
Nuclear Receptors in Myocardial and Cerebral Ischemia—Mechanisms of Action and Therapeutic Strategies

 ,
, 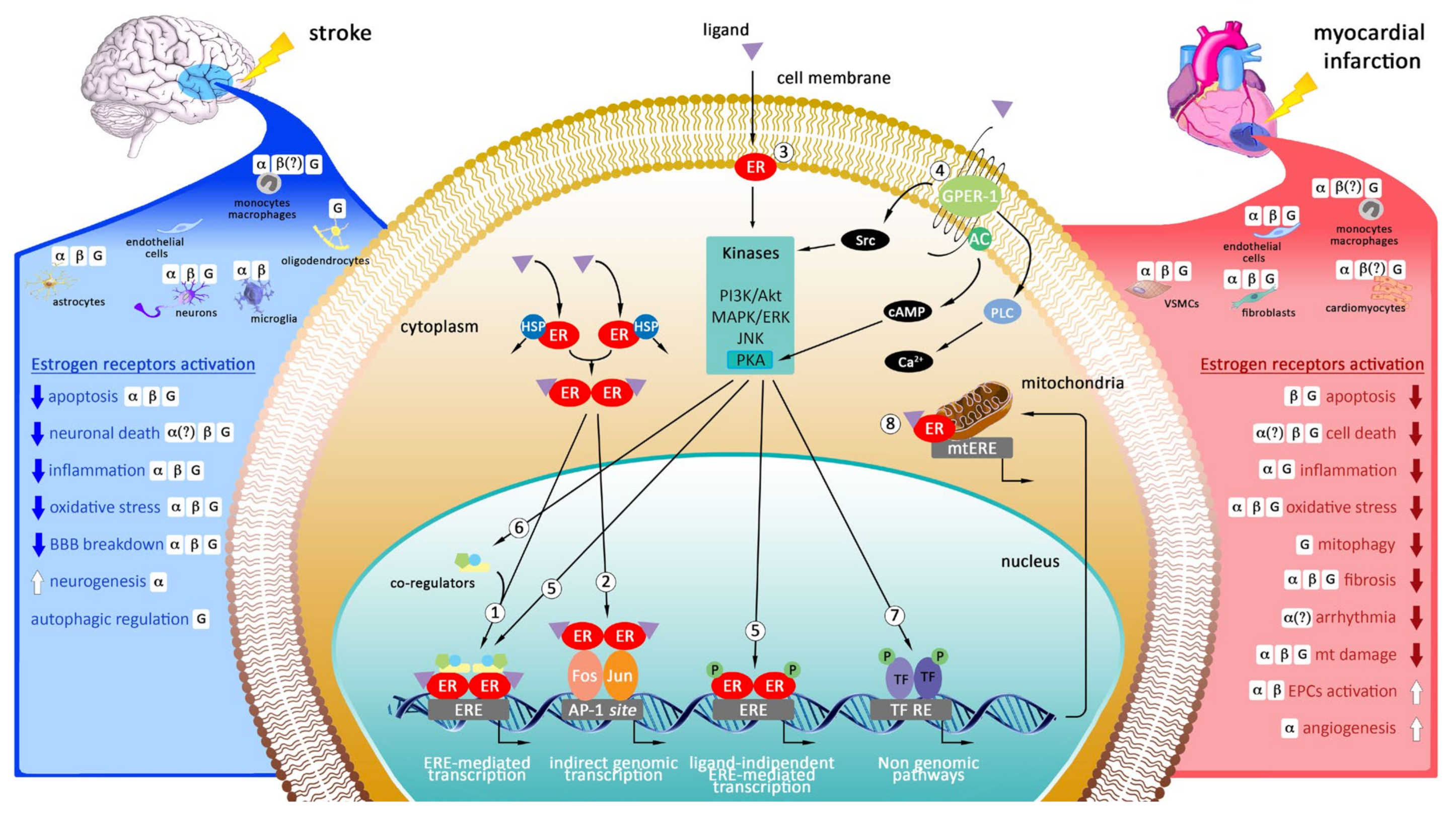{kind=link}
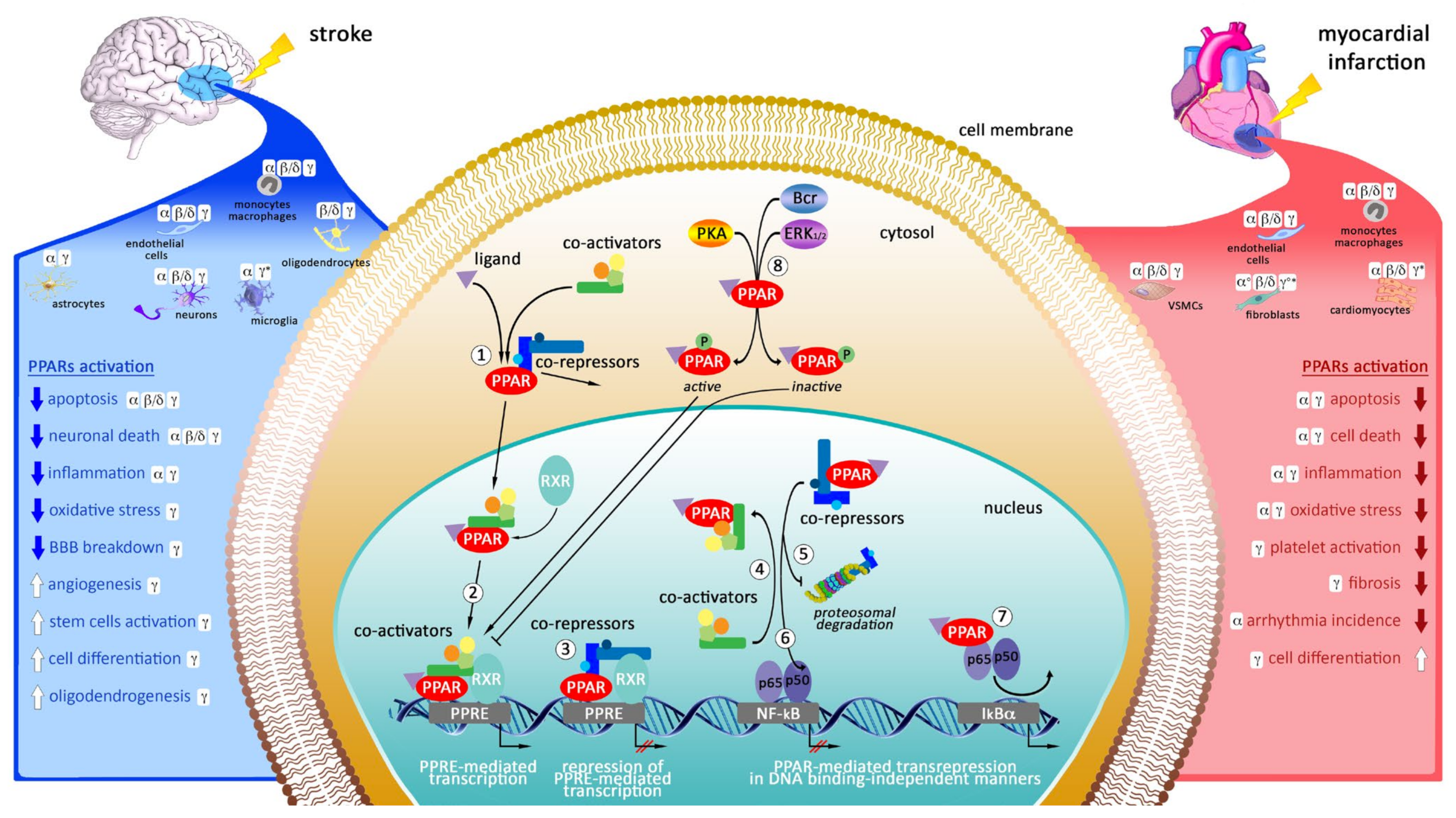{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Targeting Estrogen Receptors as Potential Therapeutic Strategy in Myocardial Infarction and Stroke
2.1. Cellular Localization of Estrogen Receptors in the Heart
2.2. Cellular Localization of Estrogen Receptors in the Brain
2.3. Genomic and Non-Genomic Mechanisms of Action of Estrogen Receptors
2.4. The Role of Estrogen Receptors in Myocardial Infarction
2.4.1. ERs Modulation in Experimental Models of Myocardial Infarction
2.4.2. GPER-1 Modulation in Experimental Models of Myocardial Infarction
2.5. The Role of Estrogen Receptors in Stroke
2.5.1. ERs Modulation in Experimental Models of Stroke
2.5.2. GPER-1 Modulation in Experimental Models of Stroke
3. Targeting of Peroxisome Proliferator-Activated Receptors (PPARs) as Potential Therapeutic Strategy in Myocardial Infarction and Stroke
3.1. Cellular Localization of PPARs in the Heart
3.2. Cellular Localization of PPARs in the Brain
3.3. Mechanisms of Action of Peroxisome Proliferator-Activated Receptors
3.4. The Modulation of PPARs in Experimental Models of Myocardial Infarction
3.5. The Modulation of PPARs in Experimental Models of Stroke
4. Targeting of Aryl Hydrocarbon Receptor (AhR) as Promising Therapeutic Strategy in Myocardial Infarction and Stroke
4.1. Cellular Localization of AhR in the Heart
4.2. Cellular Localization of AhR in the Brain
4.3. Mechanisms of Action of Aryl Hydrocarbon Receptor
4.4. The Modulation of AhR in Experimental Models of Myocardial Infarction
4.5. The Modulation of AhR in Experimental Models of Stroke
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AHR | aryl hydrocarbon receptor |
| AIP | AHR-interacting protein |
| Akt (PKB) | protein kinase B |
| ANP | pro-atrial natriuretic peptide |
| AP-1 | activator protein 1 |
| ARNT | aryl hydrocarbon nuclear translocator |
| BBB | blood brain barrier |
| CK-MB | creatine kinase-(muscle–brain) |
| CNS | central nervous system |
| CREB | cAMP response element-binding protein |
| CVDs | cardiovascular diseases |
| CXCL1 | C-X-C Motif Chemokine Ligand 1 |
| CYP1A1 | cytochrome P450 1A1 |
| DIM | 3,3′-diindolylmethane |
| DPN | diaryl-propio-nitrile |
| E2 | 17β-estradiol |
| ER | estrogen receptor |
| ERE | estrogen response element |
| ERK | extracellular signal-regulated kinase |
| ERα | estrogen receptor alpha |
| ERβ | estrogen receptor beta |
| GPER | G-protein-coupled estrogen receptor |
| GSK-3β | glycogen synthase kinase 3 beta |
| HIE | hypoxic ischemic encephalopathy |
| HSP90 | heat shock protein |
| I/R | ischemia/reperfusion |
| IL-6 | Interleukina 6 |
| JNK | c-Jun N-terminal kinase |
| KO | knock-out |
| LAD | left anterior descending |
| LPS | lipopolysaccharides |
| LV | left ventricular |
| MAPK | mitogen-activated protein kinases |
| MI | myocardial infarction |
| miRNA | microRNA |
| MMP | methyl-piperidino-pyrazole |
| mPTP | mitochondrial permeability transition pore |
| MSCs | mesenchymal stem cells |
| MT | mechanical thrombectomy |
| MTA1 | metastasis-associated protein 1 |
| mTOR | mammalian target of rapamycin kinase |
| NCoR | the nuclear receptor corepressor |
| NF-κB | nuclear factor-kappa B |
| NRF-1 | nuclear respiratory factor-1 |
| OA | Oleic acid |
| OVX | ovariectomized |
| PAI-1 | plasminogen activator inhibitor-1 |
| PCG-1 | peroxisome proliferator-activated receptor-gamma coactivator 1 |
| PI-3K | 3-Phosphatydylinosytol kinase |
| PKCε | the epsilon isoform of protein kinase C |
| PLC | phospholipase C |
| pMCAO | permanent middle cerebral artery occlusion |
| PPAR | peroxisome proliferator-activated receptor |
| PPREs | proliferator response elements |
| PPT | propyl-pyrazole-triol |
| RACK2 | receptor for activated C kinase |
| ROS | reactive oxygen species |
| rt-PA | tissue plasminogen activator |
| RXR | retinoid X receptor |
| SAHRM | selective aryl hydrocarbon receptor modulator |
| SERM | selective estrogen receptor modulator |
| SMRT | silencing mediator of retinoid and thyroid hormone receptor |
| SOD1 | superoxide dismutase |
| ta-VNS | transcutaneous auricular vagus nerve stimulation |
| TFAM | mitochondrial transcription factor A |
| Tg-ERβ | transgenic ERβ |
| tMCAO | transient middle cerebral artery occlusion |
| TNF-α | Tumor necrosis factor |
| VSMCs | vascular smooth muscle cells |
| XRE | xenobiotic response element |
References
- Virani, S.S.; Alonso, A.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Delling, F.N.; et al. Heart Disease and Stroke Statistics-2020 Update: A Report From the American Heart Association. Circulation 2020, 141, e139–e596. [Google Scholar] [CrossRef] [PubMed]
- Chiong, M.; Wang, Z.V.; Pedrozo, Z.; Cao, D.J.; Troncoso, R.; Ibacache, M.; Criollo, A.; Nemchenko, A.; Hill, J.A.; Lavandero, S. Cardiomyocyte death: Mechanisms and translational implications. Cell Death Dis. 2011, 2, e244. [Google Scholar] [CrossRef] [PubMed]
- Sekerdag, E.; Solaroglu, I.; Gursoy-Ozdemir, Y. Cell Death Mechanisms in Stroke and Novel Molecular and Cellular Treatment Options. Curr. Neuropharmacol. 2018, 16, 1396–1415. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.L.; Morrow, D.A. Acute Myocardial Infarction. N. Engl. J. Med. 2017, 376, 2053–2064. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, S.; Fukuma, S.; Ikenoue, T.; Fukuhara, S.; Kobayashi, S. Effect of Edaravone on Neurological Symptoms in Real-World Patients With Acute Ischemic Stroke. Stroke 2019, 50, 1805–1811. [Google Scholar] [CrossRef]
- Leng, T.; Xiong, Z.-G. Treatment for ischemic stroke: From thrombolysis to thrombectomy and remaining challenges. Brain Circ. 2019, 5, 8. [Google Scholar] [CrossRef]
- Regitz-Zagrosek, V.; Oertelt-Prigione, S.; Prescott, E.; Franconi, F.; Gerdts, E.; Foryst-Ludwig, A.; Maas, A.H.E.M.; Kautzky-Willer, A.; Knappe-Wegner, D.; Kintscher, U.; et al. Gender in cardiovascular diseases: Impact on clinical manifestations, management, and outcomes. Eur. Heart J. 2016, 37, 24–34. [Google Scholar] [CrossRef] [Green Version]
- Feigin, V.L.; Lawes, C.M.M.; Bennett, D.A.; Anderson, C.S. Stroke epidemiology: A review of population-based studies of incidence, prevalence, and case-fatality in the late 20th century. Lancet Neurol. 2003, 2, 43–53. [Google Scholar] [CrossRef]
- Lisabeth, L.; Bushnell, C. Menopause and stroke: An epidemiologic review. Lancet Neurol. 2012, 11, 82–91. [Google Scholar] [CrossRef] [Green Version]
- Babiker, F. Estrogenic hormone action in the heart: Regulatory network and function. Cardiovasc. Res. 2002, 53, 709–719. [Google Scholar] [CrossRef] [Green Version]
- McEwen, B.S.; Akama, K.T.; Spencer-Segal, J.L.; Milner, T.A.; Waters, E.M. Estrogen effects on the brain: Actions beyond the hypothalamus via novel mechanisms. Behav. Neurosci. 2012, 126, 4–16. [Google Scholar] [CrossRef] [Green Version]
- Iorga, A.; Cunningham, C.M.; Moazeni, S.; Ruffenach, G.; Umar, S.; Eghbali, M. The protective role of estrogen and estrogen receptors in cardiovascular disease and the controversial use of estrogen therapy. Biol. Sex Differ. 2017, 8, 33. [Google Scholar] [CrossRef]
- Suzuki, S.; Brown, C.M.; Wise, P.M. Neuroprotective effects of estrogens following ischemic stroke. Front. Neuroendocrinol. 2009, 30, 201–211. [Google Scholar] [CrossRef] [Green Version]
- Vegeto, E.; Benedusi, V.; Maggi, A. Estrogen anti-inflammatory activity in brain: A therapeutic opportunity for menopause and neurodegenerative diseases. Front. Neuroendocrinol. 2008, 29, 507–519. [Google Scholar] [CrossRef] [Green Version]
- Kajta, M.; Beyer, C. Cellular Strategies of Estrogen-Mediated Neuroprotection During Brain Development. Endocrine 2003, 21, 3–10. [Google Scholar] [CrossRef]
- Bonofiglio, D.; Gabriele, S.; Aquila, S.; Catalano, S.; Gentile, M.; Middea, E.; Giordano, F.; Andò, S. Estrogen Receptor α Binds to Peroxisome Proliferator–Activated Receptor Response Element and Negatively Interferes with Peroxisome Proliferator-Activated Receptor γ Signaling in Breast Cancer Cells. Clin. Cancer Res. 2005, 11, 6139–6147. [Google Scholar] [CrossRef] [Green Version]
- Wormke, M.; Stoner, M.; Saville, B.; Walker, K.; Abdelrahim, M.; Burghardt, R.; Safe, S. The Aryl Hydrocarbon Receptor Mediates Degradation of Estrogen Receptor α through Activation of Proteasomes. Mol. Cell. Biol. 2003, 23, 1843–1855. [Google Scholar] [CrossRef] [Green Version]
- Matthews, J.; Gustafsson, J.-Å. Estrogen receptor and aryl hydrocarbon receptor signaling pathways. Nucl. Recept. Signal. 2006, 4, nrs.04016. [Google Scholar] [CrossRef]
- Menasce, L.P.; White, G.R.M.; Harrison, C.J.; Boyle, J.M. Localization of the Estrogen Receptor Locus (ESR) to Chromosome 6q25.1 by FISH and a Simple Post-FISH Banding Technique. Genomics 1993, 17, 263–265. [Google Scholar] [CrossRef]
- Mosselman, S.; Polman, J.; Dijkema, R. ERβ: Identification and characterization of a novel human estrogen receptor. FEBS Lett. 1996, 392, 49–53. [Google Scholar] [CrossRef] [Green Version]
- Kumar, R.; Zakharov, M.N.; Khan, S.H.; Miki, R.; Jang, H.; Toraldo, G.; Singh, R.; Bhasin, S.; Jasuja, R. The Dynamic Structure of the Estrogen Receptor. J. Amino Acids 2011, 2011, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.K.; Torcaso, A.; Asimes, A.; Chung, W.C.J.; Pak, T.R. Structural and functional characteristics of oestrogen receptor β splice variants: Implications for the ageing brain. J. Neuroendocrinol. 2018, 30, e12488. [Google Scholar] [CrossRef] [Green Version]
- Kuiper, G.G.J.M.; Carlsson, B.; Grandien, K.; Enmark, E.; Häggblad, J.; Nilsson, S.; Gustafsson, J.Å. Comparison of the ligand binding specificity and transcript tissue distribution of estrogen receptors and α and β. Endocrinology 1997, 138, 863–870. [Google Scholar] [CrossRef]
- Mizukami, Y. In vivo functions of GPR30/GPER-1, a membrane receptor for estrogen: From discovery to functions in vivo. Endocr. J. 2010, 57, 101–107. [Google Scholar] [CrossRef] [Green Version]
- Amenyogbe, E.; Chen, G.; Wang, Z.; Lu, X.; Lin, M.; Lin, A.Y. A Review on Sex Steroid Hormone Estrogen Receptors in Mammals and Fish. Int. J. Endocrinol. 2020, 2020, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Taylor, A.H.; Al-Azzawi, F. Immunolocalisation of oestrogen receptor beta in human tissues. J. Mol. Endocrinol. 2000, 24, 145–155. [Google Scholar] [CrossRef]
- Mahmoodzadeh, S.; Eder, S.; Nordmeyer, J.; Ehler, E.; Huber, O.; Martus, P.; Weiske, J.; Pregla, R.; Hetzer, R.; Regitz-Zagrosek, V. Estrogen receptor alpha up-regulation and redistribution in human heart failure. FASEB J. 2006, 20, 926–934. [Google Scholar] [CrossRef] [Green Version]
- Lizotte, E.; Grandy, S.A.; Tremblay, A.; Allen, B.G.; Fiset, C. Cellular Physiology Biochemistry and Biochemistr y Expression, Distribution and Regulation of Sex Steroid Hormone Receptors in Mouse Heart. Cell. Physiol. Biochem. 2009, 8, 75–86. [Google Scholar] [CrossRef]
- Grohé, C.; Kahlert, S.; Löbbert, K.; Stimpel, M.; Karas, R.H.; Vetter, H.; Neyses, L. Cardiac myocytes and fibroblasts contain functional estrogen receptors. FEBS Lett. 1997, 416, 107–112. [Google Scholar] [CrossRef]
- Razandi, M.; Pedram, A.; Merchenthaler, I.; Greene, G.L.; Levin, E.R. Plasma membrane estrogen receptors exist and functions as dimers. Mol. Endocrinol. 2004, 18, 2854–2865. [Google Scholar] [CrossRef]
- Hodges, Y.K.; Tung, L.; Yan, X.-D.; Graham, J.D.; Horwitz, K.B.; Horwitz, L.D. Estrogen Receptors α and β. Circulation 2000, 101, 1792–1798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komi, J.; Lassila, O. Nonsteroidal anti-estrogens inhibit the functional differentiation of human monocyte-derived dendritic cells. Blood 2000, 95, 2875–2882. [Google Scholar] [CrossRef] [PubMed]
- Tomicek, N.J.; Miller-Lee, J.L.; Hunter, J.C.; Korzick, D.H. Estrogen Receptor Beta Does Not Influence Ischemic Tolerance in the Aged Female Rat Heart. Cardiovasc. Ther. 2013, 31, 32–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pugach, E.K.; Blenck, C.L.; Dragavon, J.M.; Langer, S.J.; Leinwand, L.A. Estrogen receptor profiling and activity in cardiac myocytes. Mol. Cell. Endocrinol. 2016, 431, 62–70. [Google Scholar] [CrossRef] [Green Version]
- Andersson, S.; Sundberg, M.; Pristovsek, N.; Ibrahim, A.; Jonsson, P.; Katona, B.; Clausson, C.-M.; Zieba, A.; Ramström, M.; Söderberg, O.; et al. Insufficient antibody validation challenges oestrogen receptor beta research. Nat. Commun. 2017, 8, 15840. [Google Scholar] [CrossRef]
- Pelekanou, V.; Kampa, M.; Kiagiadaki, F.; Deli, A.; Theodoropoulos, P.; Agrogiannis, G.; Patsouris, E.; Tsapis, A.; Castanas, E.; Notas, G. Estrogen anti-inflammatory activity on human monocytes is mediated through cross-talk between estrogen receptor ERα36 and GPR30/GPER1. J. Leukoc. Biol. 2016, 99, 333–347. [Google Scholar] [CrossRef]
- Nordmeyer, J.; Eder, S.; Mahmoodzadeh, S.; Martus, P.; Fielitz, J.; Bass, J.; Bethke, N.; Zurbrügg, H.R.; Pregla, R.; Hetzer, R.; et al. Upregulation of myocardial estrogen receptors in human aortic stenosis. Circulation 2004, 110, 3270–3275. [Google Scholar] [CrossRef] [Green Version]
- Broberg, A.M.; Siddiqui, A.J.; Fischer, H.; Grinnemo, K.-H.; Wardell, E.; Andersson, A.B.; Inzunza, J.; Sylvén, C.; Gustafsson, J.Å. Estrogen receptors do not influence angiogenesis after myocardial infarction. Scand. Cardiovasc. J. 2011, 45, 215–222. [Google Scholar] [CrossRef]
- Patel, V.H.; Chen, J.; Ramanjaneya, M.; Karteris, E.; Zachariades, E.; Thomas, P.; Been, M.; Randeva, H.S. G-protein coupled estrogen receptor 1 expression in rat and human heart: Protective role during ischaemic stress. Int. J. Mol. Med. 2010, 26, 193–199. [Google Scholar] [CrossRef]
- Wang, H.; Sun, X.; Chou, J.; Lin, M.; Ferrario, C.M.; Zapata-Sudo, G.; Groban, L. Cardiomyocyte-specific deletion of the G protein-coupled estrogen receptor (GPER) leads to left ventricular dysfunction and adverse remodeling: A sex-specific gene profiling analysis. Biochim. Biophys. Acta 2017, 1863, 1870–1882. [Google Scholar] [CrossRef]
- Wang, H.; Zhao, Z.; Lin, M.; Groban, L. Activation of GPR30 inhibits cardiac fibroblast proliferation. Mol. Cell. Biochem. 2015, 405, 135–148. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Wang, H.; Lin, M.; Groban, L. GPR30 decreases cardiac chymase/angiotensin II by inhibiting local mast cell number. Biochem. Biophys. Res. Commun. 2015, 459, 131–136. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Ma, H.; Barman, S.A.; Liu, A.T.; Sellers, M.; Stallone, J.N.; Prossnitz, E.R.; White, R.E.; Han, G. Activation of G protein-coupled estrogen receptor induces endothelium-independent relaxation of coronary artery smooth muscle. Am. J. Physiol. Metab. 2011, 301, E882–E888. [Google Scholar] [CrossRef] [Green Version]
- Lindsey, S.H.; Cohen, J.A.; Brosnihan, K.B.; Gallagher, P.E.; Chappell, M.C. Chronic treatment with the G protein-coupled receptor 30 agonist G-1 decreases blood pressure in ovariectomized mRen2.Lewis rats. Endocrinology 2009, 150, 3753–3758. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, D.P.; Waters, E.M.; Mermelstein, P.G.; Kramár, E.A.; Shors, T.J.; Liu, F. Rapid estrogen signaling in the brain: Implications for the fine-tuning of neuronal circuitry. J. Neurosci. 2011, 31, 16056–16063. [Google Scholar] [CrossRef] [Green Version]
- Crespo-Castrillo, A.; Arevalo, M.-A. Microglial and Astrocytic Function in Physiological and Pathological Conditions: Estrogenic Modulation. Int. J. Mol. Sci. 2020, 21, 3219. [Google Scholar] [CrossRef]
- Cersosimo, M.G.; Benarroch, E.E. Estrogen actions in the nervous system. Neurology 2015, 85, 263–273. [Google Scholar] [CrossRef]
- Llorente, R.; Marraudino, M.; Carrillo, B.; Bonaldo, B.; Simon-Areces, J.; Abellanas-Pérez, P.; Rivero-Aguilar, M.; Fernandez-Garcia, J.M.; Pinos, H.; Garcia-Segura, L.M.; et al. G Protein-Coupled Estrogen Receptor Immunoreactivity Fluctuates During the Estrous Cycle and Show Sex Differences in the Amygdala and Dorsal Hippocampus. Front. Endocrinol. 2020, 11, 1–11. [Google Scholar] [CrossRef]
- Fuentes, N.; Silveyra, P. Estrogen receptor signaling mechanisms. Adv. Protein Chem. Struct. Biol. 2019, 116, 135–170. [Google Scholar] [CrossRef]
- Björnstroöm, L.; Sjöberg, M. Mechanisms of Estrogen Receptor Signaling: Convergence of Genomic and Nongenomic Actions on Target Genes. Mol. Endocrinol. 2005, 19, 833–842. [Google Scholar] [CrossRef] [Green Version]
- Marino, M.; Galluzzo, P.; Ascenzi, P. Estrogen Signaling Multiple Pathways to Impact Gene Transcription. Curr. Genom. 2006, 7, 497–508. [Google Scholar] [CrossRef] [Green Version]
- Vrtačnik, P.; Ostanek, B.; Mencej-Bedrač, S.; Marc, J. The many faces of estrogen signaling. Biochem. Medica 2014, 24, 329–342. [Google Scholar] [CrossRef] [Green Version]
- Klinge, C.M. Estrogenic control of mitochondrial function and biogenesis. J. Cell. Biochem. 2008, 105, 1342–1351. [Google Scholar] [CrossRef] [Green Version]
- Zárate, S.; Stevnsner, T.; Gredilla, R. Role of estrogen and other sex hormones in brain aging. Neuroprotection and DNA repair. Front. Aging Neurosci. 2017, 9, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.Q.; Eshete, M.; Alworth, W.L.; Yager, J.D. Binding of MCF-7 cell mitochondrial proteins and recombinant human estrogen receptors alpha and beta to human mitochondrial DNA estrogen response elements. J. Cell. Biochem. 2004, 93, 358–373. [Google Scholar] [CrossRef]
- Prossnitz, E.R.; Barton, M. Estrogen biology: New insights into GPER function and clinical opportunities. Mol. Cell. Endocrinol. 2014, 389, 71–83. [Google Scholar] [CrossRef] [Green Version]
- Evans, P.D. Rapid signalling responses via the G protein-coupled estrogen receptor, GPER, in a hippocampal cell line. Steroids 2019, 152, 108487. [Google Scholar] [CrossRef]
- Deschamps, A.M.; Murphy, E. Activation of a novel estrogen receptor, GPER, is cardioprotective in male and female rats. Am. J. Physiol. Circ. Physiol. 2009, 297, H1806–H1813. [Google Scholar] [CrossRef] [Green Version]
- De Francesco, E.M.; Angelone, T.; Pasqua, T.; Pupo, M.; Cerra, M.C.; Maggiolini, M. GPER Mediates Cardiotropic Effects in Spontaneously Hypertensive Rat Hearts. PLoS ONE 2013, 8, e69322. [Google Scholar] [CrossRef] [Green Version]
- Roque, C.; Baltazar, G. G protein-coupled estrogen receptor 1 (GPER) activation triggers different signaling pathways on neurons and astrocytes. Neural Regen. Res. 2019, 14, 2069–2070. [Google Scholar] [CrossRef]
- Westberry, J.M.; Prewitt, A.K.; Wilson, M.E. Epigenetic regulation of the estrogen receptor alpha promoter in the cerebral cortex following ischemia in male and female rats. Neuroscience 2008, 152, 982–989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, H.F.; Hsi, E.; Liao, Y.C.; Chhor, B.; Hung, J.; Juo, S.H.H.; Lin, R.T.; Chiariotti, L. Demethylation of circulating estrogen receptor alpha gene in cerebral ischemic stroke. PLoS ONE 2015, 10, e0139608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhai, P.; Eurell, T.E.; Cotthaus, R.; Jeffery, E.H.; Bahr, J.M.; Gross, D.R. Effect of estrogen on global myocardial ischemia-reperfusion injury in female rats. Am. J. Physiol. Heart Circ. Physiol. 2000, 279, 2766–2775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Crisostomo, P.; Wairiuko, G.M.; Meldrum, D.R. Estrogen receptor-α mediates acute myocardial protection in females. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, 2204–2209. [Google Scholar] [CrossRef] [PubMed]
- Gabel, S.A.; Walker, V.R.; London, R.E.; Steenbergen, C.; Korach, K.S.; Murphy, E. Estrogen receptor beta mediates gender differences in ischemia/reperfusion injury. J. Mol. Cell. Cardiol. 2005, 38, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Wang, Y.; Weil, B.; Abarbanell, A.; Herrmann, J.; Tan, J.; Kelly, M.; Meldrum, D.R. Estrogen receptor β mediates increased activation of PI3K/Akt signaling and improved myocardial function in female hearts following acute ischemia. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 296, 972–978. [Google Scholar] [CrossRef] [Green Version]
- Pelzer, T.; Loza, P.A.A.; Hu, K.; Bayer, B.; Dienesch, C.; Calvillo, L.; Couse, J.F.; Korach, K.S.; Neyses, L.; Ertl, G. Increased mortality and aggravation of heart failure in estrogen receptor-β knockout mice after myocardial infarction. Circulation 2005, 111, 1492–1498. [Google Scholar] [CrossRef] [Green Version]
- Babiker, F.A.; Lips, D.J.; Delvaux, E.; Zandberg, P.; Janssen, B.J.A.; Prinzen, F.; van Eys, G.; Grohé, C.; Doevendans, P.A. Oestrogen modulates cardiac ischaemic remodelling through oestrogen receptor-specific mechanisms. Acta Physiol. 2007, 189, 23–31. [Google Scholar] [CrossRef]
- Luo, T.; Kim, J.K. The Role of Estrogen and Estrogen Receptors on Cardiomyocytes: An Overview. Can. J. Cardiol. 2016, 32, 1017–1025. [Google Scholar] [CrossRef] [Green Version]
- Mahmoodzadeh, S. Cardiomyocyte-specific Estrogen Receptor Alpha Increases Angiogenesis, Lymphangiogenesis and Reduces Fibrosis in the Female Mouse Heart Post-Myocardial Infarction. J. Cell Sci. Ther. 2014, 5, 153. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Z.; Kang, L.; Wang, Z.; Chen, A.; Zhao, Q.; Li, H. 17β-estradiol promotes recovery after myocardial infarction by enhancing homing and angiogenic capacity of bone marrow-derived endothelial progenitor cells through ERα-SDF-1/CXCR4 crosstalking. Acta Biochim. Biophys. Sin. 2018, 50, 1247–1256. [Google Scholar] [CrossRef]
- Hamada, H.; Kim, M.K.; Iwakura, A.; Ii, M.; Thorne, T.; Qin, G.; Asai, J.; Tsutsumi, Y.; Sekiguchi, H.; Silver, M.; et al. Estrogen receptors α and β mediate contribution of bone marrow-derived endothelial progenitor cells to functional recovery after myocardial infarction. Circulation 2006, 114, 2261–2270. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.B.; Guo, C.L. Protective effect and mechanism of estrogen receptor β on myocardial infarction in mice. Exp. Ther. Med. 2017, 14, 1315–1320. [Google Scholar] [CrossRef] [Green Version]
- Schuster, I.; Mahmoodzadeh, S.; Dworatzek, E.; Jaisser, F.; Messaoudi, S.; Morano, I.; Regitz-Zagrosek, V. Cardiomyocyte-specific overexpression of oestrogen receptor β improves survival and cardiac function after myocardial infarction in female and male mice. Clin. Sci. 2016, 130, 365–376. [Google Scholar] [CrossRef] [Green Version]
- Booth, E.A.; Obeid, N.R.; Lucchesi, B.R. Activation of estrogen receptor-α protects the in vivo rabbit heart from ischemia-reperfusion injury. Am. J. Physiol. Circ. Physiol. 2005, 289, H2039–H2047. [Google Scholar] [CrossRef] [Green Version]
- Novotny, J.L.; Simpson, A.M.; Tomicek, N.J.; Lancaster, T.S.; Korzick, D.H. Rapid estrogen receptor-α activation improves ischemic tolerance in aged female rats through a novel protein kinase Cε -dependent mechanism. Endocrinology 2009, 150, 889–896. [Google Scholar] [CrossRef] [Green Version]
- Bulut, E.C.; Abueid, L.; Ercan, F.; Süleymanoğlu, S.; Ağırbaşlı, M.; Yeğen, B.Ç. Treatment with oestrogen-receptor agonists or oxytocin in conjunction with exercise protects against myocardial infarction in ovariectomized rats. Exp. Physiol. 2016, 101, 612–627. [Google Scholar] [CrossRef]
- Lee, T.; Lin, S.; Chang, N. Both GPER and membrane oestrogen receptor-α activation protect ventricular remodelling in 17β oestradiol-treated ovariectomized infarcted rats. J. Cell. Mol. Med. 2014, 18, 2454–2465. [Google Scholar] [CrossRef]
- Jeanes, H.L.; Tabor, C.; Black, D.; Ederveen, A.; Gray, G.A. Oestrogen-mediated cardioprotection following ischaemia and reperfusion is mimicked by an oestrogen receptor (ER)α agonist and unaffected by an ERβ antagonist. J. Endocrinol. 2008, 197, 493–501. [Google Scholar] [CrossRef]
- Nikolic, I.; Liu, D.; Bell, J.A.; Collins, J.; Steenbergen, C.; Murphy, E. Treatment with an estrogen receptor-beta-selective agonist is cardioprotective. J. Mol. Cell. Cardiol. 2007, 42, 769–780. [Google Scholar] [CrossRef]
- Lin, J.; Steenbergen, C.; Murphy, E.; Sun, J. Estrogen receptor-beta activation results in S-nitrosylation of proteins involved in cardioprotection. Circulation 2009, 120, 245–254. [Google Scholar] [CrossRef] [Green Version]
- Du, M.; Shan, J.; Feng, A.; Schmull, S.; Gu, J.; Xue, S. Oestrogen Receptor β Activation Protects Against Myocardial Infarction via Notch1 Signalling. Cardiovasc. Drugs Ther. 2020, 34, 165–178. [Google Scholar] [CrossRef]
- Martinkovich, S.; Shah, D.; Planey, S.L.; Arnott, J.A. Selective estrogen receptor modulators: Tissue specificity and clinical utility. Clin. Interv. Aging 2014, 9, 1437–1452. [Google Scholar] [CrossRef] [Green Version]
- Shi, W.; Ma, H.; Liu, T.; Yan, D.; Luo, P.; Zhai, M.; Tao, J.; Huo, S.; Guo, J.; Li, C.; et al. Inhibition of Interleukin-6/glycoprotein 130 signalling by Bazedoxifene ameliorates cardiac remodelling in pressure overload mice. J. Cell. Mol. Med. 2020, 24, 4748–4761. [Google Scholar] [CrossRef] [Green Version]
- Posa, A.; Szabó, R.; Kupai, K.; Berkó, A.M.; Veszelka, M.; Szucs, G.; Börzsei, D.; Gyöngyösi, M.; Pávó, I.; Deim, Z.; et al. Cardioprotective Effect of Selective Estrogen Receptor Modulator Raloxifene Are Mediated by Heme Oxygenase in Estrogen-Deficient Rat. Oxid. Med. Cell. Longev. 2017, 2017, 2176749. [Google Scholar] [CrossRef] [Green Version]
- Lamas, A.Z.; Nascimento, A.M.; Medeiros, A.R.S.; Caliman, I.F.; Dalpiaz, P.L.M.; Firmes, L.B.; Sousa, G.J.; Oliveira, P.W.C.; Andrade, T.U.; Reis, A.M.; et al. The selective estrogen receptor modulators (SERMs) raloxifene and tamoxifen improve ANP levels and decrease nuclear translocation of NF-kB in estrogen-deficient rats. Pharmacol. Rep. 2017, 69, 798–805. [Google Scholar] [CrossRef]
- Smith, C.L.; Santen, R.J.; Komm, B.; Mirkin, S. Breast-related effects of selective estrogen receptor modulators and tissue-selective estrogen complexes. Breast Cancer Res. 2014, 16, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Rayabarapu, N.; Patel, B.M. Beneficial role of tamoxifen in isoproterenol-induced myocardial infarction. Can. J. Physiol. Pharmacol. 2014, 92, 849–857. [Google Scholar] [CrossRef]
- Chung, M.-T.; Cheng, P.-Y.; Lam, K.-K.; Chen, S.-Y.; Ting, Y.-F.; Yen, M.-H.; Lee, Y.-M. Cardioprotective effects of long-term treatment with raloxifene, a selective estrogen receptor modulator, on myocardial ischemia/reperfusion injury in ovariectomized rats. Menopause 2010, 17, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Bopassa, J.C.; Eghbali, M.; Toro, L.; Stefani, E. A novel estrogen receptor GPER inhibits mitochondria permeability transition pore opening and protects the heart against ischemia-reperfusion injury. Am. J. Physiol. Circ. Physiol. 2010, 298, H16–H23. [Google Scholar] [CrossRef] [Green Version]
- Kabir, M.E.; Singh, H.; Lu, R.; Olde, B.; Leeb-Lundberg, L.M.F.; Bopassa, J.C. G protein-coupled estrogen receptor 1 mediates acute estrogen-induced cardioprotection via MEK/ERK/GSK-3β Pathway after Ischemia/Reperfusion. PLoS ONE 2015, 10, e0135988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, S.Y.; Davidson, S.M.; Hausenloy, D.J.; Yellon, D.M. Preconditioning and postconditioning: The essential role of the mitochondrial permeability transition pore. Cardiovasc. Res. 2007, 75, 530–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Lu, L.; Tan, Y.; Jiang, L.; Zhao, M.; Gao, E.; Yu, S.; Liu, J. GPR 30 reduces myocardial infarct area and fibrosis in female ovariectomized mice by activating the PI3K/AKT pathway. Life Sci. 2019, 226, 22–32. [Google Scholar] [CrossRef]
- Azizian, H.; Khaksari, M.; Asadikaram, G.; Sepehri, G.; Najafipour, H. Therapeutic effects of tamoxifen on metabolic parameters and cytokines modulation in rat model of postmenopausal diabetic cardiovascular dysfunction: Role of classic estrogen receptors. Int. Immunopharmacol. 2018, 65, 190–198. [Google Scholar] [CrossRef]
- Feng, Y.; Madungwe, N.B.; da Cruz Junho, C.V.; Bopassa, J.C. Activation of G protein-coupled oestrogen receptor 1 at the onset of reperfusion protects the myocardium against ischemia/reperfusion injury by reducing mitochondrial dysfunction and mitophagy. Br. J. Pharmacol. 2017, 174, 4329–4344. [Google Scholar] [CrossRef]
- Rocca, C.; Femminò, S.; Aquila, G.; Granieri, M.C.; de Francesco, E.M.; Pasqua, T.; Rigiracciolo, D.C.; Fortini, F.; Cerra, M.C.; Maggiolini, M.; et al. Notch1 mediates preconditioning protection induced by GPER in normotensive and hypertensive female rat hearts. Front. Physiol. 2018, 9, 521. [Google Scholar] [CrossRef]
- McEwen, B.S.; Alves, S.E. Estrogen Actions in the Central Nervous System 1. Endocr. Rev. 1999, 20, 279–307. [Google Scholar] [CrossRef]
- Raghava, N.; Das, B.C.; Ray, S.K. Neuroprotective effects of estrogen in CNS injuries: Insights from animal models. Neurosci. Neuroecon. 2017, 6, 15–29. [Google Scholar] [CrossRef] [Green Version]
- Habib, P.; Dreymueller, D.; Ludwig, A.; Beyer, C.; Dang, J. Sex steroid hormone-mediated functional regulation of microglia-like BV-2 cells during hypoxia. J. Steroid Biochem. Mol. Biol. 2013, 138, 195–205. [Google Scholar] [CrossRef]
- Dimayuga, F.O.; Reed, J.L.; Carnero, G.A.; Wang, C.; Dimayuga, E.R.; Dimayuga, V.M.; Perger, A.; Wilson, M.E.; Keller, J.N.; Bruce-Keller, A.J. Estrogen and brain inflammation: Effects on microglial expression of MHC, costimulatory molecules and cytokines. J. Neuroimmunol. 2005, 161, 123–136. [Google Scholar] [CrossRef]
- Sampei, K.; Goto, S.; Alkayed, N.J.; Crain, B.J.; Korach, K.S.; Traystman, R.J.; Demas, G.E.; Nelson, R.J.; Hurn, P.D. Stroke in Estrogen Receptor-α–Deficient Mice. Stroke 2000, 31, 738–744. [Google Scholar] [CrossRef] [Green Version]
- Dubal, D.B. Estrogen receptor alpha, not beta, is a critical link in estradiol-mediated protection against brain injury. Proc. Natl. Acad. Sci. USA 2001, 98, 1952–1957. [Google Scholar] [CrossRef]
- Dubal, D.B.; Rau, S.W.; Shughrue, P.J.; Zhu, H.; Yu, J.; Cashion, A.B.; Suzuki, S.; Gerhold, L.M.; Bottner, M.B.; Dubal, S.B.; et al. Differential Modulation of Estrogen Receptors (ERs) in Ischemic Brain Injury: A Role for ERα in Estradiol-Mediated Protection against Delayed Cell Death. Endocrinology 2006, 147, 3076–3084. [Google Scholar] [CrossRef]
- Miller, N.R.; Jover, T.; Cohen, H.W.; Zukin, R.S.; Etgen, A.M. Estrogen can act via estrogen receptor α and β to protect hippocampal neurons against global ischemia-induced cell death. Endocrinology 2005, 146, 3070–3079. [Google Scholar] [CrossRef] [Green Version]
- Raval, A.P.; Borges-Garcia, R.; Javier Moreno, W.; Perez-Pinzon, M.A.; Bramlett, H. Periodic 17β-Estradiol Pretreatment Protects Rat Brain from Cerebral Ischemic Damage via Estrogen Receptor-β. PLoS ONE 2013, 8, e60716. [Google Scholar] [CrossRef] [Green Version]
- Shin, J.A.; Yoon, J.C.; Kim, M.; Park, E.M. Activation of classical estrogen receptor subtypes reduces tight junction disruption of brain endothelial cells under ischemia/reperfusion injury. Free Radic. Biol. Med. 2016, 92, 78–89. [Google Scholar] [CrossRef]
- Carswell, H.V.O.; Macrae, I.M.; Gallagher, L.; Harrop, E.; Horsburgh, K.J. Neuroprotection by a selective estrogen receptor β agonist in a mouse model of global ischemia. Am. J. Physiol. Circ. Physiol. 2004, 287, H1501–H1504. [Google Scholar] [CrossRef]
- Noppens, R.R.; Kofler, J.; Grafe, M.R.; Hurn, P.D.; Traystman, R.J. Estradiol after Cardiac Arrest and Cardiopulmonary Resuscitation is Neuroprotective and Mediated through Estrogen Receptor-β. J. Cereb. Blood Flow Metab. 2009, 29, 277–286. [Google Scholar] [CrossRef]
- Guo, J.; Zhang, T.; Yu, J.; Li, H.-Z.; Zhao, C.; Qiu, J.; Zhao, B.; Zhao, J.; Li, W.; Zhao, T.-Z. Neuroprotective effects of a chromatin modifier on ischemia/reperfusion neurons: Implication of its regulation of BCL2 transactivation by ERα signaling. Cell Tissue Res. 2016, 364, 475–488. [Google Scholar] [CrossRef]
- Zhang, Z.L.; Qin, P.; Liu, Y.; Zhang, L.X.; Guo, H.; Deng, Y.L.; Yizhao, L.; Hou, Y.S.; Wang, L.Y.; Miao, Y.; et al. Alleviation of ischaemia-reperfusion injury by endogenous estrogen involves maintaining Bcl-2 expression via the ERα signalling pathway. Brain Res. 2017, 1661, 15–23. [Google Scholar] [CrossRef]
- Stary, C.M.; Xu, L.; Li, L.; Sun, X.; Ouyang, Y.-B.; Xiong, X.; Zhao, J.; Giffard, R.G. Inhibition of miR-181a protects female mice from transient focal cerebral ischemia by targeting astrocyte estrogen receptor-α. Mol. Cell. Neurosci. 2017, 82, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Yang, J.; Liu, M.; Wang, L.; Hou, W.; Zhang, L.; Ma, Y. Selective activation of estrogen receptor β alleviates cerebral ischemia neuroinflammatory injury. Brain Res. 2020, 1726, 146536. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Guo, H.; Zhang, L.; Tao, L.; Yin, A.; Liu, Z.; Li, Y.; Dong, H.; Xiong, L.; Hou, W. Estrogen replacement therapy-induced neuroprotection against brain ischemia-reperfusion injury involves the activation of astrocytes via estrogen receptor β. Sci. Rep. 2016, 6, 21467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madinier, A.; Wieloch, T.; Olsson, R.; Ruscher, K. Impact of estrogen receptor beta activation on functional recovery after experimental stroke. Behav. Brain Res. 2014, 261, 282–288. [Google Scholar] [CrossRef]
- Shin, J.A.; Yang, S.J.; Jeong, S.I.; Park, H.J.; Choi, Y.H.; Park, E.M. Activation of estrogen receptor β reduces blood-brain barrier breakdown following ischemic injury. Neuroscience 2013, 235, 165–173. [Google Scholar] [CrossRef]
- Kimelberg, H.K.; Feustel, P.J.; Jin, Y.; Paquette, J.; Boulos, A.; Keller, J.; Tranmer, B.I. Acute treatment with tamoxifen reduces ischemic damage following middle cerebral artery occlusion. Neuroreport 2000, 11, 2675–2679. [Google Scholar] [CrossRef]
- Kimelberg, H.K.; Jin, Y.; Charniga, C.; Feustel, P.J. Neuroprotective activity of tamoxifen in permanent focal ischemia. J. Neurosurg. 2003, 99, 138–142. [Google Scholar] [CrossRef]
- Wakade, C.; Khan, M.M.; de Sevilla, L.M.; Zhang, Q.G.; Mahesh, V.B.; Brann, D.W. Tamoxifen neuroprotection in cerebral ischemia involves attenuation of kinase activation and superoxide production and potentiation of mitochondrial superoxide dismutase. Endocrinology 2008, 149, 367–379. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Milatovic, D.; Aschner, M.; Feustel, P.J.; Kimelberg, H.K. Neuroprotection by tamoxifen in focal cerebral ischemia is not mediated by an agonist action at estrogen receptors but is associated with antioxidant activity. Exp. Neurol. 2007, 204, 819–827. [Google Scholar] [CrossRef] [Green Version]
- Zou, W.; Fang, C.; Ji, X.; Liang, X.; Liu, Y.; Han, C.; Huang, L.; Zhang, Q.; Li, H.; Zhang, Y.; et al. Estrogen receptor (ER)-aα36 is involved in estrogen- and tamoxifen-induced neuroprotective effects in ischemic stroke models. PLoS ONE 2015, 10, e0140660. [Google Scholar] [CrossRef] [Green Version]
- Rzemieniec, J.; Litwa, E.; Wnuk, A.; Lason, W.; Gołas, A.; Krzeptowski, W.; Kajta, M. Neuroprotective action of raloxifene against hypoxia-induced damage in mouse hippocampal cells depends on ERα but not ERβ or GPR30 signalling. J. Steroid Biochem. Mol. Biol. 2015, 146, 26–37. [Google Scholar] [CrossRef]
- Rzemieniec, J.; Litwa, E.; Wnuk, A.; Lason, W.; Kajta, M. Bazedoxifene and raloxifene protect neocortical neurons undergoing hypoxia via targeting ERα and PPAR-γ. Mol. Cell. Endocrinol. 2018, 461, 64–78. [Google Scholar] [CrossRef]
- Khan, M.M.; Wakade, C.; de Sevilla, L.; Brann, D.W. Selective estrogen receptor modulators (SERMs) enhance neurogenesis and spine density following focal cerebral ischemia. J. Steroid Biochem. Mol. Biol. 2015, 146, 38–47. [Google Scholar] [CrossRef] [Green Version]
- Castelló-Ruiz, M.; Torregrosa, G.; Burguete, M.C.; Miranda, F.J.; Centeno, J.M.; López-Morales, M.A.; Gasull, T.; Alborch, E. The selective estrogen receptor modulator, bazedoxifene, reduces ischemic brain damage in male rat. Neurosci. Lett. 2014, 575, 53–57. [Google Scholar] [CrossRef]
- Jover-Mengual, T.; Castelló-Ruiz, M.; Burguete, M.C.; Jorques, M.; López-Morales, M.A.; Aliena-Valero, A.; Jurado-Rodríguez, A.; Pérez, S.; Centeno, J.M.; Miranda, F.J.; et al. Molecular mechanisms mediating the neuroprotective role of the selective estrogen receptor modulator, bazedoxifene, in acute ischemic stroke: A comparative study with 17β-estradiol. J. Steroid Biochem. Mol. Biol. 2017, 171, 296–304. [Google Scholar] [CrossRef]
- Burguete, M.C.; Jover-Mengual, T.; López-Morales, M.A.; Aliena-Valero, A.; Jorques, M.; Torregrosa, G.; Alborch, E.; Castelló-Ruiz, M.; Salom, J.B. The selective oestrogen receptor modulator, bazedoxifene, mimics the neuroprotective effect of 17β-oestradiol in diabetic ischaemic stroke by modulating oestrogen receptor expression and the MAPK/ERK1/2 signalling pathway. J. Neuroendocrinol. 2019, 31, e12751. [Google Scholar] [CrossRef]
- Zhang, B.; Subramanian, S.; Dziennis, S.; Jia, J.; Uchida, M.; Akiyoshi, K.; Migliati, E.; Lewis, A.D.; Vandenbark, A.A.; Offner, H.; et al. Estradiol and G1 Reduce Infarct Size and Improve Immunosuppression after Experimental Stroke. J. Immunol. 2010, 184, 4087–4094. [Google Scholar] [CrossRef] [Green Version]
- Zhao, T.Z.; Ding, Q.; Hu, J.; He, S.M.; Shi, F.; Ma, L.T. GPER expressed on microglia mediates the anti-inflammatory effect of estradiol in ischemic stroke. Brain Behav. 2016, 6, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Qin, P.; Deng, Y.; Ma, Z.; Guo, H.; Guo, H.; Hou, Y.; Wang, S.; Zou, W.; Sun, Y.; et al. The novel estrogenic receptor GPR30 alleviates ischemic injury by inhibiting TLR4-mediated microglial inflammation. J. Neuroinflamm. 2018, 15, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Bai, N.; Zhang, Q.; Zhang, W.; Liu, B.; Yang, F.; Brann, D.; Wang, R. G-protein-coupled estrogen receptor activation upregulates interleukin-1 receptor antagonist in the hippocampus after global cerebral ischemia: Implications for neuronal self-defense. J. Neuroinflamm. 2020, 17, 45. [Google Scholar] [CrossRef]
- Liu, S.-b.; Zhang, N.; Guo, Y.-y.; Zhao, R.; Shi, T.-y.; Feng, S.-f.; Wang, S.-q.; Yang, Q.; Li, X.-q.; Wu, Y.-m.; et al. G-Protein-Coupled Receptor 30 Mediates Rapid Neuroprotective Effects of Estrogen via Depression of NR2B-Containing NMDA Receptors. J. Neurosci. 2012, 32, 4887–4900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamprecht, M.R.; Morrison, B. GPR30 activation is neither necessary nor sufficient for acute neuroprotection by 17β-estradiol after an ischemic injury in organotypic hippocampal slice cultures. Brain Res. 2014, 1563, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Murata, T.; Dietrich, H.H.; Xiang, C.; Dacey, R.G. G Protein-Coupled Estrogen Receptor Agonist Improves Cerebral Microvascular Function After Hypoxia/Reoxygenation Injury in Male and Female Rats. Stroke 2013, 44, 779–785. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Qu, Y.; Shi, F.; Feng, D.; Tao, K.; Gao, G.; He, S.; Zhao, T. Activation of G protein-coupled estrogen receptor 1 (GPER-1) ameliorates blood-brain barrier permeability after global cerebral ischemia in ovariectomized rats. Biochem. Biophys. Res. Commun. 2016, 477, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Kosaka, Y.; Quillinan, N.; Bond, C.T.; Traystman, R.J.; Hurn, P.D.; Herson, P.S. GPER1/GPR30 Activation Improves Neuronal Survival Following Global Cerebral Ischemia Induced by Cardiac Arrest in Mice. Transl. Stroke Res. 2012, 3, 500–507. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.S.; Yue, J.; Hu, L.N.; Tian, Z.; Zhang, K.; Yang, L.; Zhang, H.N.; Guo, Y.Y.; Feng, B.; Liu, H.Y.; et al. Activation of G protein-coupled receptor 30 protects neurons by regulating autophagy in astrocytes. Glia 2020, 68, 27–43. [Google Scholar] [CrossRef] [Green Version]
- Broughton, B.R.S.; Brait, V.H.; Kim, H.A.; Lee, S.; Chu, H.X.; Gardiner-Mann, C.V.; Guida, E.; Evans, M.A.; Miller, A.A.; Arumugam, T.V.; et al. Sex-Dependent Effects of G Protein–Coupled Estrogen Receptor Activity on Outcome After Ischemic Stroke. Stroke 2014, 45, 835–841. [Google Scholar] [CrossRef] [Green Version]
- Tyagi, S.; Gupta, P.; Saini, A.; Kaushal, C.; Sharma, S. The peroxisome proliferator-activated receptor: A family of nuclear receptors role in various diseases. J. Adv. Pharm. Technol. Res. 2011, 2, 236–240. [Google Scholar] [CrossRef]
- Dong, C.; Zhou, H.; Shen, C.; Yu, L.-G.; Ding, Y.; Zhang, Y.-H.; Guo, Z.-R. Role of peroxisome proliferator-activated receptors gene polymorphisms in type 2 diabetes and metabolic syndrome. World J. Diabetes 2015, 6, 654. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.-S.; Kim, J. Peroxisome Proliferator-Activated Receptors and the Heart: Lessons from the Past and Future Directions. PPAR Res. 2015, 2015, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Higashiyama, H.; Billin, A.N.; Okamoto, Y.; Kinoshita, M.; Asano, S. Expression profiling of Peroxisome proliferator-activated receptor-delta (PPAR-delta) in mouse tissues using tissue microarray. Histochem. Cell Biol. 2007, 127, 485–494. [Google Scholar] [CrossRef]
- Rosen, E.D.; Spiegelman, B.M. PPARγ: A Nuclear Regulator of Metabolism, Differentiation, and Cell Growth. J. Biol. Chem. 2001, 276, 37731–37734. [Google Scholar] [CrossRef] [Green Version]
- Madrazo, J.A.; Kelly, D.P. The PPAR trio: Regulators of myocardial energy metabolism in health and disease. J. Mol. Cell. Cardiol. 2008, 44, 968–975. [Google Scholar] [CrossRef]
- Son, N.H.; Park, T.S.; Yamashita, H.; Yokoyama, M.; Huggins, L.A.; Okajima, K.; Homma, S.; Szabolcs, M.J.; Huang, L.S.; Goldberg, I.J. Cardiomyocyte expression of PPARγ leads to cardiac dysfunction in mice. J. Clin. Investig. 2007, 117, 2791–2801. [Google Scholar] [CrossRef] [Green Version]
- Barger, P.M.; Brandt, J.M.; Leone, T.C.; Weinheimer, C.J.; Kelly, D.P. Deactivation of peroxisome proliferator-activated receptor-α during cardiac hypertrophic growth. J. Clin. Investig. 2000, 105, 1723–1730. [Google Scholar] [CrossRef] [Green Version]
- Castiglioni, L.; Pignieri, A.; Fiaschè, M.; Giudici, M.; Crestani, M.; Mitro, N.; Abbate, M.; Zoja, C.; Rottoli, D.; Foray, C.; et al. Fenofibrate attenuates cardiac and renal alterations in young salt-loaded spontaneously hypertensive stroke-prone rats through mitochondrial protection. J. Hypertens. 2018, 36, 1129–1146. [Google Scholar] [CrossRef]
- Huss, J.M.; Levy, F.H.; Kelly, D.P. Hypoxia inhibits the peroxisome proliferator-activated receptor α/retinoid X receptor gene regulatory pathway in cardiac myocytes: A mechanism for O2-dependent modulation of mitochondrial fatty acid oxidation. J. Biol. Chem. 2001, 276, 27605–27612. [Google Scholar] [CrossRef] [Green Version]
- Finck, B.N.; Lehman, J.J.; Leone, T.C.; Welch, M.J.; Bennett, M.J.; Kovacs, A.; Han, X.; Gross, R.W.; Kozak, R.; Lopaschuk, G.D.; et al. The cardiac phenotype induced by PPARα overexpression mimics that caused by diabetes mellitus. J. Clin. Investig. 2002, 109, 121–130. [Google Scholar] [CrossRef]
- Yu, B.C.; Chang, C.K.; Ou, H.Y.; Cheng, K.C.; Cheng, J.T. Decrease of peroxisome proliferator-activated receptor delta expression in cardiomyopathy of streptozotocin-induced diabetic rats. Cardiovasc. Res. 2008, 80, 78–87. [Google Scholar] [CrossRef] [Green Version]
- Fliegner, D.; Westermann, D.; Riad, A.; Schubert, C.; Becher, E.; Fielitz, J.; Tschöpe, C.; Regitz-Zagrosek, V. Up-regulation of PPARγ in myocardial infarction. Eur. J. Heart Fail. 2008, 10, 30–38. [Google Scholar] [CrossRef] [Green Version]
- Gilde, A.J.; van der Lee, K.A.J.M.; Willemsen, P.H.M.; Chinetti, G.; van der Leij, F.R.; van der Vusse, G.J.; Staels, B.; van Bilsen, M. Peroxisome Proliferator-Activated Receptor (PPAR) α and PPARβ/δ, but not PPARγ, Modulate the Expression of Genes Involved in Cardiac Lipid Metabolism. Circ. Res. 2003, 92, 518–524. [Google Scholar] [CrossRef] [Green Version]
- Teunissen, B.E.J.; Smeets, P.J.H.; Willemsen, P.H.M.; de Windt, L.J.; van der Vusse, G.J.; van Bilsen, M. Activation of PPARδ inhibits cardiac fibroblast proliferation and the transdifferentiation into myofibroblasts. Cardiovasc. Res. 2007, 75, 519–529. [Google Scholar] [CrossRef] [Green Version]
- Chinetti, G.; Fruchart, J.C.; Staels, B. Peroxisome proliferator-activated receptors (PPARs): Nuclear receptors with functions in the vascular wall. Z. Kardiol. 2001, 90 (Suppl. 3), 125–132. [Google Scholar] [CrossRef]
- Cheang, W.S.; Fang, X.; Tian, X.Y. Pleiotropic Effects of Peroxisome Proliferator-Activated Receptor γ and δ in Vascular Diseases. Circ. J. 2013, 77, 2664–2671. [Google Scholar] [CrossRef] [Green Version]
- Adhikary, T.; Wortmann, A.; Schumann, T.; Finkernagel, F.; Lieber, S.; Roth, K.; Toth, P.M.; Diederich, W.E.; Nist, A.; Stiewe, T.; et al. The transcriptional PPARβ/δ network in human macrophages defines a unique agonist-induced activation state. Nucleic Acids Res. 2015, 43, 5033–5051. [Google Scholar] [CrossRef] [Green Version]
- Cullingford, T.E.; Bhakoo, K.; Peuchen, S.; Dolphin, C.T.; Patel, R.; Clark, J.B. Distribution of mRNAs Encoding the Peroxisome Proliferator-Activated Receptor α, β, and γ and the Retinoid X Receptor α, β, and γ in Rat Central Nervous System. J. Neurochem. 2002, 70, 1366–1375. [Google Scholar] [CrossRef]
- Warden, A.; Truitt, J.; Merriman, M.; Ponomareva, O.; Jameson, K.; Ferguson, L.B.; Mayfield, R.D.; Harris, R.A. Localization of PPAR isotypes in the adult mouse and human brain. Sci. Rep. 2016, 6, 27618. [Google Scholar] [CrossRef]
- Mandrekar-Colucci, S.; Sauerbeck, A.; Popovich, P.G.; McTigue, D.M. PPAR agonists as therapeutics for CNS trauma and neurological diseases. ASN Neuro 2013, 5, e00129. [Google Scholar] [CrossRef] [Green Version]
- Akanuma, S.; Hori, S.; Ohtsuki, S.; Fujiyoshi, M.; Terasaki, T. Expression of nuclear receptor mRNA and liver X receptor-mediated regulation of ABC transporter A1 at rat blood–brain barrier. Neurochem. Int. 2008, 52, 669–674. [Google Scholar] [CrossRef]
- Mysiorek, C.; Culot, M.; Dehouck, L.; Derudas, B.; Staels, B.; Bordet, R.; Cecchelli, R.; Fenart, L.; Berezowski, V. Peroxisome-proliferator-activated receptor-alpha activation protects brain capillary endothelial cells from oxygen-glucose deprivation-induced hyperpermeability in the blood-brain barrier. Curr. Neurovasc. Res. 2009, 6, 181–193. [Google Scholar] [CrossRef]
- Yin, K.J.; Fan, Y.; Hamblin, M.; Zhang, J.; Zhu, T.; Li, S.; Hawse, J.R.; Subramaniam, M.; Song, C.Z.; Urrutia, R.; et al. KLF11 mediates PPARγ cerebrovascular protection in ischaemic stroke. Brain 2013, 136, 1274–1287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, J.; Moller, D.E. The Mechanisms of Action of PPARs. Annu. Rev. Med. 2002, 53, 409–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, J.K.; Viswakarma, N.; Jia, Y.; Bai, L.; Vluggens, A.; Borensztajn, J.; Xu, J. Coactivators in PPAR-regulated gene expression. PPAR Res. 2010, 2010, 250126. [Google Scholar] [CrossRef] [Green Version]
- Adams, M.; Reginato, M.J.; Shao, D.; Lazar, M.A.; Chatterjee, V.K. Transcriptional Activation by Peroxisome Proliferator-activated Receptor γ Is Inhibited by Phosphorylation at a Consensus Mitogen-activated Protein Kinase Site. J. Biol. Chem. 1997, 272, 5128–5132. [Google Scholar] [CrossRef] [Green Version]
- Lazennec, G.; Canaple, L.; Saugy, D.; Wahli, W. Activation of peroxisome proliferator-activated receptors (PPARs) by their ligands and protein kinase A activators. Mol. Endocrinol. 2000, 14, 1962–1975. [Google Scholar] [CrossRef]
- Alexis, J.D.; Wang, N.; Che, W.; Lerner-Marmarosh, N.; Sahni, A.; Korshunov, V.A.; Zou, Y.; Ding, B.; Yan, C.; Berk, B.C.; et al. Bcr Kinase Activation by Angiotensin II Inhibits Peroxisome Proliferator-Activated Receptor γ Transcriptional Activity in Vascular Smooth Muscle Cells. Circ. Res. 2009, 104, 69–78. [Google Scholar] [CrossRef] [Green Version]
- Ricote, M.; Glass, C.K. PPARs and molecular mechanisms of transrepression. Biochim. Biophys. Acta 2007, 1771, 926–935. [Google Scholar] [CrossRef] [Green Version]
- Ghisletti, S.; Huang, W.; Ogawa, S.; Pascual, G.; Lin, M.-E.; Willson, T.M.; Rosenfeld, M.G.; Glass, C.K. Parallel SUMOylation-Dependent Pathways Mediate Gene- and Signal-Specific Transrepression by LXRs and PPARγ. Mol. Cell 2007, 25, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Khalid, A.M.; Hafstad, A.D.; Larsen, T.S.; Severson, D.L.; Boardman, N.; Hagve, M.; Berge, R.K.; Aasum, E. Cardioprotective effect of the PPAR ligand tetradecylthioacetic acid in type 2 diabetic mice. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, 2116–2122. [Google Scholar] [CrossRef] [Green Version]
- Chandra, M.; Miriyala, S.; Panchatcharam, M. PPAR γ and Its Role in Cardiovascular Diseases. PPAR Res. 2017, 2017, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Tokutome, M.; Matoba, T.; Nakano, Y.; Okahara, A.; Fujiwara, M.; Koga, J.I.; Nakano, K.; Tsutsui, H.; Egashira, K. Peroxisome proliferator-activated receptor-gamma targeting nanomedicine promotes cardiac healing after acute myocardial infarction by skewing monocyte/macrophage polarization in preclinical animal models. Cardiovasc. Res. 2019, 115, 419–431. [Google Scholar] [CrossRef]
- Zong, X.; Cheng, K.; Yin, G.; Wu, Z.; Su, Q.; Yu, D.; Liao, P.; Hu, W.; Chen, Y. SIRT3 is a downstream target of PPAR-α implicated in high glucose-induced cardiomyocyte injury in AC16 cells. Exp. Ther. Med. 2020, 20, 1261–1268. [Google Scholar] [CrossRef]
- Yang, M.; Xiong, J.; Zou, Q.; Wang, D.D.; Huang, C.X. Chrysin attenuates interstitial fibrosis and improves cardiac function in a rat model of acute myocardial infarction. J. Mol. Histol. 2018, 49, 555–565. [Google Scholar] [CrossRef]
- Shen, S.; Jiang, H.; Bei, Y.; Zhang, J.; Zhang, H.; Zhu, H.; Zhang, C.; Yao, W.; Wei, C.; Shang, H.; et al. Qiliqiangxin Attenuates Adverse Cardiac Remodeling after Myocardial Infarction in Ovariectomized Mice via Activation of PPARγ. Cell. Physiol. Biochem. 2017, 42, 876–888. [Google Scholar] [CrossRef]
- Mahajan, U.B.; Chandrayan, G.; Patil, C.R.; Arya, D.S.; Suchal, K.; Agrawal, Y.O.; Ojha, S.; Goyal, S.N. The protective effect of apigenin on myocardial injury in diabetic rats mediating activation of the PPAR-γ pathway. Int. J. Mol. Sci. 2017, 18, 756. [Google Scholar] [CrossRef] [Green Version]
- Lv, F.H.; Yin, H.L.; He, Y.Q.; Wu, H.M.; Kong, J.; Chai, X.Y.; Zhang, S.R. Effects of curcumin on the apoptosis of cardiomyocytes and the expression of NF-κB, PPAR-γ and Bcl-2 in rats with myocardial infarction injury. Exp. Ther. Med. 2016, 12, 3877–3884. [Google Scholar] [CrossRef] [Green Version]
- Shen, Z.X.; Yang, Q.Z.; Li, C.; Du, L.J.; Sun, X.N.; Liu, Y.; Sun, J.Y.; Gu, H.H.; Sun, Y.M.; Wang, J.; et al. Myeloid peroxisome proliferator-activated receptor gamma deficiency aggravates myocardial infarction in mice. Atherosclerosis 2018, 274, 199–205. [Google Scholar] [CrossRef]
- El-Gohary, O.A.; Allam, M.M. Effect of vitamin D on isoprenaline-induced myocardial infarction in rats: Possible role of peroxisome proliferator-activated receptor-γ. Can. J. Physiol. Pharmacol. 2017, 95, 641–646. [Google Scholar] [CrossRef]
- Chu, X.; Wang, Y.; Pang, L.; Huang, J.; Sun, X.; Chen, X. miR-130 aggravates acute myocardial infarction-induced myocardial injury by targeting PPAR-γ. J. Cell. Biochem. 2018, 119, 7235–7244. [Google Scholar] [CrossRef]
- Zhu, Z.-D.; Ye, J.-Y.; Niu, H.; Ma, Y.-M.; Fu, X.-M.; Xia, Z.-H.; Zhang, X. Effects of microRNA-292-5p on myocardial ischemia-reperfusion injury through the peroxisome proliferator-activated receptor-α/-γ signaling pathway. Gene Ther. 2018, 25, 234–248. [Google Scholar] [CrossRef]
- Zhou, H.; Li, D.; Zhu, P.; Hu, S.; Hu, N.; Ma, S.; Zhang, Y.; Han, T.; Ren, J.; Cao, F.; et al. Melatonin suppresses platelet activation and function against cardiac ischemia/reperfusion injury via PPARγ/FUNDC1/mitophagy pathways. J. Pineal Res. 2017, 63, 1–18. [Google Scholar] [CrossRef]
- Mokhtari, B.; Azizi, Y.; Abookheili, A.R.; Aboutaleb, N.; Nazarinia, D.; Naderi, N. Human amniotic membrane mesenchymal stem cells-conditioned medium attenuates myocardial ischemia-reperfusion injury in rats by targeting oxidative stress. Iran. J. Basic Med. Sci. 2020, 23, 1453–1461. [Google Scholar] [CrossRef]
- Nasser, M.I.; Qi, X.; Zhu, S.; He, Y.; Zhao, M.; Guo, H.; Zhu, P. Current situation and future of stem cells in cardiovascular medicine. Biomed. Pharmacother. 2020, 132, 110813. [Google Scholar] [CrossRef]
- Hou, J.; Wang, L.; Hou, J.; Guo, T.; Xing, Y.; Zheng, S.; Zhou, C.; Huang, H.; Long, H.; Zhong, T.; et al. Peroxisome Proliferator-Activated Receptor Gamma Promotes Mesenchymal Stem Cells to Express Connexin43 via the Inhibition of TGF-β1/Smads Signaling in a Rat Model of Myocardial Infarction. Stem Cell Rev. Rep. 2015, 11, 885–899. [Google Scholar] [CrossRef]
- Peymani, M.; Ghaedi, K.; Irani, S.; Nasr-Esfahani, M.H. Peroxisome Proliferator-Activated Receptor γ Activity is Required for Appropriate Cardiomyocyte Differentiation. Cell J. 2016, 18, 221–228. [Google Scholar] [CrossRef]
- Khan, V.; Sharma, S.; Bhandari, U.; Sharma, N.; Rishi, V.; Haque, S.E. Suppression of isoproterenol-induced cardiotoxicity in rats by raspberry ketone via activation of peroxisome proliferator activated receptor-α. Eur. J. Pharmacol. 2019, 842, 157–166. [Google Scholar] [CrossRef]
- Song, J.W.; Kim, H.J.; Lee, H.; Kim, J.W.; Kwak, Y.L. Protective effect of peroxisome proliferator-activated receptor α activation against cardiac ischemia-reperfusion injury is related to upregulation of uncoupling protein-3. Oxid. Med. Cell. Longev. 2016, 2016, 3539649. [Google Scholar] [CrossRef] [Green Version]
- Duerr, G.D.; Heinemann, J.C.; Arnoldi, V.; Feisst, A.; Kley, J.; Ghanem, A.; Welz, A.; Dewald, O. Cardiomyocyte specific peroxisome proliferator-activated receptor-α overexpression leads to irreversible damage in ischemic murine heart. Life Sci. 2014, 102, 88–97. [Google Scholar] [CrossRef]
- Wagner, K.D.; Vukolic, A.; Baudouy, D.; Michiels, J.F.; Wagner, N. Inducible conditional vascular-specific overexpression of peroxisome proliferator-activated receptor beta/delta leads to rapid cardiac hypertrophy. PPAR Res. 2016, 2016, 7631085. [Google Scholar] [CrossRef] [Green Version]
- Villapol, S. Roles of Peroxisome Proliferator-Activated Receptor Gamma on Brain and Peripheral Inflammation. Cell. Mol. Neurobiol. 2018, 38, 121–132. [Google Scholar] [CrossRef]
- Wójtowicz, S.; Strosznajder, A.K.; Jeżyna, M.; Strosznajder, J.B. The Novel Role of PPAR Alpha in the Brain: Promising Target in Therapy of Alzheimer’s Disease and Other Neurodegenerative Disorders. Neurochem. Res. 2020, 45, 972–988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strosznajder, A.K.; Wójtowicz, S.; Jeżyna, M.J.; Sun, G.Y.; Strosznajder, J.B. Recent Insights on the Role of PPAR-β/δ in Neuroinflammation and Neurodegeneration, and Its Potential Target for Therapy. Neuromol. Med. 2021, 23, 86–98. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, L.N. Peroxisome proliferator-activated receptor gamma agonists for preventing recurrent stroke and other vascular events in people with stroke or transient ischaemic attack. Cochrane Database Syst. Rev. 2019, 10, CD010693. [Google Scholar] [CrossRef]
- Woo, M.H.; Lee, H.S.; Kim, J. Effect of pioglitazone in acute ischemic stroke patients with diabetes mellitus: A nested case-control study. Cardiovasc. Diabetol. 2019, 18, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liu, S.; Chen, G. Aggravation of Cerebral Ischemia/Reperfusion Injury by Peroxisome Proliferator-Activated Receptor-Gamma Deficiency via Endoplasmic Reticulum Stress. Med. Sci. Monit. 2019, 25, 7518–7526. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Kim, Y.S.; Lee, D.H.; Lee, S.H.; Park, H.J.; Lee, D.; Kim, H. Neuroprotective effects of oleic acid in rodent models of cerebral ischaemia. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, L.; Cai, W.; Mao, L.; Liu, J.; Li, P.; Leak, R.K.; Xu, Y.; Hu, X.; Chen, J. Rosiglitazone Promotes White Matter Integrity and Long-Term Functional Recovery After Focal Cerebral Ischemia. Stroke 2015, 46, 2628–2636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, Y.; He, Q.; Kuang, G.; Jiang, Q.; Yang, J. PPAR-alpha and PPAR-beta expression changes in the hippocampus of rats undergoing global cerebral ischemia/reperfusion due to PPAR-gamma status. Behav. Brain Funct. 2014, 10, 21. [Google Scholar] [CrossRef] [Green Version]
- Guo, T.; Wang, Y.; Guo, Y.; Wu, S.; Chen, W.; Liu, N.; Wang, Y.; Geng, D. 1, 25-D 3 protects from cerebral ischemia by maintaining bbb permeability via PPAR-γ activation. Front. Cell. Neurosci. 2018, 12, 1–9. [Google Scholar] [CrossRef]
- Pan, J.; Jin, J.; Ge, H.; Yin, K.; Chen, X.; Han, L.; Chen, Y.; Qian, L.; Li, X.; Xu, Y. Malibatol A regulates microglia M1/M2 polarization in experimental stroke in a PPARγ-dependent manner. J. Neuroinflamm. 2015, 12, 51. [Google Scholar] [CrossRef] [Green Version]
- Kossatz, E.; Silva-Peña, D.; Suárez, J.; de Fonseca, F.R.; Maldonado, R.; Robledo, P. Octadecylpropyl sulfamide reduces neurodegeneration and restores the memory deficits induced by hypoxia-ischemia in mice. Front. Pharmacol. 2018, 9, 376. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Zhu, Z.Y.; Lu, B.W.; Huang, T.T.; Zhang, Y.M.; Zhou, N.Y.; Xuan, W.; Chen, Z.A.; Wen, D.X.; Yu, W.F.; et al. Rosiglitazone ameliorates tissue plasminogen activator-induced brain hemorrhage after stroke. CNS Neurosci. Ther. 2019, 25, 1343–1352. [Google Scholar] [CrossRef]
- Hasegawa, H.; Yatomi, K.; Mitome-Mishima, Y.; Miyamoto, N.; Tanaka, R.; Oishi, H.; Arai, H.; Hattori, N.; Urabe, T. Pioglitazone Prevents Hemorrhagic Infarction After Transient Focal Ischemia in Type 2 Diabetes. Neurosci. Res. 2021, 170, 314–321. [Google Scholar] [CrossRef]
- Li, Y.Y.; Guo, J.H.; Liu, Y.Q.; Dong, J.H.; Zhu, C.H. PPARγ Activation-Mediated Egr-1 Inhibition Benefits Against Brain Injury in an Experimental Ischaemic Stroke Model. J. Stroke Cerebrovasc. Dis. 2020, 29, 1–6. [Google Scholar] [CrossRef]
- Wu, X.J.; Sun, X.H.; Wang, S.W.; Chen, J.L.; Bi, Y.H.; Jiang, D.X. Mifepristone alleviates cerebral ischemiareperfusion injury in rats by stimulating PPAR γ. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 5688–5696. [Google Scholar] [CrossRef]
- Boujon, V.; Uhlemann, R.; Wegner, S.; Wright, M.B.; Laufs, U.; Endres, M.; Kronenberg, G.; Gertz, K. Dual PPARα/γ agonist aleglitazar confers stroke protection in a model of mild focal brain ischemia in mice. J. Mol. Med. 2019, 97, 1127–1138. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Xu, L.; Zeng, K.; Xu, Z.; Suo, D.; Peng, L.; Ren, T.; Sun, Z.; Yang, W.; Jin, X.; et al. Propane-2-sulfonic acid octadec-9-enyl-amide, a novel PPARα/γ dual agonist, protects against ischemia-induced brain damage in mice by inhibiting inflammatory responses. Brain. Behav. Immun. 2017, 66, 289–301. [Google Scholar] [CrossRef]
- Deng, Y.; Xiong, D.; Yin, C.; Liu, B.; Shi, J.; Gong, Q. Icariside II protects against cerebral ischemia-reperfusion injury in rats via nuclear factor-κB inhibition and peroxisome proliferator-activated receptor up-regulation. Neurochem. Int. 2016, 96, 56–61. [Google Scholar] [CrossRef]
- Xiong, D.; Deng, Y.; Huang, B.; Yin, C.; Liu, B.; Shi, J.; Gong, Q. Icariin attenuates cerebral ischemia-reperfusion injury through inhibition of inflammatory response mediated by NF-κB, PPARα and PPARγ in rats. Int. Immunopharmacol. 2016, 30, 157–162. [Google Scholar] [CrossRef]
- Shehata, A.H.F.; Ahmed, A.S.F.; Abdelrehim, A.B.; Heeba, G.H. The impact of single and combined PPAR-α and PPAR-γ activation on the neurological outcomes following cerebral ischemia reperfusion. Life Sci. 2020, 252, 117679. [Google Scholar] [CrossRef]
- Li, J.; Zhang, K.; Zhang, Q.; Zhou, X.; Wen, L.; Ma, J.; Niu, L.; Li, C. PPAR- γ Mediates Ta-VNS-Induced Angiogenesis and Subsequent Functional Recovery after Experimental Stroke in Rats. Biomed. Res. Int. 2020, 2020, 8163789. [Google Scholar] [CrossRef]
- Jiang, Y.; Li, L.; Liu, B.; Zhang, Y.; Chen, Q.; Li, C. PPARγ upregulation induced by vagus nerve stimulation exerts anti-inflammatory effect in cerebral ischemia/reperfusion rats. Med. Sci. Monit. 2015, 21, 268–275. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Zhang, Y.; Jiang, Y.; Li, L.; Li, C.; Li, J. Electrical stimulation of cerebellar fastigial nucleus protects against cerebral ischemic injury by PPARγ upregulation. Neurol. Res. 2017, 39, 23–29. [Google Scholar] [CrossRef]
- Kinouchi, T.; Kitazato, K.T.; Shimada, K.; Yagi, K.; Tada, Y.; Matsushita, N.; Kurashiki, Y.; Satomi, J.; Sata, M.; Nagahiro, S. Treatment with the PPARγ Agonist Pioglitazone in the Early Post-ischemia Phase Inhibits Pro-inflammatory Responses and Promotes Neurogenesis Via the Activation of Innate- and Bone Marrow-Derived Stem Cells in Rats. Transl. Stroke Res. 2018, 9, 306–316. [Google Scholar] [CrossRef] [Green Version]
- Gamdzyk, M.; Doycheva, D.M.; Malaguit, J.; Enkhjargal, B.; Tang, J.; Zhang, J.H. Role of PPAR-β/δ/miR-17/TXNIP pathway in neuronal apoptosis after neonatal hypoxic-ischemic injury in rats. Neuropharmacology 2018, 140, 150–161. [Google Scholar] [CrossRef]
- Pei, L.; Meng, S.; Yu, W.; Wang, Q.; Song, F.; Ma, L. Inhibition of MicroRNA-383 Ameliorates Injury after Focal Cerebral Ischemia via Targeting PPARγ. Cell. Physiol. Biochem. 2016, 39, 1339–1346. [Google Scholar] [CrossRef] [Green Version]
- Ting, S.-M.; Zhao, X.; Sun, G.; Obertas, L.; Ricote, M.; Aronowski, J. Brain Cleanup as a Potential Target for Poststroke Recovery. Stroke 2020, 51, 958–966. [Google Scholar] [CrossRef]
- Zuo, Y.; Huang, L.; Enkhjargal, B.; Xu, W.; Umut, O.; Travis, Z.D.; Zhang, G.; Tang, J.; Liu, F.; Zhang, J.H. Activation of retinoid X receptor by bexarotene attenuates neuroinflammation via PPARγ/SIRT6/FoxO3a pathway after subarachnoid hemorrhage in rats. J. Neuroinflamm. 2019, 16, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Micka, J.; Milatovich, A.; Menon, A.; Grabowski, G.A.; Puga, A.; Nebert, D.W. Human Ah receptor (AHR) gene: Localization to 7p15 and suggestive correlation of polymorphism with CYP1A1 inducibility. Pharmacogenetics 1997, 7, 95–100. [Google Scholar] [CrossRef]
- Williams, E.G.; Mouchiroud, L.; Frochaux, M.; Pandey, A.; Andreux, P.A.; Deplancke, B.; Auwerx, J. An Evolutionarily Conserved Role for the Aryl Hydrocarbon Receptor in the Regulation of Movement. PLoS Genet. 2014, 10, e1004673. [Google Scholar] [CrossRef] [Green Version]
- Lahvis, G.P.; Pyzalski, R.W.; Glover, E.; Pitot, H.C.; McElwee, M.K.; Bradfield, C.A. The aryl hydrocarbon receptor is required for developmental closure of the ductus venosus in the neonatal mouse. Mol. Pharmacol. 2005, 67, 714–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lund, A.K.; Goens, M.B.; Kanagy, N.L.; Walker, M.K. Cardiac hypertrophy in Aryl hydrocarbon receptor null mice is correlated with elevated angiotensin II, endothelin-1, and mean arterial blood pressure. Toxicol. Appl. Pharmacol. 2003, 193, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Lund, A.K.; Peterson, S.L.; Timmins, G.S.; Walker, M.K. Endothelin-1-Mediated Increase in Reactive Oxygen Species and NADPH Oxidase Activity in Hearts of Aryl Hydrocarbon Receptor (AhR) Null Mice. Toxicol. Sci. 2005, 88, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, Y.; Haarmann-Stemmann, T.; Kado, N.Y.; Vogel, C.F.A. Interleukin 33 Expression Induced by Aryl Hydrocarbon Receptor in Macrophages. Toxicol. Sci. 2019, 170, 404–414. [Google Scholar] [CrossRef]
- Lew, B.J.; Manickam, R.; Lawrence, B.P. Activation of the Aryl hydrocarbon receptor during pregnancy in the mouse alters mammary development through direct effects on stromal and epithelial tissues. Biol. Reprod. 2011, 84, 1094–1102. [Google Scholar] [CrossRef] [Green Version]
- Quintana, F.J.; Basso, A.S.; Iglesias, A.H.; Korn, T.; Farez, M.F.; Bettelli, E.; Caccamo, M.; Oukka, M.; Weiner, H.L. Control of Treg and TH17 cell differentiation by the aryl hydrocarbon receptor. Nature 2008, 453, 65–71. [Google Scholar] [CrossRef]
- Gutiérrez-Vázquez, C.; Quintana, F.J. Regulation of the Immune Response by the Aryl Hydrocarbon Receptor. Immunity 2018, 48, 19–33. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Hisaka, K.; Carreira, V.; Ko, C.I.; Fan, Y.; Zhang, X.; Biesiada, J.; Medvedovic, M.; Puga, A. Ah receptor activation by dioxin disrupts activin, BMP, and WNT signals during the early differentiation of mouse embryonic stem cells and inhibits cardiomyocyte functions. Toxicol. Sci. 2016, 149, 346–357. [Google Scholar] [CrossRef] [Green Version]
- Singh, K.P.; Bennett, J.A.; Casado, F.L.; Walrath, J.L.; Welle, S.L.; Gasiewicz, T.A. Loss of aryl hydrocarbon receptor promotes gene changes associated with premature hematopoietic stem cell exhaustion and development of a myeloproliferative disorder in aging mice. Stem Cells Dev. 2014, 23, 95–106. [Google Scholar] [CrossRef] [Green Version]
- Chevallier, A.; Mialot, A.; Petit, J.M.; Fernandez-Salguero, P.; Barouki, R.; Coumoul, X.; Beraneck, M. Oculomotor Deficits in Aryl Hydrocarbon Receptor Null Mouse. PLoS ONE 2013, 8, e53520. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.C.; Chang, L.H.; Huang, S.S.; Huang, Y.J.; Chih, C.L.; Kuo, H.C.; Lee, Y.H.; Lee, I.H. Aryl hydrocarbon receptor modulates stroke-induced astrogliosis and neurogenesis in the adult mouse brain. J. Neuroinflamm. 2019, 16, 1–13. [Google Scholar] [CrossRef]
- Lin, C.H.; Chen, C.C.; Chou, C.M.; Wang, C.Y.; Hung, C.C.; Chen, J.Y.; Chang, H.W.; Chen, Y.C.; Yeh, G.C.; Lee, Y.H. Knockdown of the aryl hydrocarbon receptor attenuates excitotoxicity and enhances NMDA-induced BDNF expression in cortical neurons. J. Neurochem. 2009, 111, 777–789. [Google Scholar] [CrossRef]
- Dever, D.P.; Adham, Z.O.; Thompson, B.; Genestine, M.; Cherry, J.; Olschowka, J.A.; DiCicco-Bloom, E.; Opanashuk, L.A. Aryl hydrocarbon receptor deletion in cerebellar granule neuron precursors impairs neurogenesis. Dev. Neurobiol. 2016, 76, 533–550. [Google Scholar] [CrossRef]
- Zhu, C.; Chen, Y.L.; Wang, X.J.; Hu, X.S.; Yu, Z.B.; Han, S.P. ShRNA-mediated gene silencing of AHR promotes the differentiation of P19 mouse embryonic carcinoma cells into cardiomyocytes. Mol. Med. Rep. 2012, 6, 513–518. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Chen, J.; Ko, C.I.; Fan, Y.; Carreira, V.; Chen, Y.; Xia, Y.; Medvedovic, M.; Puga, A. Disruption of aryl hydrocarbon receptor homeostatic levels during embryonic stem cell differentiation alters expression of homeobox transcription factors that control cardiomyogenesis. Environ. Health Perspect. 2013, 121, 1334–1343. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Wang, Y.; Ma, Z.; Liang, Q.; Tang, X.; Tan, H.; Xiao, C.; Gao, Y. Ginsenoside Rb1 inhibits doxorubicin-triggered H9C2 cell apoptosis via aryl hydrocarbon receptor. Biomol. Ther. 2017, 25, 202–212. [Google Scholar] [CrossRef] [Green Version]
- Xue, Y.; Shui, X.; Su, W.; He, Y.; Lu, X.; Zhang, Y.; Yan, G.; Huang, S.; Lei, W.; Chen, C. Baicalin inhibits inflammation and attenuates myocardial ischaemic injury by aryl hydrocarbon receptor. J. Pharm. Pharmacol. 2015, 67, 1756–1764. [Google Scholar] [CrossRef]
- Wu, P.Y.; Yu, I.S.; Lin, Y.C.; Chang, Y.T.; Chen, C.C.; Lin, K.H.; Tseng, T.H.; Kargren, M.; Tai, Y.L.; Shen, T.L.; et al. Activation of aryl hydrocarbon receptor by kynurenine impairs progression and metastasis of neuroblastoma. Cancer Res. 2019, 79, 5550–5562. [Google Scholar] [CrossRef] [Green Version]
- Thatcher, T.H.; Williams, M.A.; Pollock, S.J.; Mccarthy, C.E.; Lacy, S.H.; Phipps, R.P.; Sime, P.J. Endogenous ligands of the aryl hydrocarbon receptor regulate lung dendritic cell function. Immunology 2016, 147, 41–54. [Google Scholar] [CrossRef] [Green Version]
- Safe, S.; Jin, U.H.; Park, H.; Chapkin, R.S.; Jayaraman, A. Aryl hydrocarbon receptor (AHR) ligands as selective ahr modulators (SAHRMS). Int. J. Mol. Sci. 2020, 21, 6654. [Google Scholar] [CrossRef]
- Vasquez, A.; Atallah-Yunes, N.; Smith, F.C.; You, X.; Chase, S.E.; Silverstone, A.E.; Vikstrom, K.L. A role for the aryl hydrocarbon receptor in cardiac physiology and function as demonstrated by AhR knockout mice. Cardiovasc. Toxicol. 2003, 3, 153–163. [Google Scholar] [CrossRef]
- Thackaberry, E.A.; Gabaldon, D.M.; Walker, M.K.; Smith, S.M. Aryl hydrocarbon receptor null mice develop cardiac hypertrophy and increased hypoxia-inducible factor-1α in the absence of cardiac hypoxia. Cardiovasc. Toxicol. 2002, 2, 263–273. [Google Scholar] [CrossRef]
- Stegeman, J.J.; Hahn, M.E.; Weisbrod, R.; Woodin, B.R.; Joy, J.S.; Najibi, S.; Cohen, R.A. Induction of cytochrome P4501A1 by aryl hydrocarbon receptor agonists in porcine aorta endothelial cells in culture and cytochrome P4501A1 activity in intact cells. Mol. Pharmacol. 1995, 47, 296–306. [Google Scholar]
- Kerzee, J.K.; Ramos, K.S. Constitutive and inducible expression of Cyp1a1 and Cyp1b1 in vascular smooth muscle cells: Role of the Ahr bHLH/PAS transcription factor. Circ. Res. 2001, 89, 573–582. [Google Scholar] [CrossRef] [Green Version]
- Thum, T.; Borlak, J. Gene expression in distinct regions of the heart. Lancet 2000, 355, 979–983. [Google Scholar] [CrossRef]
- Lighthouse, J.K.; Burke, R.M.; Velasquez, L.S.; Dirkx, R.A.; Aiezza, A.; Moravec, C.S.; Alexis, J.D.; Rosenberg, A.; Small, E.M. Exercise promotes a cardioprotective gene program in resident cardiac fibroblasts. JCI Insight 2019, 4, e92098. [Google Scholar] [CrossRef] [Green Version]
- Goudot, C.; Coillard, A.; Villani, A.-C.; Gueguen, P.; Cros, A.; Sarkizova, S.; Tang-Huau, T.-L.; Bohec, M.; Baulande, S.; Hacohen, N.; et al. Aryl Hydrocarbon Receptor Controls Monocyte Differentiation into Dendritic Cells versus Macrophages. Immunity 2017, 47, 582–596.e6. [Google Scholar] [CrossRef] [Green Version]
- Carreira, V.S.; Fan, Y.; Wang, Q.; Zhang, X.; Kurita, H.; Ko, C.I.; Naticchioni, M.; Jiang, M.; Koch, S.; Medvedovic, M.; et al. Ah receptor signaling controls the expression of cardiac development and homeostasis genes. Toxicol. Sci. 2015, 147, 425–435. [Google Scholar] [CrossRef] [Green Version]
- Kimura, E.; Tohyama, C. Embryonic and postnatal expression of aryl hydrocarbon receptor mRNA in mouse brain. Front. Neuroanat. 2017, 11, 4. [Google Scholar] [CrossRef] [Green Version]
- Filbrandt, C.R.; Wu, Z.; Zlokovic, B.; Opanashuk, L.; Gasiewicz, T.A. Presence and functional activity of the aryl hydrocarbon receptor in isolated murine cerebral vascular endothelial cells and astrocytes. Neurotoxicology 2004, 25, 605–616. [Google Scholar] [CrossRef]
- Lee, Y.H.; Lin, C.H.; Hsu, P.C.; Sun, Y.Y.; Huang, Y.J.; Zhuo, J.H.; Wang, C.Y.; Gan, Y.L.; Hung, C.C.; Kuan, C.Y.; et al. Aryl hydrocarbon receptor mediates both proinflammatory and anti-inflammatory effects in lipopolysaccharide-activated microglia. Glia 2015, 63, 1138–1154. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Fujikawa, M.; Oguro, A.; Itoh, K.; Vogel, C.F.A.; Ishihara, Y. Involvement of the Microglial Aryl Hydrocarbon Receptor in Neuroinflammation and Vasogenic Edema after Ischemic Stroke. Cells 2021, 10, 718. [Google Scholar] [CrossRef] [PubMed]
- Kajta, M.; Wnuk, A.; Rzemieniec, J.; Lason, W.; Mackowiak, M.; Chwastek, E.; Staniszewska, M.; Nehring, I.; Wojtowicz, A.K. Triclocarban Disrupts the Epigenetic Status of Neuronal Cells and Induces AHR/CAR-Mediated Apoptosis. Mol. Neurobiol. 2019, 56, 3113–3131. [Google Scholar] [CrossRef] [PubMed]
- Wnuk, A.; Rzemieniec, J.; Przepiórska, K.; Wesołowska, J.; Wójtowicz, A.K.; Kajta, M. Autophagy-related neurotoxicity is mediated via AHR and CAR in mouse neurons exposed to DDE. Sci. Total Environ. 2020, 742, 140599. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-H.; Juan, S.-H.; Wang, C.Y.; Sun, Y.-Y.; Chou, C.-M.; Chang, S.-F.; Hu, S.-Y.; Lee, W.-S.; Lee, Y.-H. Neuronal activity enhances aryl hydrocarbon receptor-mediated gene expression and dioxin neurotoxicity in cortical neurons. J. Neurochem. 2008, 104, 1415–1429. [Google Scholar] [CrossRef]
- Wu, P.-Y.; Chuang, P.-Y.; Chang, G.-D.; Chan, Y.-Y.; Tsai, T.-C.; Wang, B.-J.; Lin, K.-H.; Hsu, W.-M.; Liao, Y.-F.; Lee, H. Novel Endogenous Ligands of Aryl Hydrocarbon Receptor Mediate Neural Development and Differentiation of Neuroblastoma. ACS Chem. Neurosci. 2019, 10, 4031–4042. [Google Scholar] [CrossRef]
- Ramos-García, N.A.; Orozco-Ibarra, M.; Estudillo, E.; Elizondo, G.; Apo, E.G.; Macías, L.G.C.; Sosa-Ortiz, A.L.; Torres-Ramos, M.A. Aryl hydrocarbon receptor in post-mortem hippocampus and in serum from young, elder, and Alzheimer’s patients. Int. J. Mol. Sci. 2020, 21, 1983. [Google Scholar] [CrossRef] [Green Version]
- Rothhammer, V.; Quintana, F.J. The aryl hydrocarbon receptor: An environmental sensor integrating immune responses in health and disease. Nat. Rev. Immunol. 2019, 19, 184–197. [Google Scholar] [CrossRef]
- Mulero-Navarro, S.; Fernandez-Salguero, P.M. New trends in Aryl hydrocarbon receptor biology. Front. Cell Dev. Biol. 2016, 4, 45. [Google Scholar] [CrossRef] [Green Version]
- Juricek, L.; Coumoul, X. The aryl hydrocarbon receptor and the nervous system. Int. J. Mol. Sci. 2018, 19, 2504. [Google Scholar] [CrossRef] [Green Version]
- Ohtake, F.; Takeyama, K.i.; Matsumoto, T.; Kitagawa, H.; Yamamoto, Y.; Nohara, K.; Tohyama, C.; Krust, A.; Mimura, J.; Chambon, P.; et al. Modulation of oestrogen receptor signalling by association with the activated dioxin receptor. Nature 2003, 423, 545–550. [Google Scholar] [CrossRef]
- Kimura, A.; Naka, T.; Nohara, K.; Fujii-Kuriyama, Y.; Kishimoto, T. Aryl hydrocarbon receptor regulates Stat1 activation and participates in the development of Th17 cells. Proc. Natl. Acad. Sci. USA 2008, 105, 9721–9726. [Google Scholar] [CrossRef] [Green Version]
- Lee, B.J.; Kim, B.; Lee, K. Air pollution exposure and cardiovascular disease. Toxicol. Res. 2014, 30, 71–75. [Google Scholar] [CrossRef]
- Miller, M.R.; Newby, D.E. Air pollution and cardiovascular disease: Car sick. Cardiovasc. Res. 2020, 116, 279–294. [Google Scholar] [CrossRef]
- Marris, C.R.; Kompella, S.N.; Miller, M.R.; Incardona, J.P.; Brette, F.; Hancox, J.C.; Sørhus, E.; Shiels, H.A. Polyaromatic hydrocarbons in pollution: A heart-breaking matter. J. Physiol. 2020, 598, 227–247. [Google Scholar] [CrossRef]
- Ko, C.I.; Fan, Y.; de Gannes, M.; Wang, Q.; Xia, Y.; Puga, A. Repression of the Aryl Hydrocarbon Receptor Is Required to Maintain Mitotic Progression and Prevent Loss of Pluripotency of Embryonic Stem Cells. Stem Cells 2016, 34, 2825–2839. [Google Scholar] [CrossRef] [Green Version]
- Vilahur, G.; Cubedo, J.; Casani, L.; Padro, T.; Sabate-Tenas, M.; Badimon, J.J.; Badimon, L. Reperfusion-triggered stress protein response in the myocardium is blocked by post-conditioning. Systems biology pathway analysis highlights the key role of the canonical aryl-hydrocarbon receptor pathway. Eur. Heart J. 2013, 34, 2082–2093. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Li, Y.; Sun, R.; Zhou, S.; Li, M.; Feng, M.; Xie, Y. Dual character of flavonoids in attenuating and aggravating ischemia-reperfusion-induced myocardial injury. Exp. Ther. Med. 2017, 14, 1307–1314. [Google Scholar] [CrossRef] [Green Version]
- Cuartero, M.I.; Ballesteros, I.; de la Parra, J.; Harkin, A.L.; Abautret-Daly, A.; Sherwin, E.; Fernández-Salguero, P.; Corbí, Á.L.; Lizasoain, I.; Moro, M.A. L-kynurenine/aryl hydrocarbon receptor pathway mediates brain damage after experimental stroke. Circulation 2014, 130, 2040–2051. [Google Scholar] [CrossRef] [Green Version]
- Kwon, J.I.; Heo, H.; Ham, S.J.; Chae, Y.J.; Lee, D.W.; Kim, S.T.; Min, J.; Sung, Y.S.; Kim, K.W.; Choi, Y.; et al. Aryl hydrocarbon receptor antagonism before reperfusion attenuates cerebral ischaemia/reperfusion injury in rats. Sci. Rep. 2020, 10, 14906. [Google Scholar] [CrossRef]
- Rzemieniec, J.; Litwa, E.; Wnuk, A.; Lason, W.; Krzeptowski, W.; Kajta, M. Selective Aryl Hydrocarbon Receptor Modulator 3,3′-Diindolylmethane Impairs AhR and ARNT Signaling and Protects Mouse Neuronal Cells Against Hypoxia. Mol. Neurobiol. 2016, 53, 5591–5606. [Google Scholar] [CrossRef]
- Rzemieniec, J.; Wnuk, A.; Lasoń, W.; Bilecki, W.; Kajta, M. The neuroprotective action of 3,3′-diindolylmethane against ischemia involves an inhibition of apoptosis and autophagy that depends on HDAC and AhR/CYP1A1 but not ERα/CYP19A1 signaling. Apoptosis 2019, 24, 435–452. [Google Scholar] [CrossRef] [Green Version]
- Rzemieniec, J.; Bratek, E.; Wnuk, A.; Przepiórska, K.; Salińska, E.; Kajta, M. Neuroprotective effect of 3,3’-Diindolylmethane against perinatal asphyxia involves inhibition of the AhR and NMDA signaling and hypermethylation of specific genes. Apoptosis 2020, 25, 747–762. [Google Scholar] [CrossRef]
- Matsumoto, K.; Kinoshita, K.; Yoshimizu, A.; Kurauchi, Y.; Hisatsune, A.; Seki, T.; Katsuki, H. Laquinimod and 3,3′-diindolylemethane alleviate neuropathological events and neurological deficits in a mouse model of intracerebral hemorrhage. J. Neuroimmunol. 2020, 342, 577195. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rzemieniec, J.; Castiglioni, L.; Gelosa, P.; Muluhie, M.; Mercuriali, B.; Sironi, L. Nuclear Receptors in Myocardial and Cerebral Ischemia—Mechanisms of Action and Therapeutic Strategies. Int. J. Mol. Sci. 2021, 22, 12326. https://doi.org/10.3390/ijms222212326
Rzemieniec J, Castiglioni L, Gelosa P, Muluhie M, Mercuriali B, Sironi L. Nuclear Receptors in Myocardial and Cerebral Ischemia—Mechanisms of Action and Therapeutic Strategies. International Journal of Molecular Sciences. 2021; 22(22):12326. https://doi.org/10.3390/ijms222212326
Chicago/Turabian StyleRzemieniec, Joanna, Laura Castiglioni, Paolo Gelosa, Majeda Muluhie, Benedetta Mercuriali, and Luigi Sironi. 2021. "Nuclear Receptors in Myocardial and Cerebral Ischemia—Mechanisms of Action and Therapeutic Strategies" International Journal of Molecular Sciences 22, no. 22: 12326. https://doi.org/10.3390/ijms222212326