
Ventricular TLR4 Levels Abrogate TLR2-Mediated Adverse Cardiac Remodeling upon Pressure Overload in Mice

 , , ,
, , ,
Abstract
:1. Introduction
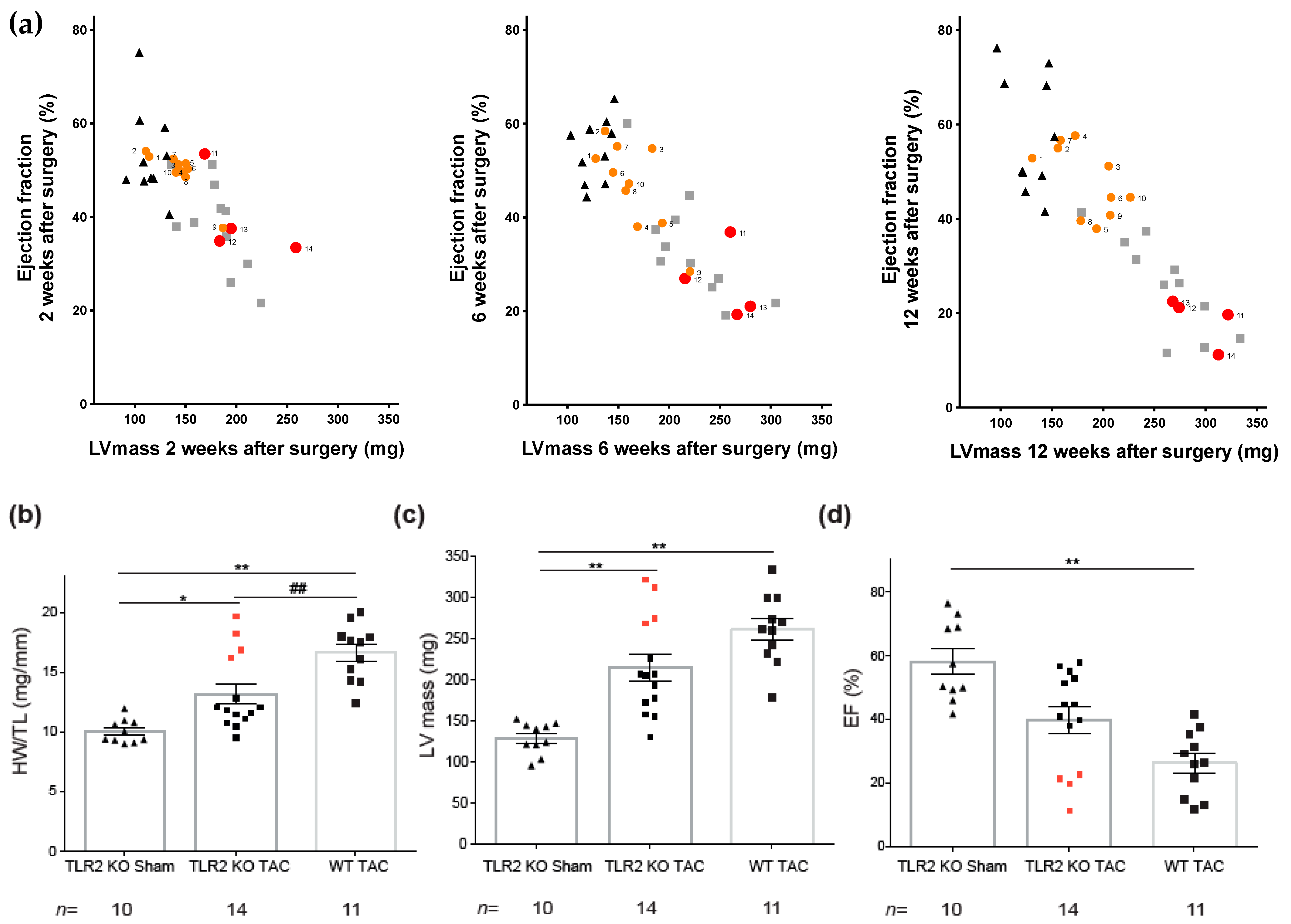
2. Results
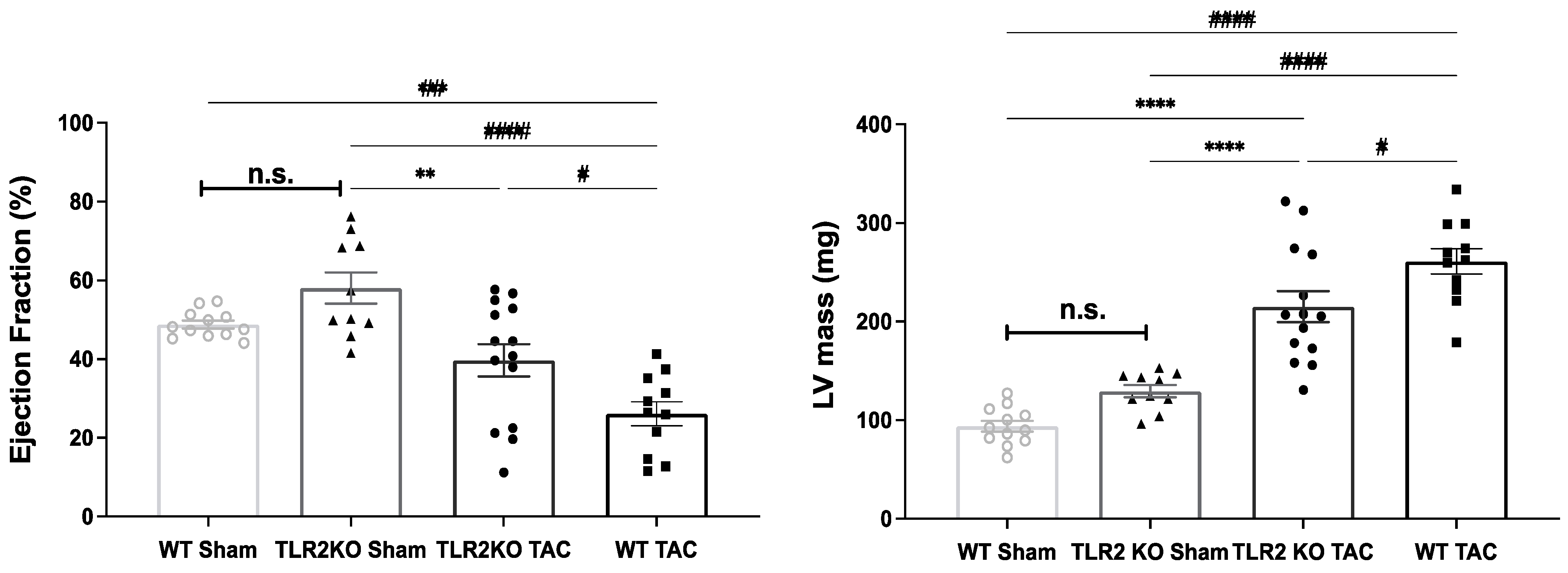
2.1. TLR2 Deficiency Improves Cardiac Function and Contractility
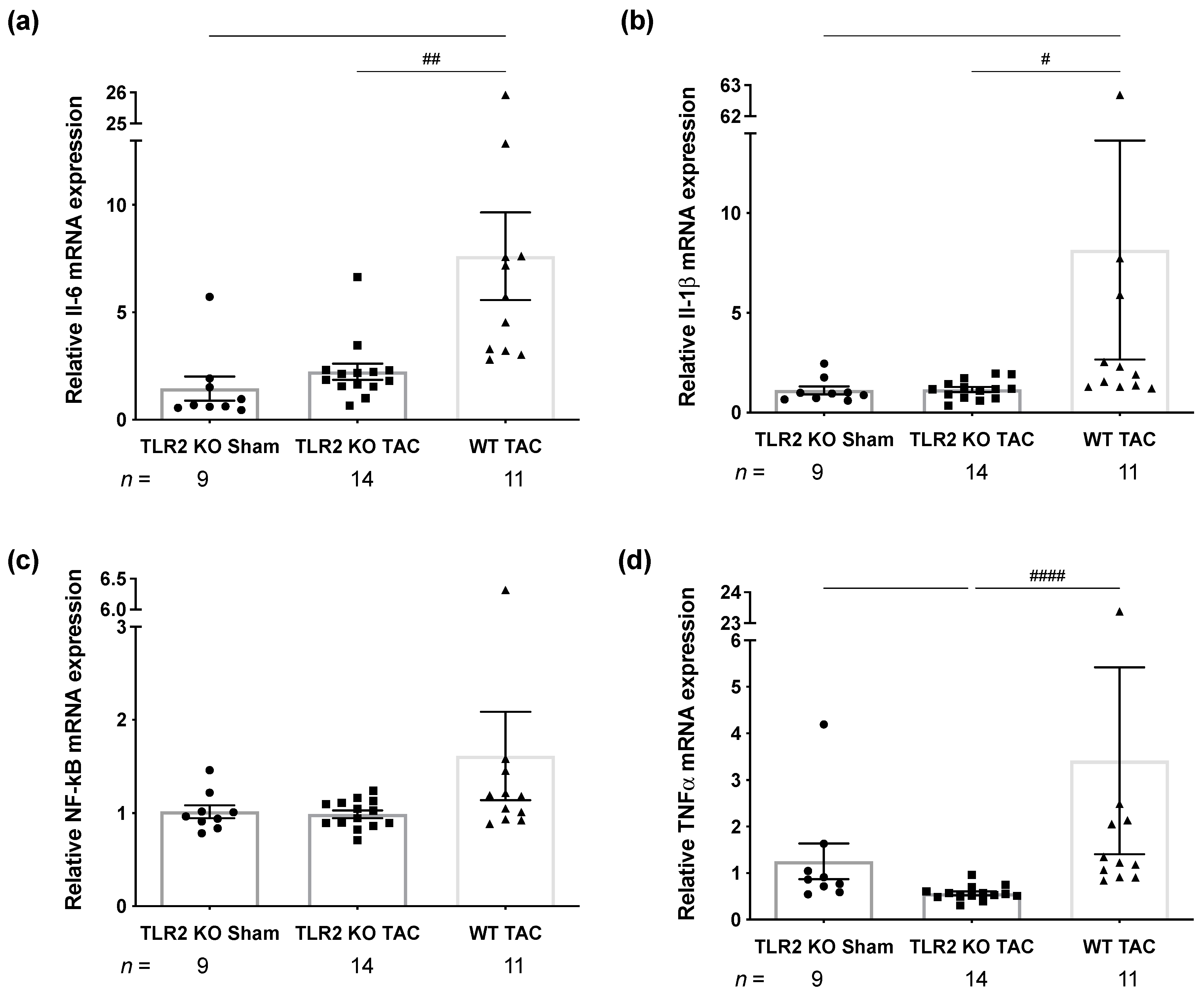
2.2. TLR2 Deficiency Attenuates Inflammation
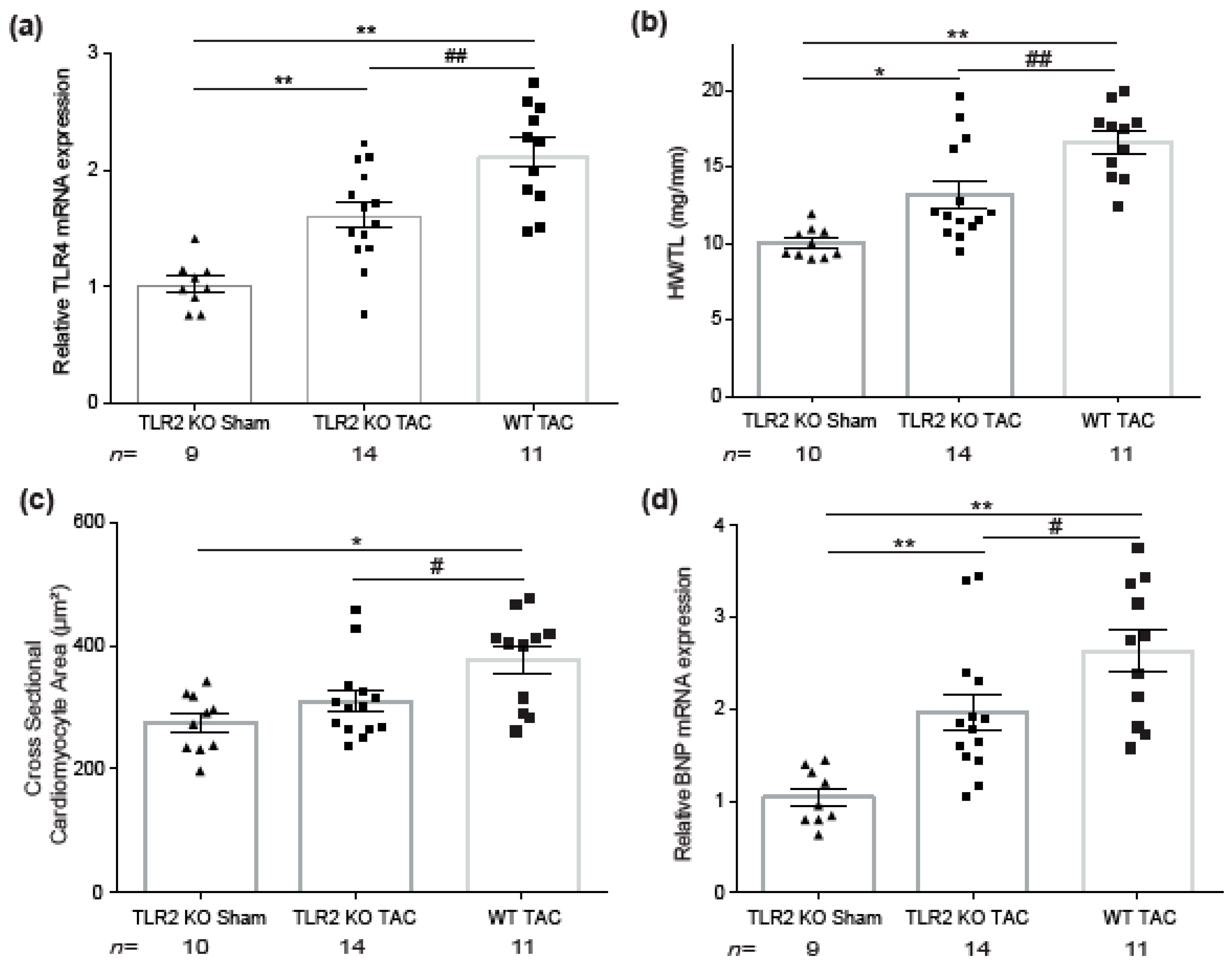
2.3. TLR2 Deficiency in Mice Ameliorates Hypertrophic Remodeling and Level of TLR4
2.4. Increased TLR2 and TLR4 mRNA Gene Expression in Mice Subjected to Chronic Pressure Overload
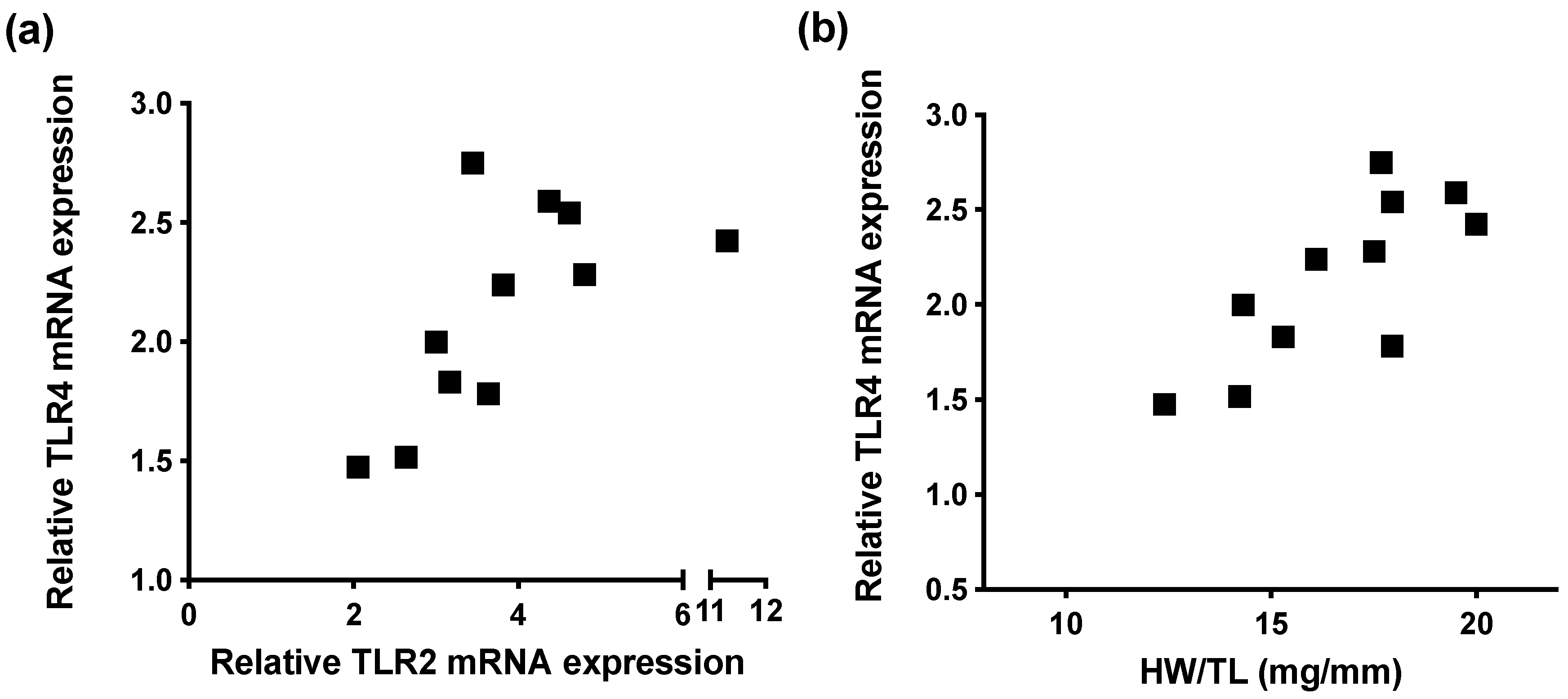
2.5. High TLR4 Levels in TLR2 KO Animals Predict Adverse Cardiac Remodeling after TAC
3. Discussion
Study Limitations
4. Materials and Methods
4.1. Experimental Design for Mice
4.2. Genotyping
4.3. Electrocardiography and Echocardiography
4.4. Immunohistochemistry
4.5. Real-Time Quantitative PCR (RT-qPCR)
4.6. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ponikowski, P.; Voors, A.A.; Anker, S.D.; Bueno, H.; Cleland, J.G.F.; Coats, A.J.S.; Falk, V.; González-Juanatey, J.R.; Harjola, V.-P.; Jankowska, E.A.; et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur. Heart J. 2016, 37, 2129–2200. [Google Scholar] [CrossRef]
- Mozaffarian, D.; Benjamin, E.J.; Go, A.S.; Arnett, D.K.; Blaha, M.J.; Cushman, M.; Das, S.R.; de Ferranti, S.; Després, J.-P.; Fullerton, H.J.; et al. Executive summary: Heart disease and stroke statistics-2016 update: A Report from the American Heart Association. Circulation 2016, 133, 447–454. [Google Scholar] [CrossRef] [PubMed]
- Swynghedauw, B. Molecular Mechanisms of Myocardial Remodeling. Physiol. Rev. 1999, 79, 215–262. [Google Scholar] [CrossRef]
- Dick, S.A.; Epelman, S. Chronic Heart Failure and Inflammation: What Do We Really Know? Circ. Res. 2016, 119, 159–176. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.; Feng, Z. The Role of Toll-Like Receptor Signaling in the Progression of Heart Failure. Mediat. Inflamm. 2018, 2018, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Botos, I.; Segal, D.M.; Davies, D.R. The Structural Biology of Toll-like Receptors. Structure 2011, 19, 447–459. [Google Scholar] [CrossRef] [Green Version]
- Mann, D.L. The Emerging Role of Innate Immunity in the Heart and Vascular System. Circ. Res. 2011, 108, 1133–1145. [Google Scholar] [CrossRef]
- Kawasaki, T.; Kawai, T. Toll-Like Receptor Signaling Pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arslan, F.; Smeets, M.B.; O’Neill, L.A.; Keogh, B.; McGuirk, P.; Timmers, L.; Tersteeg, C.; Hoefer, I.E.; Doevendans, P.A.; Pasterkamp, G.; et al. Myocardial Ischemia/Reperfusion Injury Is Mediated by Leukocytic Toll-Like Receptor-2 and Reduced by Systemic Administration of a Novel Anti–Toll-Like Receptor-2 Antibody. Circulation 2010, 121, 80–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shishido, T.; Nozaki, N.; Yamaguchi, S.; Shibata, Y.; Nitobe, J.; Miyamoto, T.; Takahashi, H.; Arimoto, T.; Maeda, K.; Yamakawa, M.; et al. Toll-Like Receptor-2 Modulates Ventricular Remodeling After Myocardial Infarction. Circulation 2003, 108, 2905–2910. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Li, Y.-L.; Zhang, C.-C.; Cui, W.; Wang, X.; Xia, Y.; Du, J.; Li, H.-H. Inhibition of Toll-like receptor 2 reduces cardiac fibrosis by attenuating macrophage-mediated inflammation. Cardiovasc. Res. 2013, 101, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Methe, H.; Kim, J.-O.; Kofler, S.; Weis, M.; Nabauer, M.; Koglin, J. Expansion of Circulating Toll-Like Receptor 4–Positive Monocytes in Patients With Acute Coronary Syndrome. Circulation 2005, 111, 2654–2661. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Shao, L.; Ma, J. Toll-Like Receptors 2 and 4 Predict New-Onset Atrial Fibrillation in Acute Myocardial Infarction Patients. Int. Heart J. 2018, 59, 64–70. [Google Scholar] [CrossRef] [Green Version]
- Avlas, O.; Bragg, A.; Fuks, A.; Nicholson, J.D.; Farkash, A.; Porat, E.; Aravot, D.; Levy-Drummer, R.S.; Cohen, C.; Shainberg, A.; et al. TLR4 Expression Is Associated with Left Ventricular Dysfunction in Patients Undergoing Coronary Artery Bypass Surgery. PLoS ONE 2015, 10, e0120175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Wang, Y.; Cao, Z.; Wang, M.; Liu, X.; Gao, T.; Hu, Q.; Yuan, W.; Lin, L. Up-regulated TLR 4 in cardiomyocytes exacerbates heart failure after long-term myocardial infarction. J. Cell. Mol. Med. 2015, 19, 2728–2740. [Google Scholar] [CrossRef]
- Ehrentraut, H.; Weber, C.; Ehrentraut, S.; Schwederski, M.; Boehm, O.; Knuefermann, P.; Meyer, R.; Baumgarten, G. The toll-like receptor 4-antagonist eritoran reduces murine cardiac hypertrophy. Eur. J. Heart Fail. 2011, 13, 602–610. [Google Scholar] [CrossRef] [Green Version]
- Timmers, L.; Sluijter, J.P.; van Keulen, J.K.; Hoefer, I.E.; Nederhoff, M.G.; Goumans, M.-J.; Doevendans, P.A.; van Echteld, C.J.; Joles, J.A.; Quax, P.; et al. Toll-Like Receptor 4 Mediates Maladaptive Left Ventricular Remodeling and Impairs Cardiac Function After Myocardial Infarction. Circ. Res. 2008, 102, 257–264. [Google Scholar] [CrossRef] [Green Version]
- Ehrentraut, H.; Ehrentraut, S.; Boehm, O.; El Aissati, S.; Foltz, F.; Goelz, L.; Goertz, D.; Kebir, S.; Weisheit, C.; Wolf, M.; et al. Tlr4 Deficiency Protects against Cardiac Pressure Overload Induced Hyperinflammation. PLoS ONE 2015, 10, e0142921. [Google Scholar] [CrossRef] [Green Version]
- Frantz, S.; Hu, K.; Bayer, B.; Gerondakis, S.; Strotmann, J.; Adamek, A.; Ertl, G.; Bauersachs, J. Absence of NF-κB subunit p50 improves heart failure after myocardial infarction. FASEB J. 2006, 20, 1918–1920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.Q.; Tang, R.; Li, L.; Szucsik, A.; Javan, H.; Saegusa, N.; Spitzer, K.W.; Selzman, C.H. Cardiomyocyte-specific p65 NF-κB deletion protects the injured heart by preservation of calcium handling. Am. J. Physiol. Circ. Physiol. 2013, 305, H1089–H1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Groot, D.; Hoefer, I.E.; Grundmann, S.; Schoneveld, A.; Haverslag, R.T.; van Keulen, J.K.; Bot, P.T.; Timmers, L.; Piek, J.J.; Pasterkamp, G.; et al. Arteriogenesis requires toll-like receptor 2 and 4 expression in bone-marrow derived cells. J. Mol. Cell. Cardiol. 2011, 50, 25–32. [Google Scholar] [CrossRef]
- Wang, Y.-C.; Zhou, Y.; Fang, H.; Lin, S.; Wang, P.-F.; Ma, R.-P.X.; Chen, J.; Xiong, X.-Y.; Lv, F.-L.; Liang, Q.-L.; et al. Toll-like receptor 2/4 heterodimer mediates inflammatory injury in intracerebral hemorrhage. Ann. Neurol. 2014, 75, 876–889. [Google Scholar] [CrossRef] [PubMed]
- Dong, R.; Wang, Z.-F.; Zhao, C.; Gu, H.-R.; Hu, Z.-W.; Xie, J.; Wu, Y.-Q. Toll-Like Receptor 4 Knockout Protects Against Isoproterenol-Induced Cardiac Fibrosis: The role of autophagy. J. Cardiovasc. Pharmacol. Ther. 2015, 20, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Spurthi, K.M.; Sarikhani, M.; Mishra, S.; Desingu, P.A.; Yadav, S.; Rao, S.; Maity, S.; Tamta, A.K.; Kumar, S.; Majumdar, S.; et al. Toll-like receptor 2 deficiency hyperactivates the FoxO1 transcription factor and induces aging-associated cardiac dysfunction in mice. J. Biol. Chem. 2018, 293, 13073–13089. [Google Scholar] [CrossRef] [Green Version]
- Faure, E.; Thomas, L.; Xu, H.; Medvedev, A.E.; Equils, O.; Arditi, M. Bacterial Lipopolysaccharide and IFN-γ Induce Toll-Like Receptor 2 and Toll-Like Receptor 4 Expression in Human Endothelial Cells: Role of NF-κB Activation. J. Immunol. 2001, 166, 2018–2024. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Lafuse, W.P.; Zwilling, B.S. NFκB and Sp1 Elements Are Necessary for Maximal Transcription of Toll-like Receptor 2 Induced byMycobacterium avium. J. Immunol. 2001, 167, 6924–6932. [Google Scholar] [CrossRef] [Green Version]
- Johnson, C.M.; Tapping, R.I.; Chambers, J.E.; Jessop, C.E.; Bulleid, N.J. Microbial Products Stimulate Human Toll-like Receptor 2 Expression through Histone Modification Surrounding a Proximal NF-κB-binding Site. J. Biol. Chem. 2007, 282, 31197–31205. [Google Scholar] [CrossRef] [Green Version]
- Bualeong, T.; Kebir, S.; Hof, D.; Goelz, L.; Graewe, M.; Ehrentraut, S.F.; Knuefermann, P.; Baumgarten, G.; Meyer, R.; Ehrentraut, H. Tlr2 deficiency does not limit the development of left ventricular hypertrophy in a model of transverse aortic constriction induced pressure overload. J. Negat. Results Biomed. 2016, 15, 9. [Google Scholar] [CrossRef] [Green Version]
- Fan, W.; Morinaga, H.; Kim, J.J.; Bae, E.; Spann, N.J.; Heinz, S.; Glass, C.K.; Olefsky, J.M. FoxO1 regulates Tlr4 inflammatory pathway signalling in macrophages. EMBO J. 2010, 29, 4223–4236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.; Zhang, X.; Bao, H.; Mi, S.; Cai, W.; Yan, H.; Wang, Q.; Wang, Z.; Yan, J.; Fan, G.; et al. Toll-Like Receptor (TLR) 2 and TLR4 Differentially Regulate Doxorubicin Induced Cardiomyopathy in Mice. PLoS ONE 2012, 7, e40763. [Google Scholar] [CrossRef]
- Birks, E.J.; Felkin, L.E.; Banner, N.R.; Khaghani, A.; Barton, P.; Yacoub, M.H. Increased toll-like receptor 4 in the myocardium of patients requiring left ventricular assist devices. J. Heart Lung Transplant. 2004, 23, 228–235. [Google Scholar] [CrossRef]
- Good, D.W.; George, T.; Watts, B.A. Toll-like Receptor 2 Is Required for LPS-induced Toll-like Receptor 4 Signaling and Inhibition of Ion Transport in Renal Thick Ascending Limb. J. Biol. Chem. 2012, 287, 20208–20220. [Google Scholar] [CrossRef] [Green Version]
- van Zoelen, M.A.; Yang, H.; Florquin, S.; Meijers, J.C.; Akira, S.; Arnold, B.; Nawroth, P.P.; Bierhaus, A.; Tracey, K.J.; van der Poll, T. Role of toll-like receptors 2 and 4, and the receptor for advanced glycation end products in high-mobility group box 1-induced inflammation in vivo. Shock 2009, 31, 280–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boulaksil, M.; Winckels, S.K.; Engelen, M.A.; Stein, M.; Van Veen, T.A.; Jansen, J.A.; Linnenbank, A.C.; Bierhuizen, M.F.; Groenewegen, W.A.; Van Oosterhout, M.F.; et al. Heterogeneous Connexin43 distribution in heart failure is associated with dispersed conduction and enhanced susceptibility to ventricular arrhythmias. Eur. J. Heart Fail. 2010, 12, 913–921. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.-W.; Fontes, M.S.C.; Wang, X.; Chong, S.Y.; Kessler, E.L.; Zhang, Y.-N.; De Haan, J.J.; Arslan, F.; De Jager, S.C.A.; Timmers, L.; et al. Leukocytic Toll-Like Receptor 2 Deficiency Preserves Cardiac Function And Reduces Fibrosis In Sustained Pressure Overload. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Fontes, M.S.C.; Raaijmakers, A.J.A.; Van Doorn, T.; Kok, B.; Nieuwenhuis, S.; Van Der Nagel, R.; Vos, M.A.; De Boer, T.P.; van Rijen, H.; Bierhuizen, M.F.A. Changes in Cx43 and NaV1.5 Expression Precede the Occurrence of Substantial Fibrosis in Calcineurin-Induced Murine Cardiac Hypertrophy. PLoS ONE 2014, 9, e87226. [Google Scholar] [CrossRef]
- Helms, S.A.; Azhar, G.; Zuo, C.; Theus, S.A.; Bartke, A.; Wei, J.Y. Smaller cardiac cell size and reduced extra-cellular collagen might be beneficial for hearts of Ames dwarf mice. Int. J. Biol. Sci. 2010, 6, 475–490. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sham | TAC | |||
|---|---|---|---|---|
| TLR2 KO | WT | TLR2 KO | WT | |
| n | 10 | 12 | 14 | 11 |
| Echocardiography | ||||
| Pressure gradient (mmHg) | 3.5 ± 0.3 | 61.6 ± 3.6 ** | 61.7 ± 4.4 ** | |
| LVAW,s (mm) | 1.3 ± 0.1 | 1.5 ± 0.0 | 1.4 ± 0.1 | |
| LVAW,d (mm) | 0.9 ± 0.1 | 1.1 ± 0.0 ** | 1.2 ± 0.1 ** | |
| LVPW,s (mm) | 1.1 ± 0.1 | 1.3 ± 0.1 | 1.3 ± 0.1 | |
| LVPW,d (mm) | 0.8 ± 0.0 | 1.1 ± 0.1 ** | 1.1 ± 0.1 ** | |
| LVID,s (mm) | 2.8 ± 0.2 | 3.6 ± 0.2 * | 4.2 ± 0.2 ** | |
| LVID,d (mm) | 4.1 ± 0.1 | 4.4 ± 0.1 | 4.8 ± 0.2 ** | |
| LV Vol,s (μL) | 31.4 ± 4.0 | 56.5 ± 8.7 | 81.7 ± 9.2 ** | |
| LV Vol,d (μL) | 72.6 ± 3.3 | 88.3 ± 7.4 | 108.1 ± 8.4 ** | |
| LV mass (mg) | 129.6 ± 6.1 | 93.9 ± 19.1 | 215.2 ± 15.7 ** | 261.1 ± 12.8 ** |
| EF (%) | 58.0 ± 4.0 | 48.8 ± 3.4 | 39.7 ± 4.1 | 26.1 ± 3.1 ** |
| FS (%) | 30.8 ± 2.7 | 19.6 ± 2.2 | 12.2 ± 1.5 ** | |
| SV (μL) | 41.2 ± 1.7 | 31.7 ± 2.1* | 26.4 ± 2.3 ** | |
| CO (mL/min) | 19.0 ± 1.2 | 16.2 ± 1.1 | 13.3 ± 1.1 ** | |
| Electrocardiography | ||||
| Heart rate (bpm) | 427.8 ± 18.4 | 479.8 ± 14.9 | 480.2 ± 15.5 | |
| RR (ms) | 142.4 ± 5.7 | 126.6 ± 3.9 * | 126.3 ± 4.2 | |
| PR (ms) | 42.7 ± 0.8 | 42.8 ± 1.4 | 42.8 ± 1.5 | |
| P (ms) | 9.8 ± 0.2 | 10.3 ± 0.3 | 11.2 ± 0.5 | |
| QRS (ms) | 10.6 ± 0.3 | 11.4 ± 0.3 | 12.7 ± 0.4 **# | |
| QTc (ms) | 42.2 ± 0.6 | 51.1 ± 1.9 | 57.0 ± 2.3 **## | |
| Sham | TAC | ||
|---|---|---|---|
| TLR2 KO | TLR2 KO | WT | |
| n | 10 | 14 | 11 |
| Body weight (g) | 35.5 ± 0.8 | 33.9 ± 0.6 | 33.0 ± 0.5 * |
| Heart weight (mg) | 184.2 ± 5.6 | 242.5 ± 15.9 | 304.6 ± 13.0 ** |
| Tibia length (mm) | 18.4 ± 0.1 | 18.4 ± 0.0 | 18.3 ± 0.1 |
| HW/TL (mg/mm) | 10.0 ± 0.3 | 13.2 ± 0.8 * | 16.6 ± 0.7 **## |
| LuW/TL (mg/mm) | 9.7 ± 0.2 | 11.8 ± 1.4 | 13.7 ± 2.0 |
| LiW/TL (mg/mm) | 83.5 ± 2.5 | 73.6 ± 2.6 | 80.7 ± 3.6 |
| KW/TL (mg/mm) | 10.6 ± 0.3 | 8.9 ± 0.2 ** | 9.7 ± 0.3 *# |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kessler, E.L.; Wang, J.-W.; Kok, B.; Brans, M.A.; Nederlof, A.; van Stuijvenberg, L.; Huang, C.; Vink, A.; Arslan, F.; Efimov, I.R.; et al. Ventricular TLR4 Levels Abrogate TLR2-Mediated Adverse Cardiac Remodeling upon Pressure Overload in Mice. Int. J. Mol. Sci. 2021, 22, 11823. https://doi.org/10.3390/ijms222111823
Kessler EL, Wang J-W, Kok B, Brans MA, Nederlof A, van Stuijvenberg L, Huang C, Vink A, Arslan F, Efimov IR, et al. Ventricular TLR4 Levels Abrogate TLR2-Mediated Adverse Cardiac Remodeling upon Pressure Overload in Mice. International Journal of Molecular Sciences. 2021; 22(21):11823. https://doi.org/10.3390/ijms222111823
Chicago/Turabian StyleKessler, Elise L., Jiong-Wei Wang, Bart Kok, Maike A. Brans, Angelique Nederlof, Leonie van Stuijvenberg, Chenyuan Huang, Aryan Vink, Fatih Arslan, Igor R. Efimov, and et al. 2021. "Ventricular TLR4 Levels Abrogate TLR2-Mediated Adverse Cardiac Remodeling upon Pressure Overload in Mice" International Journal of Molecular Sciences 22, no. 21: 11823. https://doi.org/10.3390/ijms222111823