
Differential Expression Patterns of TDP-43 in Single Moderate versus Repetitive Mild Traumatic Brain Injury in Mice


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
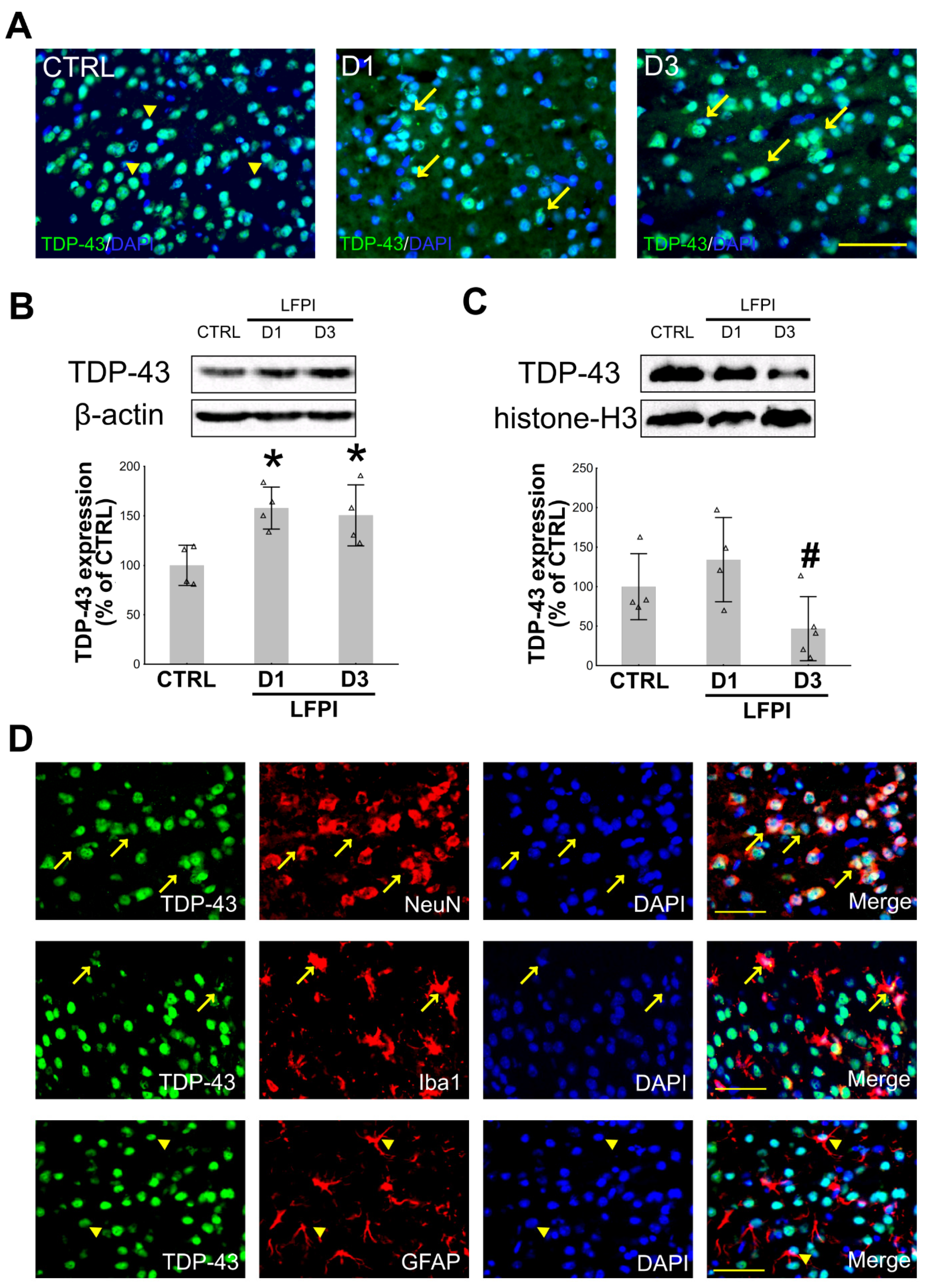
2.1. Single Moderate Traumatic Brain Injury Causes Changes in the TDP-43 Expression Pattern in the Cortical Neurons and Microglia of the Injured Mice
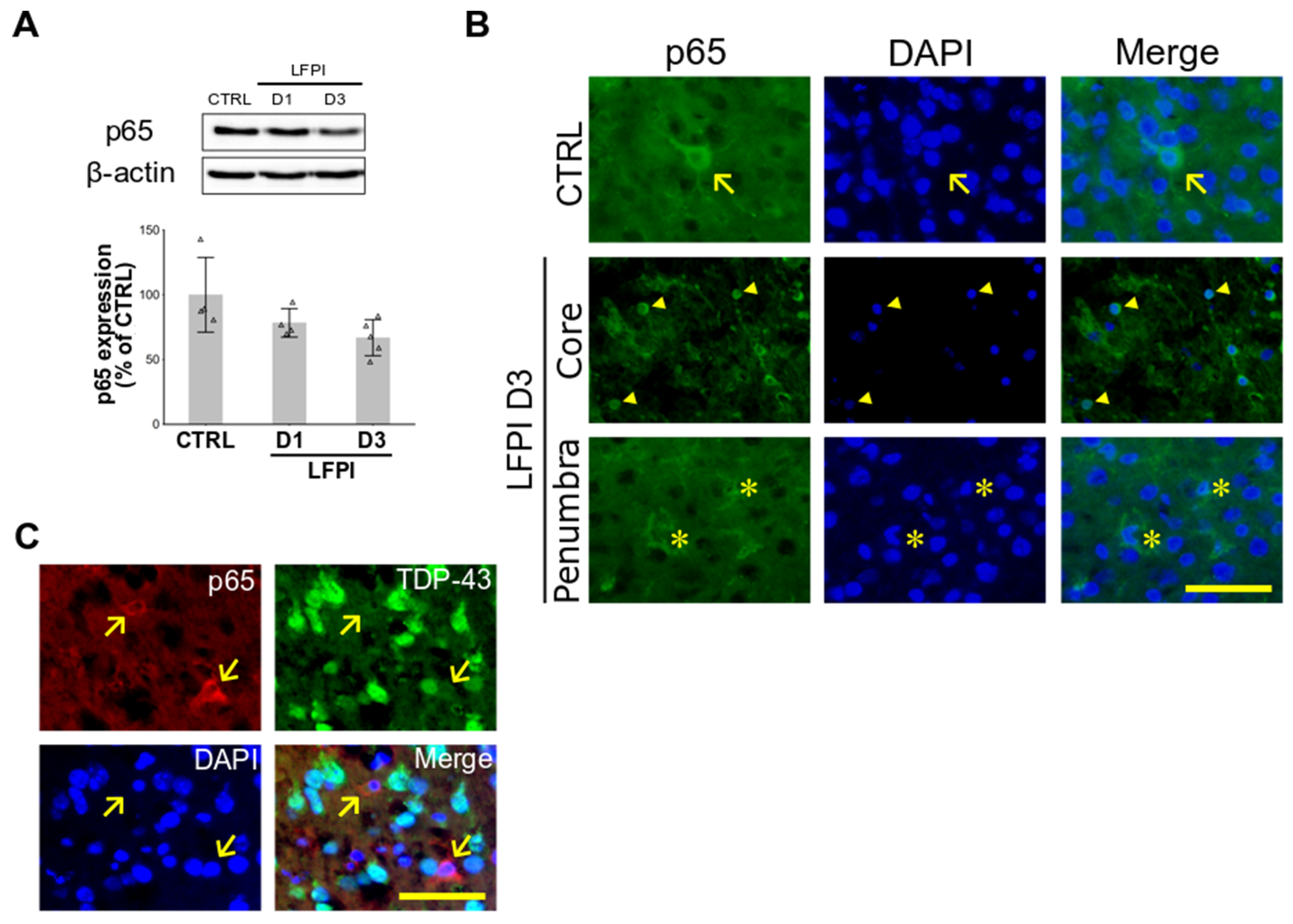
2.2. Single Moderate Brain Trauma Triggers Changes in the Cortical Expression of p65 Subunit of NF-κB That Are Area- and Cell Type-Dependent
2.3. Single Moderate Brain Trauma Causes a Decrease in Postsynaptic Protein PSD-95 Levels in the Ipsilateral Cortices of Injured Animals
2.4. Single Moderate, but Not Repetitive Mild Traumatic Brain Injury Induces Pathological Post-Translational Changes of TDP-43 in the Mouse Hippocampi
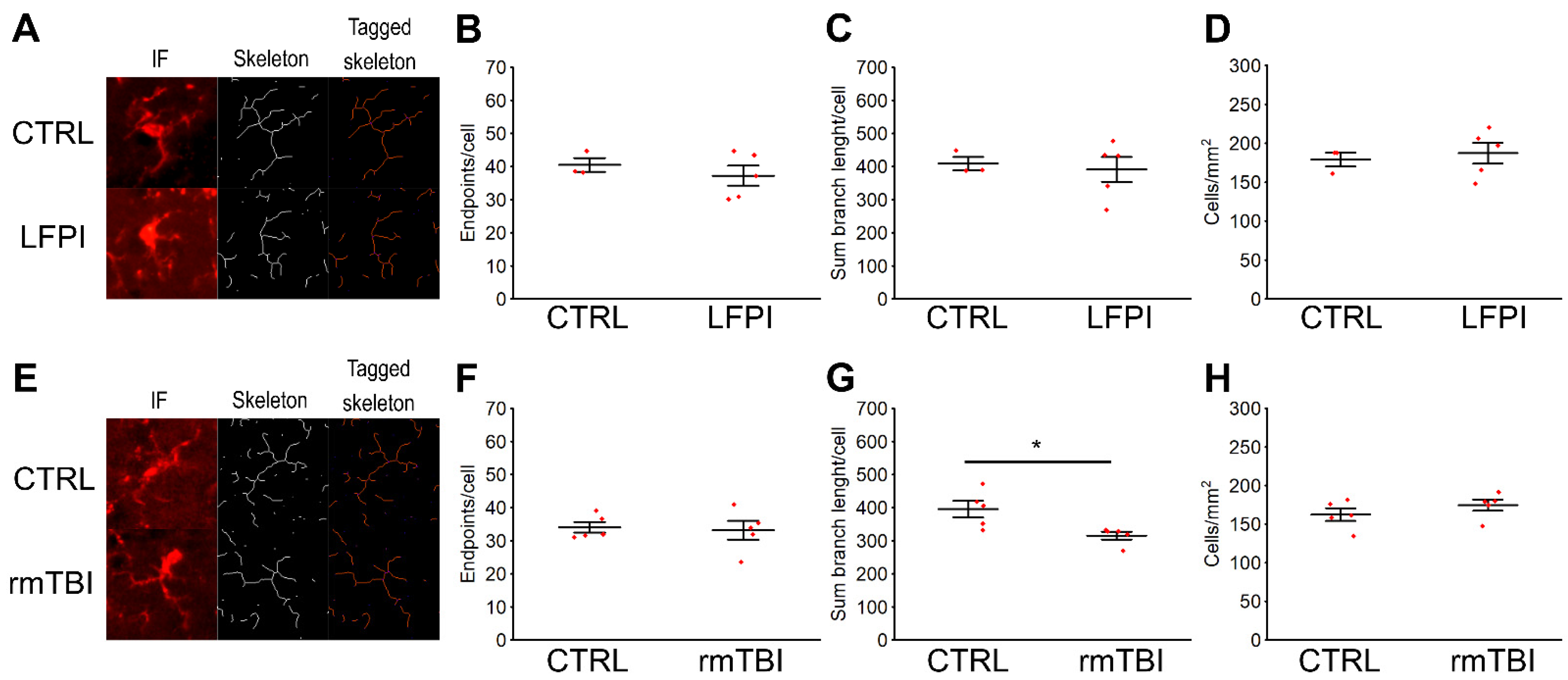
2.5. Post-Translational Modifications of TDP-43 Protein in the Hippocampus of Mice Exposed to a Single Moderate Traumatic Brain Injury Are Not Accompanied by Changes in the Microglia Activation Pattern
3. Discussion
4. Materials and Methods
4.1. Animals
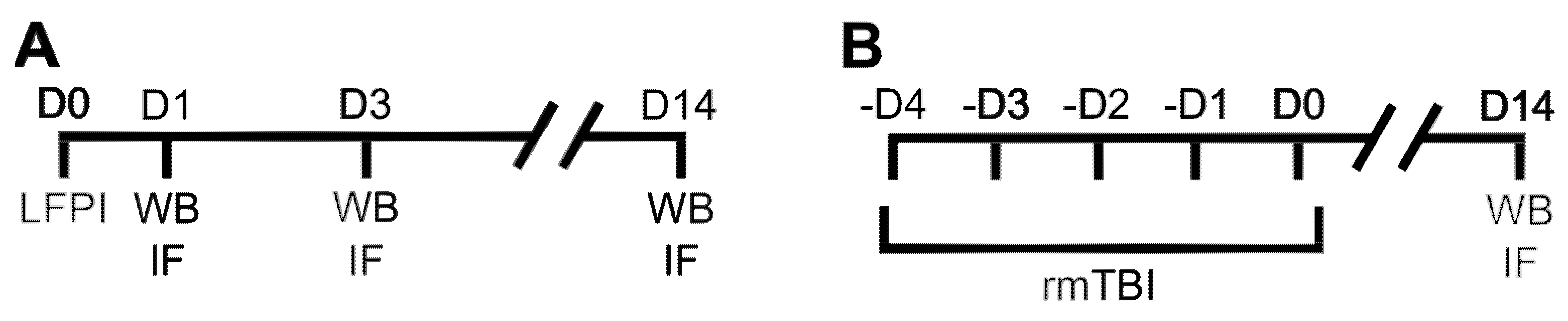
4.2. Experimental Traumatic Brain Injuries
4.3. Tissue Collection
4.4. Western Blotting
4.5. Immunofluorescence, Histochemistry and Imaging
4.6. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ghajar, J. Traumatic Brain Injury. Lancet 2000, 356, 923–929. [Google Scholar] [CrossRef]
- Tagliaferri, F.; Compagnone, C.; Korsic, M.; Servadei, F.; Kraus, J. A Systematic Review of Brain Injury Epidemiology in Europe. Acta Neurochir. 2006, 148, 255–268. [Google Scholar] [CrossRef] [PubMed]
- Peeters, W.; van den Brande, R.; Polinder, S.; Brazinova, A.; Steyerberg, E.W.; Lingsma, H.F.; Maas, A.I.R. Epidemiology of Traumatic Brain Injury in Europe. Acta Neurochir. 2015, 157, 1683–1696. [Google Scholar] [CrossRef] [Green Version]
- The Lancet Neurology. The Changing Landscape of Traumatic Brain Injury Research. Lancet Neurol. 2012, 11, 651. [Google Scholar] [CrossRef]
- Masel, B.E.; DeWitt, D.S. Traumatic Brain Injury: A Disease Process, Not an Event. J. Neurotrauma 2010, 27, 1529–1540. [Google Scholar] [CrossRef] [Green Version]
- Semple, B.D.; Lee, S.; Sadjadi, R.; Fritz, N.; Carlson, J.; Griep, C.; Ho, V.; Jang, P.; Lamb, A.; Popolizio, B.; et al. Repetitive Concussions in Adolescent Athletes–Translating Clinical and Experimental Research into Perspectives on Rehabilitation Strategies. Front. Neurol. 2015, 6, 69. [Google Scholar] [CrossRef] [Green Version]
- Vile, A.R.; Atkinson, L. Chronic Traumatic Encephalopathy: The Cellular Sequela to Repetitive Brain Injury. J. Clin. Neurosci. 2017, 41, 24–29. [Google Scholar] [CrossRef]
- DeKosky, S.T.; Ikonomovic, M.D.; Gandy, S. Traumatic Brain Injury-–Football, Warfare, and Long-Term Effects. N. Engl. J. Med. 2010, 363, 1293–1296. [Google Scholar] [CrossRef]
- Ojo, J.O.; Mouzon, B.; Algamal, M.; Leary, P.; Lynch, C.; Abdullah, L.; Evans, J.; Mullan, M.; Bachmeier, C.; Stewart, W.; et al. Chronic Repetitive Mild Traumatic Brain Injury Results in Reduced Cerebral Blood Flow, Axonal Injury, Gliosis, and Increased T-Tau and Tau Oligomers. J. Neuropathol. Exp. Neurol. 2016, 75, 636–655. [Google Scholar] [CrossRef] [Green Version]
- Zieman, G.; Bridwell, A.; Cárdenas, J.F. Traumatic Brain Injury in Domestic Violence Victims: A Retrospective Study at the Barrow Neurological Institute. J. Neurotrauma 2017, 34, 876–880. [Google Scholar] [CrossRef]
- McKee, A.C.; Cantu, R.C.; Nowinski, C.J.; Hedley-Whyte, E.T.; Gavett, B.E.; Budson, A.E.; Santini, V.E.; Lee, H.-S.; Kubilus, C.A.; Stern, R.A. Chronic Traumatic Encephalopathy in Athletes: Progressive Tauopathy after Repetitive Head Injury. J. Neuropathol. Exp. Neurol. 2009, 68, 709–735. [Google Scholar] [CrossRef]
- McKee, A.C.; Stein, T.D.; Nowinski, C.J.; Stern, R.A.; Daneshvar, D.H.; Alvarez, V.E.; Lee, H.-S.; Hall, G.; Wojtowicz, S.M.; Baugh, C.M.; et al. The Spectrum of Disease in Chronic Traumatic Encephalopathy. Brain 2013, 136, 43–64. [Google Scholar] [CrossRef]
- LoBue, C.; Denney, D.; Hynan, L.S.; Rossetti, H.C.; Lacritz, L.H.; Hart, J.; Womack, K.B.; Woon, F.L.; Cullum, C.M. Self-Reported Traumatic Brain Injury and Mild Cognitive Impairment: Increased Risk and Earlier Age of Diagnosis. J. Alzheimers Dis. JAD 2016, 51, 727–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackenzie, I.R.A.; Rademakers, R. The Role of TDP-43 in Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Curr. Opin. Neurol. 2008, 21, 693–700. [Google Scholar] [CrossRef] [Green Version]
- Ravanidis, S.; Kattan, F.-G.; Doxakis, E. Unraveling the Pathways to Neuronal Homeostasis and Disease: Mechanistic Insights into the Role of RNA-Binding Proteins and Associated Factors. Int. J. Mol. Sci. 2018, 19, 2280. [Google Scholar] [CrossRef] [Green Version]
- Barmada, S.J.; Skibinski, G.; Korb, E.; Rao, E.J.; Wu, J.Y.; Finkbeiner, S. Cytoplasmic Mislocalization of TDP-43 Is Toxic to Neurons and Enhanced by a Mutation Associated with Familial Amyotrophic Lateral Sclerosis. J. Neurosci. Off. J. Soc. Neurosci. 2010, 30, 639–649. [Google Scholar] [CrossRef] [Green Version]
- Lucke-Wold, B.P.; Turner, R.C.; Logsdon, A.F.; Bailes, J.E.; Huber, J.D.; Rosen, C.L. Linking Traumatic Brain Injury to Chronic Traumatic Encephalopathy: Identification of Potential Mechanisms Leading to Neurofibrillary Tangle Development. J. Neurotrauma 2014, 31, 1129–1138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapeli, K.; Martinez, F.J.; Yeo, G.W. Genetic Mutations in RNA-Binding Proteins and Their Roles in ALS. Hum. Genet. 2017, 136, 1193–1214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moisse, K.; Mepham, J.; Volkening, K.; Welch, I.; Hill, T.; Strong, M.J. Cytosolic TDP-43 Expression Following Axotomy Is Associated with Caspase 3 Activation in NFL-/- Mice: Support for a Role for TDP-43 in the Physiological Response to Neuronal Injury. Brain Res. 2009, 1296, 176–186. [Google Scholar] [CrossRef] [PubMed]
- Saykally, J.N.; Ratliff, W.A.; Keeley, K.L.; Pick, C.G.; Mervis, R.F.; Citron, B.A. Repetitive Mild Closed Head Injury Alters Protein Expression and Dendritic Complexity in a Mouse Model. J. Neurotrauma 2018, 35, 139–148. [Google Scholar] [CrossRef]
- Rajič Bumber, J.; Pilipović, K.; Janković, T.; Dolenec, P.; Gržeta, N.; Križ, J.; Župan, G. Repetitive Traumatic Brain Injury Is Associated With TDP-43 Alterations, Neurodegeneration, and Glial Activation in Mice. J. Neuropathol. Exp. Neurol. 2021, 80, 2–14. [Google Scholar] [CrossRef] [PubMed]
- Correia, A.S.; Patel, P.; Dutta, K.; Julien, J.-P. Inflammation Induces TDP-43 Mislocalization and Aggregation. PLoS ONE 2015, 10, e0140248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swarup, V.; Phaneuf, D.; Dupré, N.; Petri, S.; Strong, M.; Kriz, J.; Julien, J.-P. Deregulation of TDP-43 in Amyotrophic Lateral Sclerosis Triggers Nuclear Factor ΚB-Mediated Pathogenic Pathways. J. Exp. Med. 2011, 208, 2429–2447. [Google Scholar] [CrossRef] [PubMed]
- Snow, W.M.; Stoesz, B.M.; Kelly, D.M.; Albensi, B.C. Roles for NF-ΚB and Gene Targets of NF-ΚB in Synaptic Plasticity, Memory, and Navigation. Mol. Neurobiol. 2014, 49, 757–770. [Google Scholar] [CrossRef]
- Engelmann, C.; Haenold, R. Transcriptional Control of Synaptic Plasticity by Transcription Factor NF-ΚB. Neural Plast. 2016, 2016, 7027949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uchida, S.; Shumyatsky, G.P. Synaptically Localized Transcriptional Regulators in Memory Formation. Neuroscience 2018, 370, 4–13. [Google Scholar] [CrossRef]
- Dresselhaus, E.C.; Meffert, M.K. Cellular Specificity of NF-ΚB Function in the Nervous System. Front. Immunol. 2019, 10, 1043. [Google Scholar] [CrossRef]
- Nonaka, M.; Chen, X.H.; Pierce, J.E.; Leoni, M.J.; McIntosh, T.K.; Wolf, J.A.; Smith, D.H. Prolonged Activation of NF-KappaB Following Traumatic Brain Injury in Rats. J. Neurotrauma 1999, 16, 1023–1034. [Google Scholar] [CrossRef]
- Hang, C.H.; Shi, J.-X.; Li, J.-S.; Wu, W.; Yin, H.X. Concomitant Upregulation of Nuclear Factor-KB Activity, Proinflammatory Cytokines and ICAM-1 in the Injured Brain after Cortical Contusion Trauma in a Rat Model. Neurol. India 2005, 53, 312–317. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.-C.; Sun, Q.; Li, W.; Zhang, D.-D.; Ma, B.; Li, S.; Li, W.-D.; Zhou, M.-L.; Hang, C.-H. Biphasic Activation of Nuclear Factor Kappa B and Expression of P65 and C-Rel after Traumatic Brain Injury in Rats. Inflamm. Res. 2014, 63, 109–115. [Google Scholar] [CrossRef]
- Logsdon, A.F.; Lucke-Wold, B.P.; Nguyen, L.; Matsumoto, R.R.; Turner, R.C.; Rosen, C.L.; Huber, J.D. Salubrinal Reduces Oxidative Stress, Neuroinflammation and Impulsive-like Behavior in a Rodent Model of Traumatic Brain Injury. Brain Res. 2016, 1643, 140–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, S.-J.; Cao, L.-X.; Cheng, S.-B.; Dai, Q.-F.; Lin, J.-H.; Pu, L.; Chen, W.-H.; Zhang, Y.-J.; Chen, S.-L.; Zhang, Y.-M. Effect of Acupuncture on the TLR2/4-NF-ΚB Signalling Pathway in a Rat Model of Traumatic Brain Injury. Acupunct. Med. J. Br. Med. Acupunct. Soc. 2018, 36, 247–253. [Google Scholar] [CrossRef]
- Boersma, M.C.H.; Dresselhaus, E.C.; De Biase, L.M.; Mihalas, A.B.; Bergles, D.E.; Meffert, M.K. A Requirement for Nuclear Factor-KappaB in Developmental and Plasticity-Associated Synaptogenesis. J. Neurosci. Off. J. Soc. Neurosci. 2011, 31, 5414–5425. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.-Q.; Yi, S.; Chen, B.-B.; Cui, P.-P.; Wang, Y.; Li, Y.-Z. MTOR/NF-ΚB Signaling Pathway Protects Hippocampal Neurons from Injury Induced by Intermittent Hypoxia in Rats. Int. J. Neurosci. 2021, 131, 994–1003. [Google Scholar] [CrossRef] [PubMed]
- El-Husseini, A.E.; Schnell, E.; Chetkovich, D.M.; Nicoll, R.A.; Bredt, D.S. PSD-95 Involvement in Maturation of Excitatory Synapses. Science 2000, 290, 1364–1368. [Google Scholar] [CrossRef] [PubMed]
- Handley, E.E.; Pitman, K.A.; Dawkins, E.; Young, K.M.; Clark, R.M.; Jiang, T.C.; Turner, B.J.; Dickson, T.C.; Blizzard, C.A. Synapse Dysfunction of Layer V Pyramidal Neurons Precedes Neurodegeneration in a Mouse Model of TDP-43 Proteinopathies. Cereb. Cortex 2017, 27, 3630–3647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fogarty, M.J.; Klenowski, P.M.; Lee, J.D.; Drieberg-Thompson, J.R.; Bartlett, S.E.; Ngo, S.T.; Hilliard, M.A.; Bellingham, M.C.; Noakes, P.G. Cortical Synaptic and Dendritic Spine Abnormalities in a Presymptomatic TDP-43 Model of Amyotrophic Lateral Sclerosis. Sci. Rep. 2016, 6, 37968. [Google Scholar] [CrossRef] [Green Version]
- Wang, I.-F.; Wu, L.-S.; Shen, C.-K.J. TDP-43: An Emerging New Player in Neurodegenerative Diseases. Trends Mol. Med. 2008, 14, 479–485. [Google Scholar] [CrossRef]
- Ling, S.-C. Synaptic Paths to Neurodegeneration: The Emerging Role of TDP-43 and FUS in Synaptic Functions. Neural Plast. 2018, 2018, 8413496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, C.-E.; Jin, L.-W.; Chu, Y.-P.; Wei, W.-Y.; Ho, P.-C.; Tsai, K.-J. TDP-43 Proteinopathy Impairs MRNP Granule Mediated Postsynaptic Translation and MRNA Metabolism. Theranostics 2021, 11, 330–345. [Google Scholar] [CrossRef]
- Koza, P.; Beroun, A.; Konopka, A.; Górkiewicz, T.; Bijoch, L.; Torres, J.C.; Bulska, E.; Knapska, E.; Kaczmarek, L.; Konopka, W. Neuronal TDP-43 Depletion Affects Activity-Dependent Plasticity. Neurobiol. Dis. 2019, 130, 104499. [Google Scholar] [CrossRef]
- Brioschi, S.; d’Errico, P.; Amann, L.S.; Janova, H.; Wojcik, S.M.; Meyer-Luehmann, M.; Rajendran, L.; Wieghofer, P.; Paolicelli, R.C.; Biber, K. Detection of Synaptic Proteins in Microglia by Flow Cytometry. Front. Mol. Neurosci. 2020, 13, 149. [Google Scholar] [CrossRef]
- Johnson, V.E.; Stewart, W.; Trojanowski, J.Q.; Smith, D.H. Acute and Chronically Increased Immunoreactivity to Phosphorylation-Independent but Not Pathological TDP-43 after a Single Traumatic Brain Injury in Humans. Acta Neuropathol. 2011, 122, 715–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Lin, F.; Robertson, C.S.; Wang, K.K.W. Dual Vulnerability of TDP-43 to Calpain and Caspase-3 Proteolysis after Neurotoxic Conditions and Traumatic Brain Injury. J. Cereb. Blood Flow Metab. 2014, 34, 1444–1452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.-K.; Lee, Y.-C.; Huang, C.-Y.; Liliang, P.-C.; Lu, K.; Chen, H.-J.; Li, Y.-C.; Tsai, K.-J. Traumatic Brain Injury Causes Frontotemporal Dementia and TDP-43 Proteolysis. Neuroscience 2015, 300, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.L.; Sun, M.; Brady, R.D.; Liu, S.; Llanos, R.; Cheung, S.; Wright, D.K.; Casillas-Espinosa, P.M.; Sashindranath, M.; O’Brien, T.J.; et al. Transactive Response DNA-Binding Protein 43 Abnormalities after Traumatic Brain Injury. J. Neurotrauma 2019, 36, 87–99. [Google Scholar] [CrossRef]
- Thammisetty, S.S.; Pedragosa, J.; Weng, Y.C.; Calon, F.; Planas, A.; Kriz, J. Age-Related Deregulation of TDP-43 after Stroke Enhances NF-ΚB-Mediated Inflammation and Neuronal Damage. J. Neuroinflamm. 2018, 15, 312. [Google Scholar] [CrossRef]
- Wright, D.K.; Liu, S.; van der Poel, C.; McDonald, S.J.; Brady, R.D.; Taylor, L.; Yang, L.; Gardner, A.J.; Ordidge, R.; O’Brien, T.J.; et al. Traumatic Brain Injury Results in Cellular, Structural and Functional Changes Resembling Motor Neuron Disease. Cereb. Cortex 2017, 27, 4503–4515. [Google Scholar] [CrossRef]
- Wiesner, D.; Tar, L.; Linkus, B.; Chandrasekar, A.; Olde Heuvel, F.; Dupuis, L.; Tsao, W.; Wong, P.C.; Ludolph, A.; Roselli, F. Reversible Induction of TDP-43 Granules in Cortical Neurons after Traumatic Injury. Exp. Neurol. 2018, 299, 15–25. [Google Scholar] [CrossRef]
- Heyburn, L.; Abutarboush, R.; Goodrich, S.; Urioste, R.; Batuure, A.; Statz, J.; Wilder, D.; Ahlers, S.T.; Long, J.B.; Sajja, V.S.S.S. Repeated Low-Level Blast Overpressure Leads to Endovascular Disruption and Alterations in TDP-43 and Piezo2 in a Rat Model of Blast TBI. Front. Neurol. 2019, 10, 766. [Google Scholar] [CrossRef] [Green Version]
- McKee, A.C.; Gavett, B.E.; Stern, R.A.; Nowinski, C.J.; Cantu, R.C.; Kowall, N.W.; Perl, D.P.; Hedley-Whyte, E.T.; Price, B.; Sullivan, C.; et al. TDP-43 Proteinopathy and Motor Neuron Disease in Chronic Traumatic Encephalopathy. J. Neuropathol. Exp. Neurol. 2010, 69, 918–929. [Google Scholar] [CrossRef]
- Carbonell, W.S.; Grady, M.S. Regional and Temporal Characterization of Neuronal, Glial, and Axonal Response after Traumatic Brain Injury in the Mouse. Acta Neuropathol. 1999, 98, 396–406. [Google Scholar] [CrossRef]
- Ransohoff, R.M. A Polarizing Question: Do M1 and M2 Microglia Exist? Nat. Neurosci. 2016, 19, 987–991. [Google Scholar] [CrossRef] [PubMed]
- Thompson, H.J.; Lifshitz, J.; Marklund, N.; Grady, M.S.; Graham, D.I.; Hovda, D.A.; McIntosh, T.K. Lateral Fluid Percussion Brain Injury: A 15-Year Review and Evaluation. J. Neurotrauma 2005, 22, 42–75. [Google Scholar] [CrossRef] [PubMed]
- Mouzon, B.; Chaytow, H.; Crynen, G.; Bachmeier, C.; Stewart, J.; Mullan, M.; Stewart, W.; Crawford, F. Repetitive Mild Traumatic Brain Injury in a Mouse Model Produces Learning and Memory Deficits Accompanied by Histological Changes. J. Neurotrauma 2012, 29, 2761–2773. [Google Scholar] [CrossRef] [PubMed]
- Washington, P.M.; Villapol, S.; Burns, M.P. Polypathology and Dementia after Brain Trauma: Does Brain Injury Trigger Distinct Neurodegenerative Diseases, or Should It Be Classified Together as Traumatic Encephalopathy? Exp. Neurol. 2016, 275, 381–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daneshvar, D.H.; Goldstein, L.E.; Kiernan, P.T.; Stein, T.D.; McKee, A.C. Post-Traumatic Neurodegeneration and Chronic Traumatic Encephalopathy. Mol. Cell. Neurosci. 2015, 66, 81–90. [Google Scholar] [CrossRef]
- LoBue, C.; Schaffert, J.; Cullum, C.M.; Peters, M.E.; Didehbani, N.; Hart, J.; White, C.L. Clinical and Neuropsychological Profile of Patients with Dementia and Chronic Traumatic Encephalopathy. J. Neurol. Neurosurg. Psychiatry 2020, 91, 586–592. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Beers, D.R.; Bell, S.; Wang, J.; Wen, S.; Baloh, R.H.; Appel, S.H. TDP-43 Activates Microglia through NF-ΚB and NLRP3 Inflammasome. Exp. Neurol. 2015, 273, 24–35. [Google Scholar] [CrossRef]
- Bales, K.R.; Du, Y.; Dodel, R.C.; Yan, G.M.; Hamilton-Byrd, E.; Paul, S.M. The NF-KappaB/Rel Family of Proteins Mediates Abeta-Induced Neurotoxicity and Glial Activation. Brain Res. Mol. Brain Res. 1998, 57, 63–72. [Google Scholar] [CrossRef]
- Lam, A.G.; Koppal, T.; Akama, K.T.; Guo, L.; Craft, J.M.; Samy, B.; Schavocky, J.P.; Watterson, D.M.; Van Eldik, L.J. Mechanism of Glial Activation by S100B: Involvement of the Transcription Factor NFkappaB. Neurobiol. Aging 2001, 22, 765–772. [Google Scholar] [CrossRef]
- Frakes, A.E.; Ferraiuolo, L.; Haidet-Phillips, A.M.; Schmelzer, L.; Braun, L.; Miranda, C.J.; Ladner, K.J.; Bevan, A.K.; Foust, K.D.; Godbout, J.P.; et al. Microglia Induce Motor Neuron Death via the Classical NF-ΚB Pathway in Amyotrophic Lateral Sclerosis. Neuron 2014, 81, 1009–1023. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Cynader, M.S.; Jia, W. TDP-43 Inhibits NF-ΚB Activity by Blocking P65 Nuclear Translocation. PLoS ONE 2015, 10, e0142296. [Google Scholar] [CrossRef] [Green Version]
- Walker, K.R.; Tesco, G. Molecular Mechanisms of Cognitive Dysfunction Following Traumatic Brain Injury. Front. Aging Neurosci. 2013, 5, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sultana, R.; Banks, W.A.; Butterfield, D.A. Decreased Levels of PSD95 and Two Associated Proteins and Increased Levels of BCl2 and Caspase 3 in Hippocampus from Subjects with Amnestic Mild Cognitive Impairment: Insights into Their Potential Roles for Loss of Synapses and Memory, Accumulation of Aβ, and Neurodegeneration in a Prodromal Stage of Alzheimer’s Disease. J. Neurosci. Res. 2010, 88, 469–477. [Google Scholar] [CrossRef] [Green Version]
- Gobbel, G.T.; Bonfield, C.; Carson-Walter, E.B.; Adelson, P.D. Diffuse Alterations in Synaptic Protein Expression Following Focal Traumatic Brain Injury in the Immature Rat. Childs Nerv. Syst. 2007, 23, 1171–1179. [Google Scholar] [CrossRef] [PubMed]
- Ansari, M.A.; Roberts, K.N.; Scheff, S.W. A Time Course of Contusion-Induced Oxidative Stress and Synaptic Proteins in Cortex in a Rat Model of TBI. J. Neurotrauma 2008, 25, 513–526. [Google Scholar] [CrossRef] [PubMed]
- Wakade, C.; Sukumari-Ramesh, S.; Laird, M.D.; Dhandapani, K.M.; Vender, J.R. Delayed Reduction in Hippocampal Postsynaptic Density Protein-95 Expression Temporally Correlates with Cognitive Dysfunction Following Controlled Cortical Impact in Mice. J. Neurosurg. 2010, 113, 1195–1201. [Google Scholar] [CrossRef]
- Campbell, J.N.; Low, B.; Kurz, J.E.; Patel, S.S.; Young, M.T.; Churn, S.B. Mechanisms of Dendritic Spine Remodeling in a Rat Model of Traumatic Brain Injury. J. Neurotrauma 2012, 29, 218–234. [Google Scholar] [CrossRef]
- Wang, C.-F.; Zhao, C.-C.; Jiang, G.; Gu, X.; Feng, J.-F.; Jiang, J.-Y. The Role of Posttraumatic Hypothermia in Preventing Dendrite Degeneration and Spine Loss after Severe Traumatic Brain Injury. Sci. Rep. 2016, 6, 37063. [Google Scholar] [CrossRef] [Green Version]
- Jalin, A.M.A.A.; Jin, R.; Wang, M.; Li, G. EPPS Treatment Attenuates Traumatic Brain Injury in Mice by Reducing Aβ Burden and Ameliorating Neuronal Autophagic Flux. Exp. Neurol. 2019, 314, 20–33. [Google Scholar] [CrossRef]
- Girgis, F.; Pace, J.; Sweet, J.; Miller, J.P. Hippocampal Neurophysiologic Changes after Mild Traumatic Brain Injury and Potential Neuromodulation Treatment Approaches. Front. Syst. Neurosci. 2016, 10, 8. [Google Scholar] [CrossRef] [PubMed]
- Jamshidi, P.; Kim, G.; Shahidehpour, R.K.; Bolbolan, K.; Gefen, T.; Bigio, E.H.; Mesulam, M.-M.; Geula, C. Distribution of TDP-43 Pathology in Hippocampal Synaptic Relays Suggests Transsynaptic Propagation in Frontotemporal Lobar Degeneration. J. Neuropathol. Exp. Neurol. 2020, 79, 585–591. [Google Scholar] [CrossRef] [PubMed]
- Roof, R.L.; Hall, E.D. Estrogen-Related Gender Difference in Survival Rate and Cortical Blood Flow after Impact-Acceleration Head Injury in Rats. J. Neurotrauma 2000, 17, 1155–1169. [Google Scholar] [CrossRef]
- Brann, D.W.; Dhandapani, K.; Wakade, C.; Mahesh, V.B.; Khan, M.M. Neurotrophic and Neuroprotective Actions of Estrogen: Basic Mechanisms and Clinical Implications. Steroids 2007, 72, 381–405. [Google Scholar] [CrossRef] [Green Version]
- Brazinova, A.; Rehorcikova, V.; Taylor, M.S.; Buckova, V.; Majdan, M.; Psota, M.; Peeters, W.; Feigin, V.; Theadom, A.; Holkovic, L.; et al. Epidemiology of Traumatic Brain Injury in Europe: A Living Systematic Review. J. Neurotrauma 2021, 38, 1411–1440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obernier, J.A.; Baldwin, R.L. Establishing an Appropriate Period of Acclimatization Following Transportation of Laboratory Animals. ILAR J. 2006, 47, 364–369. [Google Scholar] [CrossRef] [Green Version]
- McIntosh, T.K.; Vink, R.; Noble, L.; Yamakami, I.; Fernyak, S.; Soares, H.; Faden, A.L. Traumatic Brain Injury in the Rat: Characterization of a Lateral Fluid-Percussion Model. Neuroscience 1989, 28, 233–244. [Google Scholar] [CrossRef]
- Carbonell, W.S.; Maris, D.O.; McCall, T.; Grady, M.S. Adaptation of the Fluid Percussion Injury Model to the Mouse. J. Neurotrauma 1998, 15, 217–229. [Google Scholar] [CrossRef]
- Kane, M.J.; Angoa-Pérez, M.; Briggs, D.I.; Viano, D.C.; Kreipke, C.W.; Kuhn, D.M. A Mouse Model of Human Repetitive Mild Traumatic Brain Injury. J. Neurosci. Methods 2012, 203, 41–49. [Google Scholar] [CrossRef] [Green Version]
- Dolenec, P.; Pilipović, K.; Janković, T.; Župan, G. Pattern of Neuronal and Axonal Damage, Glial Response, and Synaptic Changes in Rat Cerebellum within the First Week Following Traumatic Brain Injury. J. Neuropathol. Exp. Neurol. 2020, 79, 1163–1182. [Google Scholar] [CrossRef] [PubMed]
- Dolenec, P.; Pilipović, K.; Rajič, J.; Župan, G. Temporal Pattern of Neurodegeneration, Programmed Cell Death, and Neuroplastic Responses in the Thalamus after Lateral Fluid Percussion Brain Injury in the Rat. J. Neuropathol. Exp. Neurol. 2015, 74, 512–526. [Google Scholar] [CrossRef] [Green Version]
- Yadavilli, S.; Hegde, V.; Deutsch, W.A. Translocation of Human Ribosomal Protein S3 to Sites of DNA Damage Is Dependant on ERK-Mediated Phosphorylation Following Genotoxic Stress. DNA Repair 2007, 6, 1453–1462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrison, H.; Young, K.; Qureshi, M.; Rowe, R.K.; Lifshitz, J. Quantitative Microglia Analyses Reveal Diverse Morphologic Responses in the Rat Cortex after Diffuse Brain Injury. Sci. Rep. 2017, 7, 13211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pilipović, K.; Rajič Bumber, J.; Dolenec, P.; Gržeta, N.; Janković, T.; Križ, J.; Župan, G. Long-Term Effects of Repetitive Mild Traumatic Injury on the Visual System in Wild-Type and TDP-43 Transgenic Mice. Int. J. Mol. Sci. 2021, 22, 6584. [Google Scholar] [CrossRef]
- Ruijter, J.M.; Thygesen, H.H.; Schoneveld, O.J.; Das, A.T.; Berkhout, B.; Lamers, W.H. Factor Correction as a Tool to Eliminate Between-Session Variation in Replicate Experiments: Application to Molecular Biology and Retrovirology. Retrovirology 2006, 3, 2. [Google Scholar] [CrossRef] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Janković, T.; Dolenec, P.; Rajič Bumber, J.; Gržeta, N.; Kriz, J.; Župan, G.; Pilipović, K. Differential Expression Patterns of TDP-43 in Single Moderate versus Repetitive Mild Traumatic Brain Injury in Mice. Int. J. Mol. Sci. 2021, 22, 12211. https://doi.org/10.3390/ijms222212211
Janković T, Dolenec P, Rajič Bumber J, Gržeta N, Kriz J, Župan G, Pilipović K. Differential Expression Patterns of TDP-43 in Single Moderate versus Repetitive Mild Traumatic Brain Injury in Mice. International Journal of Molecular Sciences. 2021; 22(22):12211. https://doi.org/10.3390/ijms222212211
Chicago/Turabian StyleJanković, Tamara, Petra Dolenec, Jelena Rajič Bumber, Nika Gržeta, Jasna Kriz, Gordana Župan, and Kristina Pilipović. 2021. "Differential Expression Patterns of TDP-43 in Single Moderate versus Repetitive Mild Traumatic Brain Injury in Mice" International Journal of Molecular Sciences 22, no. 22: 12211. https://doi.org/10.3390/ijms222212211