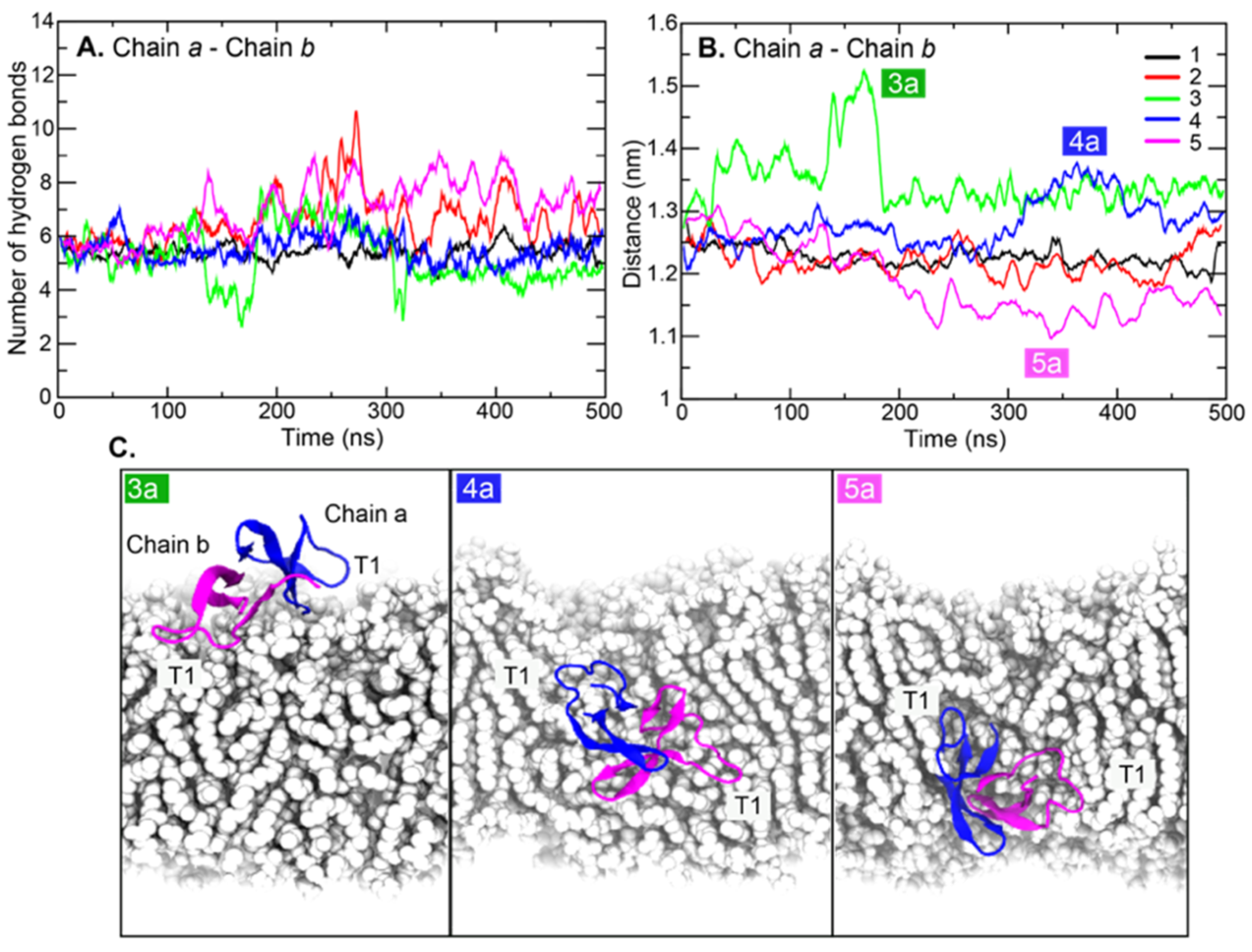
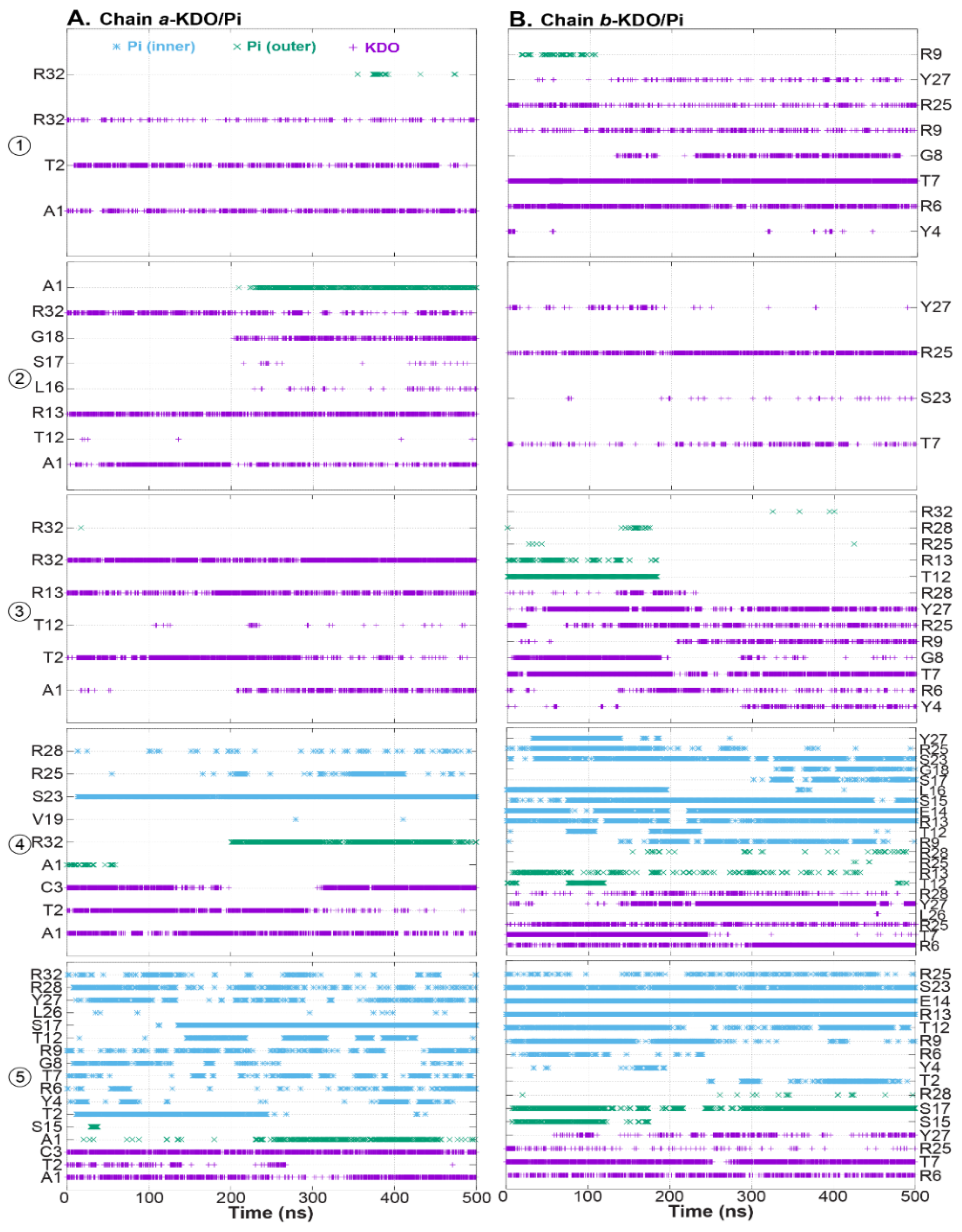
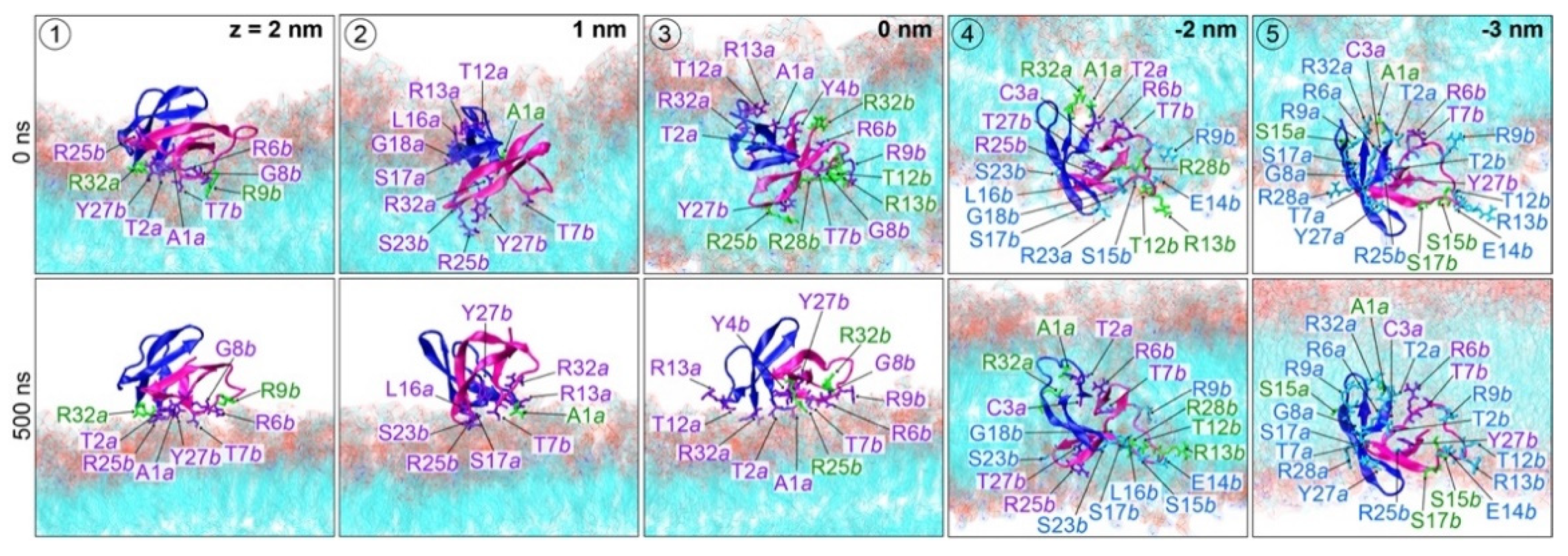
2. Results and Discussion
3. Materials and Methods
3.1. HD5–Membrane Complex Setup
3.2. Molecular Dynamics Simulations
3.3. MM/PBSA Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hooper, L.V.; Littman, D.R.; Macpherson, A.J. Interactions between the microbiota and the immune system. Science 2012, 336, 1268–1273. [Google Scholar] [CrossRef] [Green Version]
- Davies, J.; Davies, D. Origins and evolution of antibiotic resistance. Microbiol. Mol. Biol. Rev. 2010, 74, 417–433. [Google Scholar] [CrossRef] [Green Version]
- Akar-Ghibril, N. Defects of the Innate Immune System and Related Immune Deficiencies. Clin. Rev. Allergy Immunol. 2021, 21, 8885. [Google Scholar] [CrossRef]
- Cruz, J.; Ortiz, C.; Guzman, F.; Fernandez-Lafuente, R.; Torres, R. Antimicrobial peptides: Promising compounds against pathogenic microorganisms. Curr. Med. Chem. 2014, 21, 2299–2321. [Google Scholar] [CrossRef] [PubMed]
- Ganz, T. Defensins: Antimicrobial peptides of innate immunity. Nat. Rev. Immunol. 2003, 3, 710–720. [Google Scholar] [CrossRef] [PubMed]
- Durr, U.H.; Sudheendra, U.S.; Ramamoorthy, A. LL-37, the only human member of the cathelicidin family of antimicrobial peptides. Biochim. Biophys. Acta 2006, 1758, 1408–1425. [Google Scholar] [CrossRef] [Green Version]
- Hazlett, L.; Wu, M. Defensins in innate immunity. Cell Tissue Res. 2011, 343, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Lu, W. Defensins: A Double-Edged Sword in Host Immunity. Front. Immunol. 2020, 11, 764. [Google Scholar] [CrossRef]
- de Leeuw, E.; Rajabi, M.; Zou, G.; Pazgier, M.; Lu, W. Selective arginines are important for the antibacterial activity and host cell interaction of human alpha-defensin 5. FEBS Lett. 2009, 583, 2507–2512. [Google Scholar] [CrossRef] [Green Version]
- Selsted, M.E.; Ouellette, A.J. Mammalian defensins in the antimicrobial immune response. Nat. Immunol. 2005, 6, 551–557. [Google Scholar] [CrossRef] [PubMed]
- Rajabi, M.; Ericksen, B.; Wu, X.J.; de Leeuw, E.; Zhao, L.; Pazgier, M.; Lu, W.Y. Functional Determinants of Human Enteric alpha-Defensin HD5 crucial for hydrophobicity at dimer interface. J. Biol. Chem. 2012, 287, 21615–21627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Jung, S.W.; Cho, A.E. Molecular insights into the adsorption mechanism of human β-defensin-3 on bacterial membranes. Langmuir 2016, 32, 1782–1790. [Google Scholar] [CrossRef]
- Yeasmin, R.; Brewer, A.; Fine, L.R.; Zhang, L. Molecular Dynamics Simulations of Human Beta-Defensin Type 3 Crossing Different Lipid Bilayers. ACS Omega 2021, 6, 13926–13939. [Google Scholar] [CrossRef] [PubMed]
- Ericksen, B.; Wu, Z.; Lu, W.; Lehrer, R.I. Antibacterial activity and specificity of the six human α-defensins. Antimicrob. Agents Chemother. 2005, 49, 269–275. [Google Scholar] [CrossRef] [Green Version]
- Wommack, A.J.; Robson, S.A.; Wanniarachchi, Y.A.; Wan, A.; Turner, C.J.; Wagner, G.; Nolan, E.M. NMR solution structure and condition-dependent oligomerization of the antimicrobial peptide human defensin 5. Biochemistry 2012, 51, 9624–9637. [Google Scholar] [CrossRef]
- Lehrer, R.I.; Jung, G.; Ruchala, P.; Andre, S.; Gabius, H.J.; Lu, W. Multivalent binding of carbohydrates by the human alpha-defensin, HD5. J. Immunol. 2009, 183, 480–490. [Google Scholar] [CrossRef] [Green Version]
- Chairatana, P.; Niramitranon, J.; Pongprayoon, P. Dynamics of human defensin 5 (HD5) self-assembly in solution: Molecular simulations/insights. Comput. Biol. Chem. 2019, 83, 107091. [Google Scholar] [CrossRef] [PubMed]
- Chileveru, H.R.; Lim, S.A.; Chairatana, P.; Wommack, A.J.; Chiang, I.L.; Nolan, E.M. Visualizing attack of Escherichia coli by the antimicrobial peptide human defensin 5. Biochemistry 2015, 54, 1767–1777. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Zhao, G.; Wang, S.; Chen, Y.; Gong, Y.; Chen, S.; Xu, Y.; Hu, M.; Wang, X.; Zeng, H.; et al. A Simplified Derivative of Human Defensin 5 with Potent and Efficient Activity against Multidrug-Resistant Acinetobacter baumannii. Antimicrob. Agents Chemother. 2018, 62, e01504-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Shen, M.; Zhang, N.; Wang, S.; Xu, Y.; Chen, S.; Chen, F.; Yang, K.; He, T.; Wang, A.; et al. Reduction Impairs the Antibacterial Activity but Benefits the LPS Neutralization Ability of Human Enteric Defensin 5. Sci. Rep. 2016, 6, 22875. [Google Scholar] [CrossRef] [Green Version]
- Awang, T.; Pongprayoon, P. The adsorption of human defensin 5 on bacterial membranes: Simulation studies. J. Mol. Model. 2018, 24, 273. [Google Scholar] [CrossRef]
- Jung, S.W.; Lee, J.; Cho, A.E. Elucidating the Bacterial Membrane Disruption Mechanism of Human α-Defensin 5: A Theoretical Study. J. Phys. Chem. B 2017, 121, 741–748. [Google Scholar] [CrossRef] [PubMed]
- Ganz, T. Defensins: Antimicrobial peptides of vertebrates. C. R. Biol. 2004, 327, 539–549. [Google Scholar] [CrossRef]
- Awang, T.; Pongprayoon, P. The penetration of human defensin 5 (HD5) through bacterial outer membrane: Simulation studies. J. Mol. Model. 2021, 27, 291. [Google Scholar] [CrossRef] [PubMed]
- Wimley, W.C.; Selsted, M.E.; White, S.H. Interactions between human defensins and lipid bilayers: Evidence for formation of multimeric pores. Protein Sci. 1994, 3, 1362–1373. [Google Scholar] [CrossRef] [Green Version]
- Hill, C.P.; Yee, J.; Selsted, M.E.; Eisenberg, D. Crystal structure of defensin HNP-3, an amphiphilic dimer: Mechanisms of membrane permeabilization. Science 1991, 251, 1481–1485. [Google Scholar] [CrossRef] [PubMed]
- Colavita, I.; Nigro, E.; Sarnataro, D.; Scudiero, O.; Granata, V.; Daniele, A.; Zagari, A.; Pessi, A.; Salvatore, F. Membrane protein 4F2/CD98 is a cell surface receptor involved in the internalization and trafficking of human β-Defensin 3 in epithelial cells. Chem. Biol. 2015, 22, 217–228. [Google Scholar] [CrossRef] [Green Version]
- Hsu, P.-C.; Jefferies, D.; Khalid, S. Molecular Dynamics Simulations Predict the Pathways via Which Pristine Fullerenes Penetrate Bacterial Membranes. J. Phys. Chem. B 2016, 120, 11170–11179. [Google Scholar] [CrossRef] [PubMed]
- Oostenbrink, C.; Villa, A.; Mark, A.E.; Van Gunsteren, W.F. A biomolecular force field based on the free enthalpy of hydration and solvation: The GROMOS force-field parameter sets 53A5 and 53A6. J. Comput. Chem. 2004, 25, 1656–1676. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Sankaran-Walters, S.; Hart, R.; Dills, C. Guardians of the Gut: Enteric Defensins. Front. Microbiol. 2017, 8, 647. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | Membrane | Water | ||||
|---|---|---|---|---|---|---|
| Protein | Chain a | Chain b | Protein | Chain a | Chain b | |
| 1 | 10.82 ± 3.54 | 1.97 ± 1.34 | 8.85 ± 3.28 | 134.16 ± 7.39 | 70.30 ± 5.45 | 63.86 ± 5.61 |
| 2 | 9.71 ± 2.41 | 6.18 ± 1.93 | 3.54 ± 1.80 | 137.60 ± 8.29 | 62.98 ± 5.65 | 74.62 ± 5.85 |
| 3 | 14.08 ± 4.90 | 5.29 ± 1.86 | 8.56 ± 4.69 | 130.99 ± 10.48 | 63.71 ± 5.92 | 67.28 ± 9.16 |
| 4 | 23.99± 3.44 | 9.05 ± 1.94 | 14.95 ± 2.97 | 103.01 ± 8.13 | 47.32 ± 4.69 | 55.70 ± 6.36 |
| 5 | 23.33 ± 3.84 | 9.30 ± 2.46 | 14.03 ± 2.96 | 99.48 ± 8.26 | 44.70 ± 5.40 | 54.78 ± 6.04 |
| Binding Energy of HD5-Membrane (×103 kJ/mol) | Position | ||||
|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | |
| ∆EvdW | −0.06 ± 0.01 | −0.21 ± 0.02 | −0.15 ± 0.02 | −1.03 ± 0.06 | −0.42 ± 0.04 |
| ∆EElec | −45.74 ± 1.15 | −51.50 ± 1.12 | −49.30 ± 0.95 | −52.72 ± 0.47 | −41.80 ± 0.60 |
| ∆Epolar solv | 1.02 ± 0.39 | 2.00 ± 0.39 | 0.28 ± 0.42 | 2.65 ± 0.37 | 2.40 ± 0.69 |
| ∆Enon-polar solv | 0.25 ± 0.03 | 0.23 ± 0.02 | 0.24 ± 0.01 | 0.14 ± 0.03 | 0.19 ± 0.02 |
| ∆Gtotal | −44.54 ± 1.11 | −49.49 ± 0.82 | −48.93 ± 0.76 | −50.97 ± 0.57 | −39.63 ± 1.03 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Awang, T.; Chairatana, P.; Vijayan, R.; Pongprayoon, P. Evaluation of the Binding Mechanism of Human Defensin 5 in a Bacterial Membrane: A Simulation Study. Int. J. Mol. Sci. 2021, 22, 12401. https://doi.org/10.3390/ijms222212401
Awang T, Chairatana P, Vijayan R, Pongprayoon P. Evaluation of the Binding Mechanism of Human Defensin 5 in a Bacterial Membrane: A Simulation Study. International Journal of Molecular Sciences. 2021; 22(22):12401. https://doi.org/10.3390/ijms222212401
Chicago/Turabian StyleAwang, Tadsanee, Phoom Chairatana, Ranjit Vijayan, and Prapasiri Pongprayoon. 2021. "Evaluation of the Binding Mechanism of Human Defensin 5 in a Bacterial Membrane: A Simulation Study" International Journal of Molecular Sciences 22, no. 22: 12401. https://doi.org/10.3390/ijms222212401