
The Influences of Sulphation, Salt Type, and Salt Concentration on the Structural Heterogeneity of Glycosaminoglycans



Abstract
:1. Introduction
2. Materials and Methods
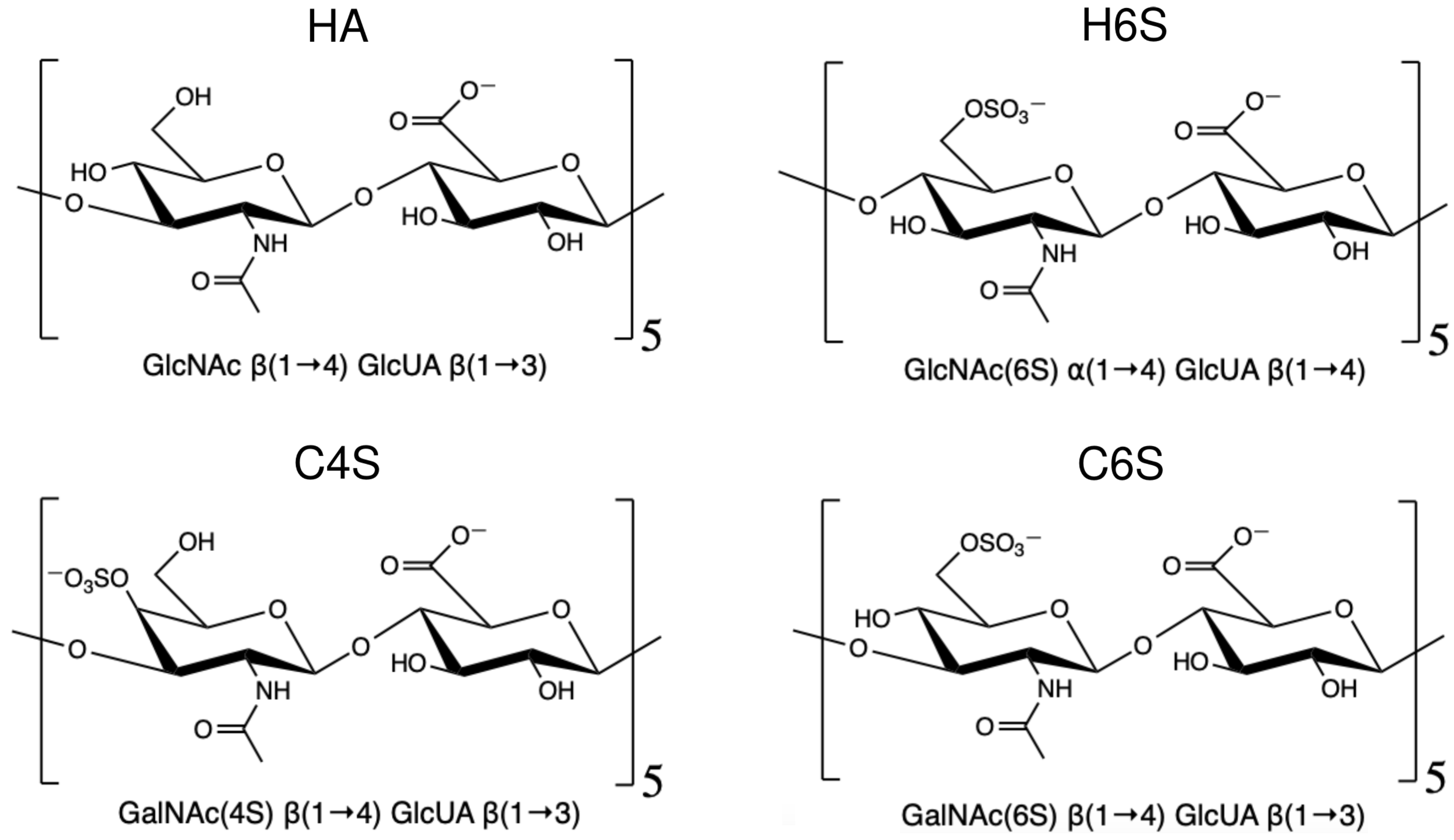
2.1. Model Systems
2.2. Simulation Protocols
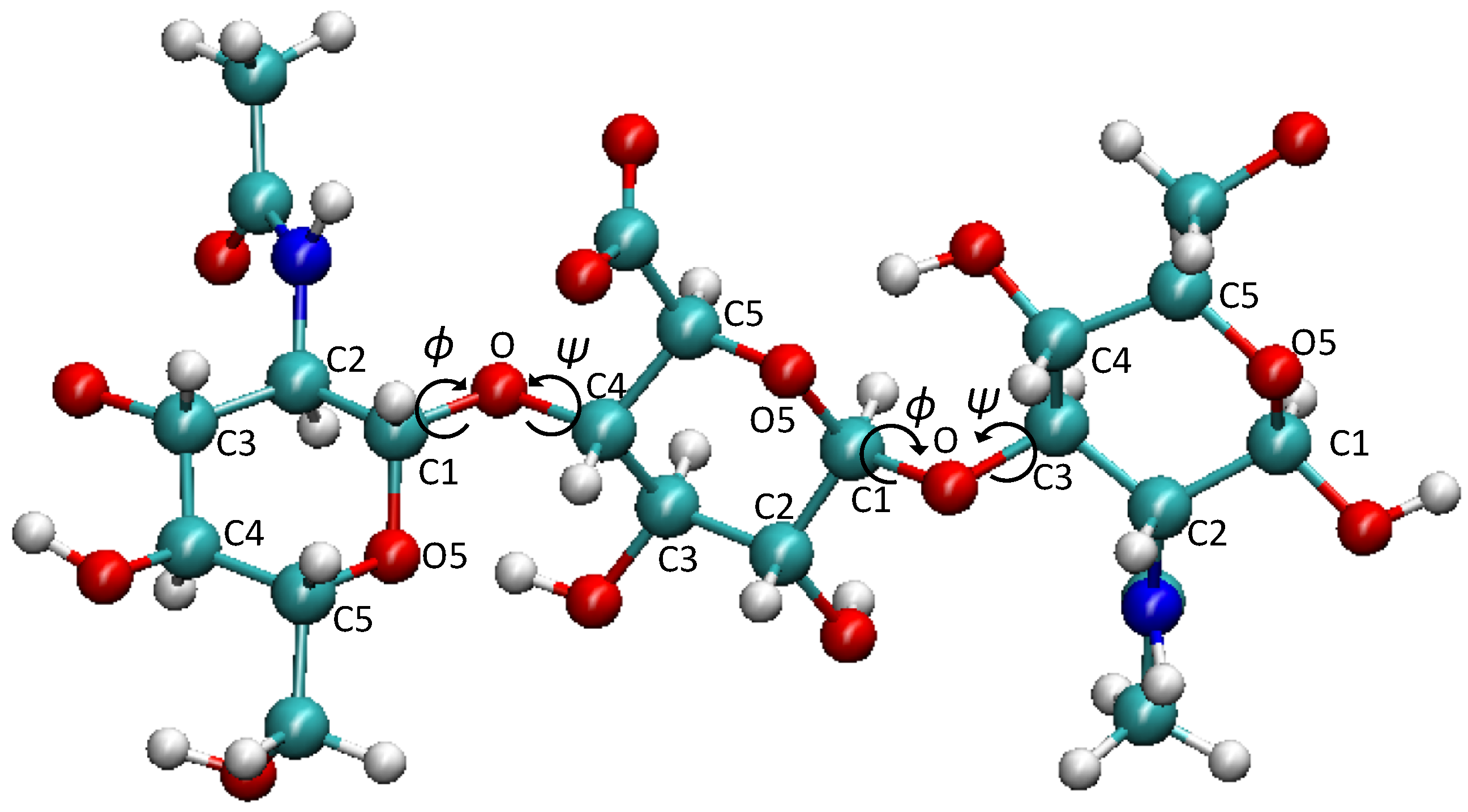
2.3. Conformational Analysis
2.4. Free Energy Landscape
3. Results and Discussions
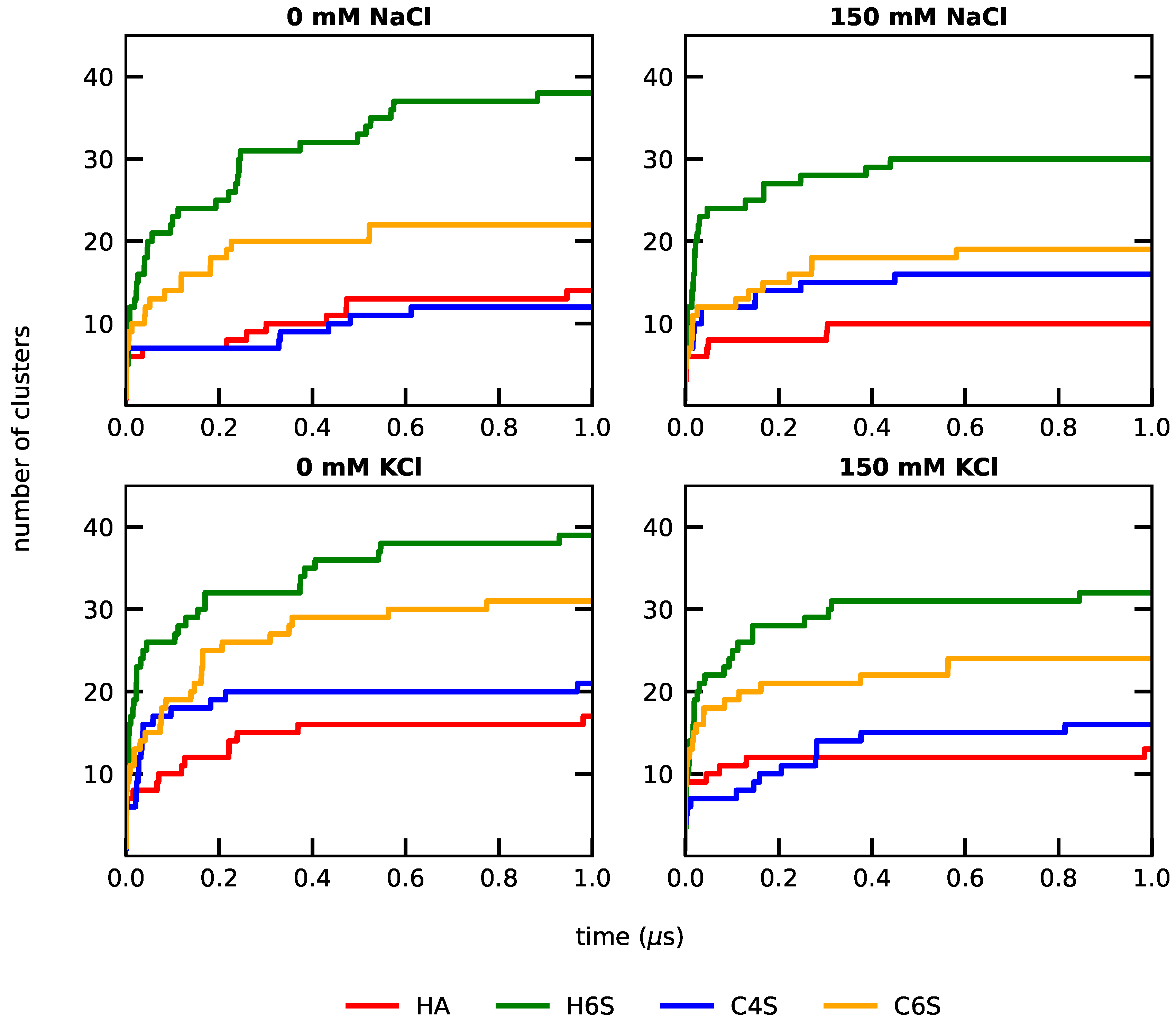
3.1. Characterization of the GAGs’ Structural Data
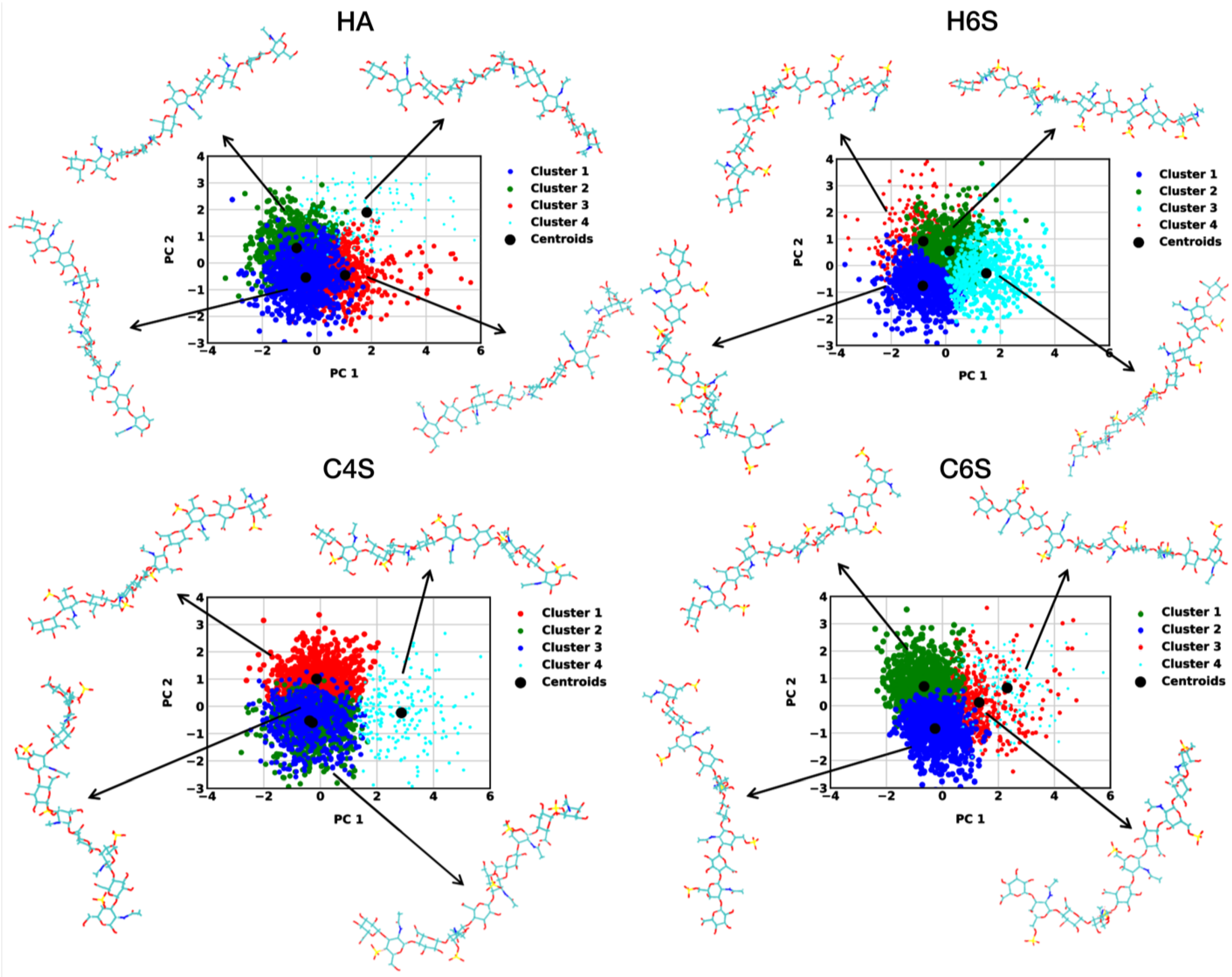
3.1.1. RMSD-Based Conformational Clustering
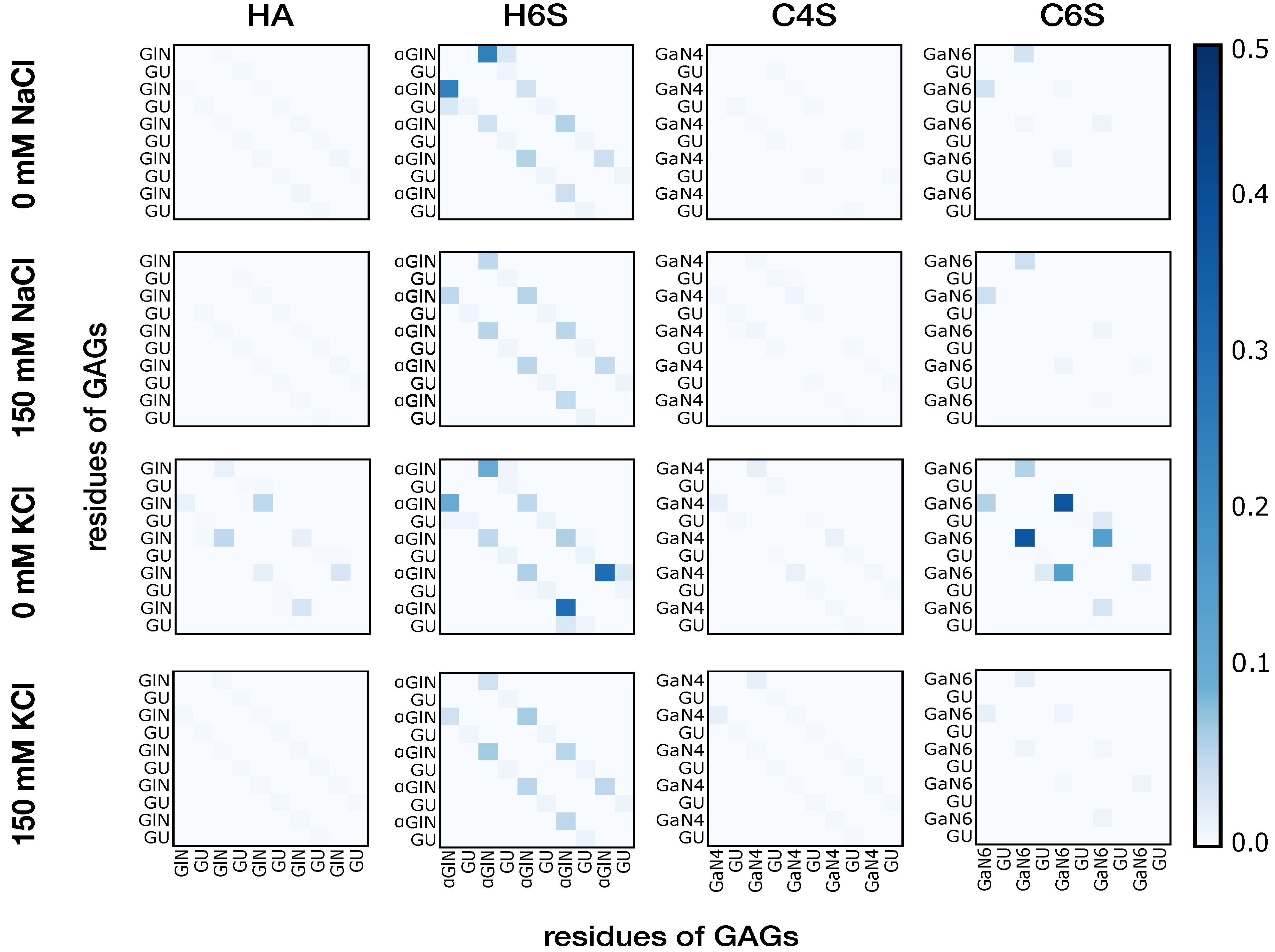
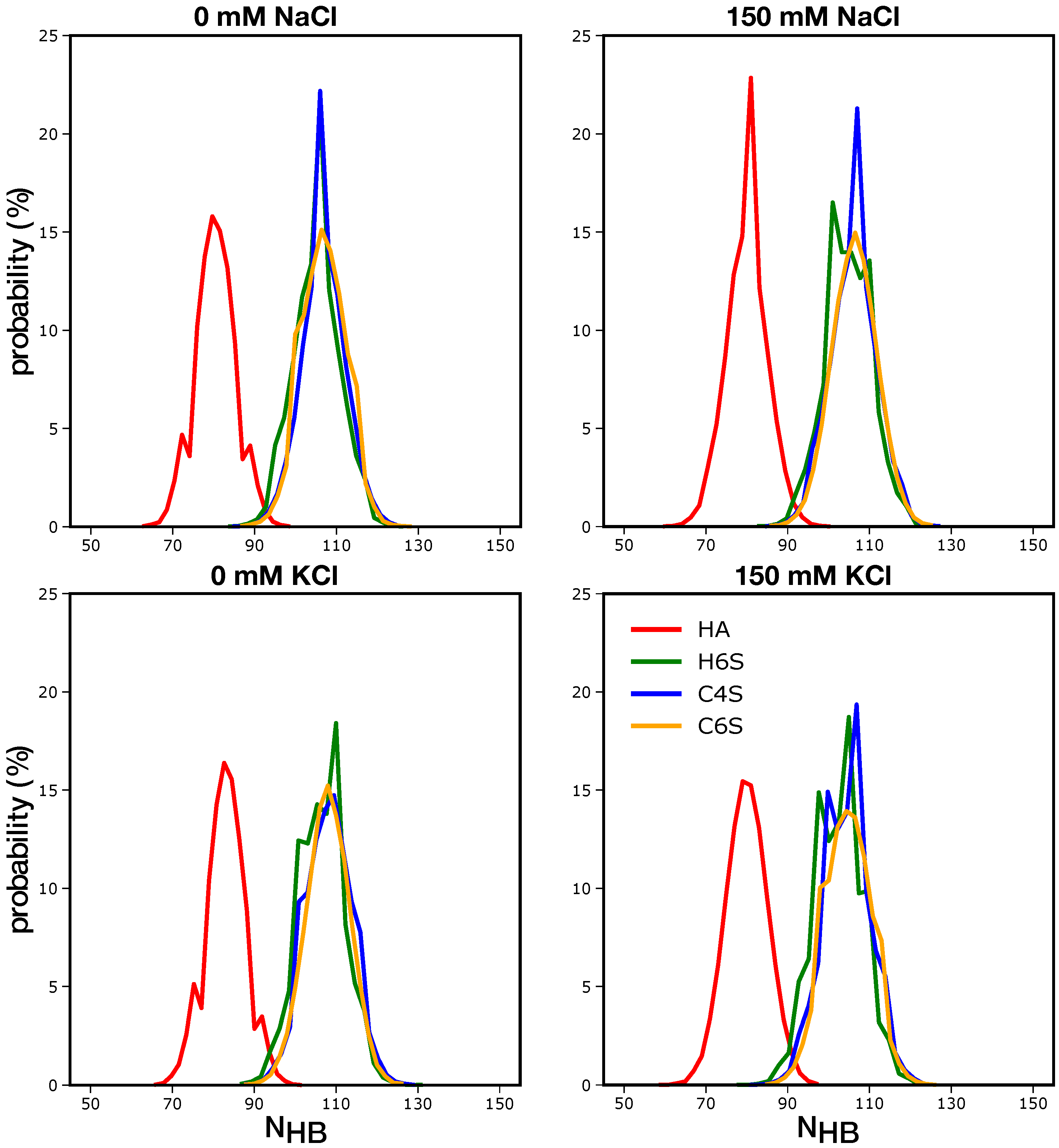
3.1.2. Intramolecular Interactions in GAGs
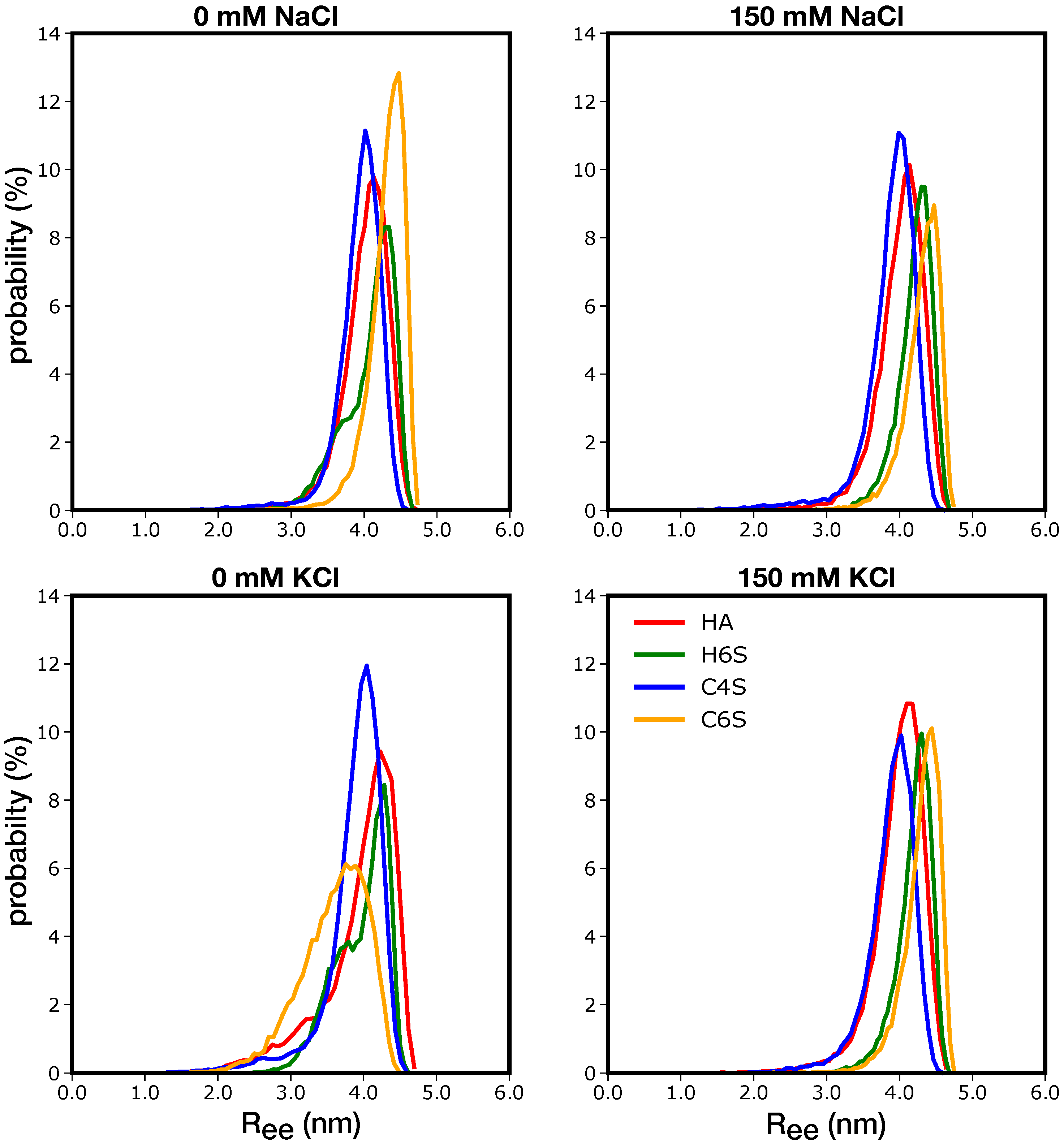
3.1.3. Shape of the GAGs
3.2. Characterization of the GAGs Interactions with Water and Ions
3.2.1. GAG–Water Interactions
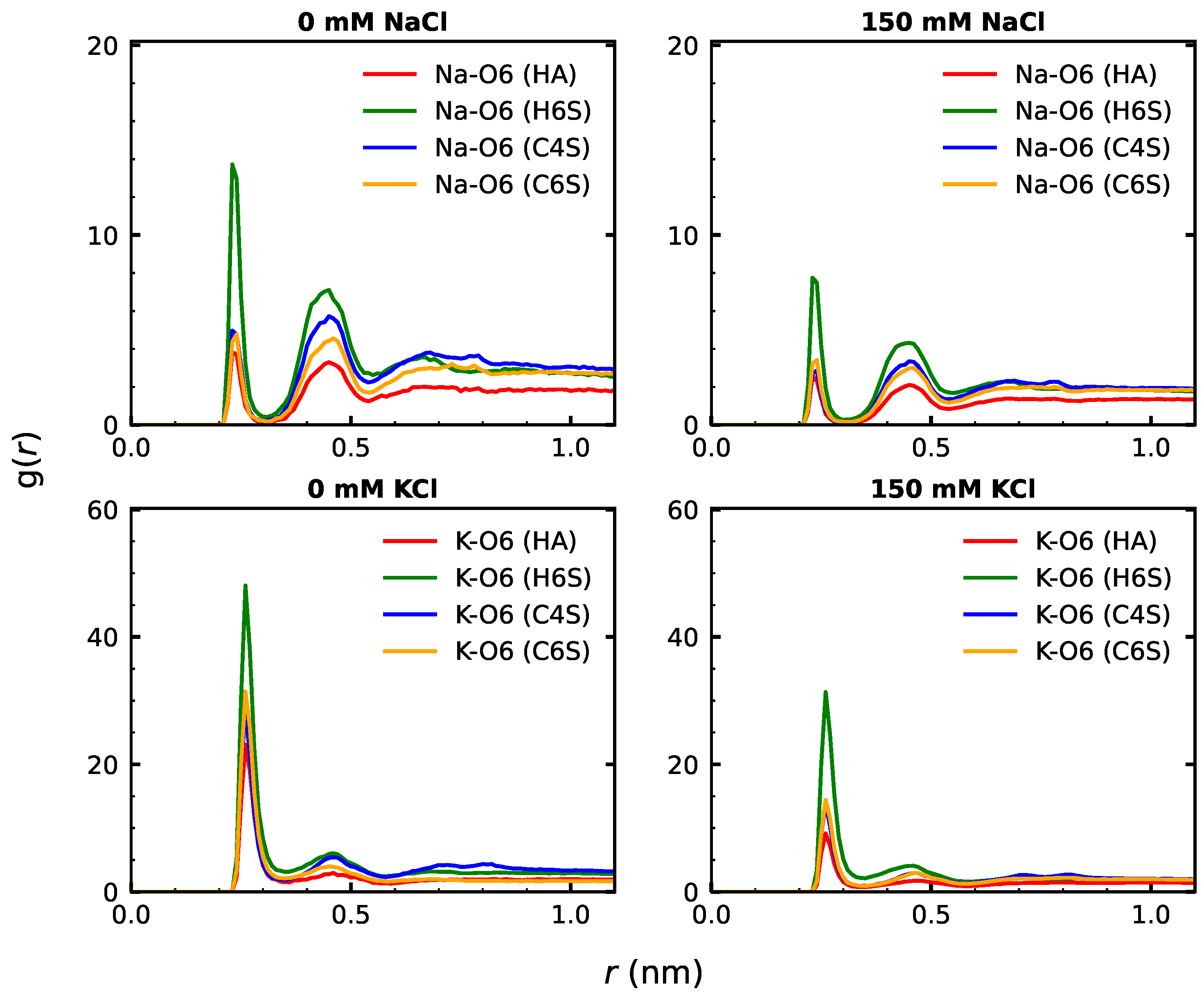
3.2.2. GAG–Ion Interactions
3.3. Free Energy Profile Based on Collective Fluctuations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nicholson, C.; Syková, E. Extracellular space structure revealed by diffusion analysis. Trends Neurosci. 1998, 21, 207–215. [Google Scholar] [CrossRef]
- Gandhi, N.S.; Mancera, R.L. The structure of glycosaminoglycans and their interactions with proteins. Chem. Biol. Drug Des. 2008, 72, 455–482. [Google Scholar] [CrossRef]
- Dityatev, A.; Schachner, M.; Sonderegger, P. The dual role of the extracellular matrix in synaptic plasticity and homeostasis. Nat. Rev. Neurosci. 2010, 11, 735–746. [Google Scholar] [CrossRef]
- Jackson, R.L.; Busch, S.J.; Cardin, A.D. Glycosaminoglycans: Molecular properties, protein interactions, and role in physiological processes. Physiol. Rev. 1991, 71, 481–539. [Google Scholar] [CrossRef]
- Gulati, K.; Poluri, K.M. Mechanistic and therapeutic overview of glycosaminoglycans: The unsung heroes of biomolecular signaling. Glycoconj. J. 2016, 33, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Motamedi-Shad, N.; Monsellier, E.; Chiti, F. Amyloid formation by the model protein muscle acylphosphatase is accelerated by heparin and heparan sulphate through a scaffolding-based mechanism. J. Biochem. 2009, 146, 805–814. [Google Scholar] [CrossRef]
- Ariga, T.; Miyatake, T.; Yu, R.K. Role of proteoglycans and glycosaminoglycans in the pathogenesis of Alzheimer’s disease and related disorders: Amyloidogenesis and therapeutic strategies—A review. J. Neurosci. Res. 2010, 88, 2303–2315. [Google Scholar] [CrossRef]
- Gigante, A.; Callegari, L. The role of intra-articular hyaluronan (Sinovial®) in the treatment of osteoarthritis. Rheumatol. Int. 2011, 31, 427–444. [Google Scholar] [CrossRef]
- Migliore, A.; Granata, M. Intra-articular use of hyaluronic acid in the treatment of osteoarthritis. Clin. Interv. Aging 2008, 3, 365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simanek, V.; Kren, V.; Ulrichová, J.; Gallo, J. The efficacy of glucosamine and chondroitin sulfate in the treatment of osteoarthritis: Are these saccharides drugs or nutraceuticals. Biomed. Pap. Med. Fac. Univ Palacky. Olomouc. Czech. Repub. 2005, 149, 51–56. [Google Scholar] [CrossRef] [Green Version]
- Salvatore, S.; Heuschkel, R.; Tomlin, S.; Davies, S.; Edwards, S.; Walker-Smith, J.; French, I.; Murch, S. A pilot study of N-acetyl glucosamine, a nutritional substrate for glycosaminoglycan synthesis, in paediatric chronic inflammatory bowel disease. Aliment. Pharmacol. Ther. 2000, 14, 1567–1579. [Google Scholar] [CrossRef] [PubMed]
- Pomin, V.H. Keratan sulfate: An up-to-date review. Int. J. Biol. Macromol. 2015, 72, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Zhang, Z.; Linhardt, R.J. Glycosaminoglycans. In Handb. Glycomics; Elsevier: Amsterdam, The Netherlands, 2010; pp. 59–80. [Google Scholar] [CrossRef]
- Mycroft-West, C.; Su, D.; Li, Y.; Guimond, S.; Rudd, T.; Elli, S.; Miller, G.; Nunes, Q.; Procter, P.; Bisio, A.; et al. Glycosaminoglycans induce conformational change in the SARS-CoV-2 Spike S1 Receptor Binding Domain. BioRxiv 2020. [Google Scholar] [CrossRef]
- Fouissac, E.; Milas, M.; Rinaudo, M.; Borsali, R. Influence of the ionic strength on the dimensions of sodium hyaluronate. Macromolecules 1992, 25, 5613–5617. [Google Scholar] [CrossRef]
- Hayashi, K.; Tsutsumi, K.; Nakajima, F.; Norisuye, T.; Teramoto, A. Chain-stiffness and excluded-volume effects in solutions of sodium hyaluronate at high ionic strength. Macromolecules 1995, 28, 3824–3830. [Google Scholar] [CrossRef]
- Mendichi, R.; Šoltés, L.; Giacometti Schieroni, A. Evaluation of radius of gyration and intrinsic viscosity molar mass dependence and stiffness of hyaluronan. Biomacromolecules 2003, 4, 1805–1810. [Google Scholar] [CrossRef]
- Musilová, L.; Kašpárková, V.; Mráček, A.; Minařík, A.; Minařík, M. The behavior of hyaluronan solutions in the presence of Hofmeister ions: A light scattering, viscometry and surface tension study. Carbohydr. Polym. 2019, 212, 395–402. [Google Scholar] [CrossRef]
- Gama, C.I.; Tully, S.E.; Sotogaku, N.; Clark, P.M.; Rawat, M.; Vaidehi, N.; Goddard, W.A.; Nishi, A.; Hsieh-Wilson, L.C. Sulfation patterns of glycosaminoglycans encode molecular recognition and activity. Nat. Chem. Biol. 2006, 2, 467–473. [Google Scholar] [CrossRef] [Green Version]
- Soares da Costa, D.; Reis, R.L.; Pashkuleva, I. Sulfation of glycosaminoglycans and its implications in human health and disorders. Annu. Rev. Biomed. Eng. 2017, 19, 1–26. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Cremer, P.S. Interactions between macromolecules and ions: The Hofmeister series. Curr. Opin. Chem. Biol. 2006, 10, 658–663. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.A.; Meade, K.; Pickford, C.E.; Holley, R.J.; Merry, C.L. Glycosaminoglycans as regulators of stem cell differentiation. Biochem. Soc. Trans. 2011, 39, 383–387. [Google Scholar] [CrossRef]
- Gasimli, L.; Hickey, A.M.; Yang, B.; Li, G.; dela Rosa, M.; Nairn, A.V.; Kulik, M.J.; Dordick, J.S.; Moremen, K.W.; Dalton, S.; et al. Changes in glycosaminoglycan structure on differentiation of human embryonic stem cells towards mesoderm and endoderm lineages. Biochim. Biophys. Acta, Gen. Subj. 2014, 1840, 1993–2003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudd, T.; Skidmore, M.; Guerrini, M.; Hricovini, M.; Powell, A.; Siligardi, G.; Yates, E. The conformation and structure of GAGs: Recent progress and perspectives. Curr. Opin. Struct. Biol. 2010, 20, 567–574. [Google Scholar] [CrossRef]
- Kutálková, E.; Hrnčiřík, J.; Witasek, R.; Ingr, M. Effect of solvent and ions on the structure and dynamics of a hyaluronan molecule. Carbohydr. Polym. 2020, 234, 115919. [Google Scholar] [CrossRef] [PubMed]
- Faller, C.E.; Guvench, O. Sulfation and cation effects on the conformational properties of the glycan backbone of chondroitin sulfate disaccharides. J. Phys. Chem. B 2015, 119, 6063–6073. [Google Scholar] [CrossRef] [Green Version]
- Whitmore, E.K.; Vesenka, G.; Sihler, H.; Guvench, O. Efficient Construction of Atomic-Resolution Models of Non-Sulfated Chondroitin Glycosaminoglycan Using Molecular Dynamics Data. Biomolecules 2020, 10, 537. [Google Scholar] [CrossRef] [Green Version]
- Whitmore, E.K.; Martin, D.; Guvench, O. Constructing 3-dimensional atomic-resolution models of nonsulfated glycosaminoglycans with arbitrary lengths using conformations from molecular dynamics. Int. J. Mol. Sci. 2020, 21, 7699. [Google Scholar] [CrossRef] [PubMed]
- Guvench, O.; Whitmore, E.K. Sulfation and Calcium Favor Compact Conformations of Chondroitin in Aqueous Solutions. ACS Omega 2021, 6, 13204–13217. [Google Scholar] [CrossRef]
- Giubertoni, G.; Pérez de Alba Ortíz, A.; Bano, F.; Zhang, X.; Linhardt, R.J.; Green, D.E.; DeAngelis, P.L.; Koenderink, G.H.; Richter, R.P.; Ensing, B.; et al. Strong reduction of the chain rigidity of hyaluronan by selective binding of Ca2+ ions. Macromolecules 2021, 54, 1137–1146. [Google Scholar] [CrossRef]
- Winter, W.T.; Arnott, S.; Isaac, D.H.; Atkins, E.D. Chondroitin 4-Sulfate: The Structure of a Sulfated Glycosaminoglycan. J. Mol. Biol. 1978, 125, 1–19. [Google Scholar] [CrossRef]
- Cael, J.J.; Winter, W.T.; Arnott, S. Calcium Chondroitin 4-Sulfate: Molecular Conformation and Organization of Polysaccharide Chains in a Proteoglycan. J. Mol. Biol. 1978, 125, 21–42. [Google Scholar] [CrossRef]
- Rigden, D.J.; Jedrzejas, M.J. Structures of Streptococcus pneumoniae Hyaluronate Lyase in Complex with Chondroitin and Chondroitin Sulfate Disaccharides. Insights into Specificity and Mechanism of Action. J. Biol. Chem. 2003, 278, 50596–50606. [Google Scholar] [CrossRef] [Green Version]
- Lunin, V.V.; Li, Y.; Linhardt, R.J.; Miyazono, H.; Kyogashima, M.; Kaneko, T.; Bell, A.W.; Cygler, M. High-Resolution Crystal Structure of Arthrobacter aurescens Chondroitin AC Lyase: An Enzyme-Substrate Complex Defines the Catalytic Mechanism. J. Mol. Biol. 2004, 337, 367–386. [Google Scholar] [CrossRef]
- Michel, G.; Pojasek, K.; Li, Y.; Sulea, T.; Linhardt, R.J.; Raman, R.; Prabhakar, V.; Sasisekharan, R.; Cygler, M. The Structure of Chondroitin B Lyase Complexed with Glycosaminoglycan Oligosaccharides Unravels a Calcium-Dependent Catalytic Machinery. J. Biol. Chem. 2004, 279, 32882–32896. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Kienetz, M.; Cherney, M.M.; James, M.N.; Bromme, D. The Crystal and Molecular Structures of a Cathepsin K:Chondroitin Sulfate Complex. J. Mol. Biol. 2008, 383, 78–91. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.; Gittis, A.G.; Nguyen, P.; Gowda, D.C.; Miller, L.H.; Garboczi, D.N. Structure of the DBL3x Domain of Pregnancy-Associated Malaria Protein VAR2CSA Complexed with Chondroitin Sulfate A. Nat. Struct. Mol. Biol. 2008, 15, 932–938. [Google Scholar] [CrossRef] [PubMed]
- Cherney, M.M.; Lecaille, F.; Kienitz, M.; Nallaseth, F.S.; Li, Z.; James, M.N.; Bromme, D. Structure-Activity Analysis of Cathepsin K/Chondroitin 4-Sulfate Interactions. J. Biol. Chem. 2011, 286, 8988–8998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sattelle, B.M.; Shakeri, J.; Roberts, I.S.; Almond, A. A 3D-Structural Model of Unsulfated Chondroitin from High-Field NMR: 4-Sulfation Has Little Effect on Backbone Conformation. Carbohydr. Res. 2010, 345, 291–302. [Google Scholar] [CrossRef]
- Yu, F.; Wolff, J.J.; Amster, I.J.; Prestegard, J.H. Conformational Preferences of Chondroitin Sulfate Oligomers Using Partially Oriented Nmr Spectroscopy of 13C-Labeled Acetyl Groups. J. Am. Chem. Soc. 2007, 129, 13288–13297. [Google Scholar] [CrossRef] [PubMed]
- Sattelle, B.M.; Hansen, S.U.; Gardiner, J.; Almond, A. Free energy landscapes of iduronic acid and related monosaccharides. J. Am. Chem. Soc. 2010, 132, 13132–13134. [Google Scholar] [CrossRef]
- Sattelle, B.M.; Shakeri, J.; Almond, A. Does microsecond sugar ring flexing encode 3D-shape and bioactivity in the heparanome? Biomacromolecules 2013, 14, 1149–1159. [Google Scholar] [CrossRef]
- Samsonov, S.A.; Gehrcke, J.P.; Pisabarro, M.T. Flexibility and explicit solvent in molecular-dynamics-based docking of protein–glycosaminoglycan systems. J. Chem. Inf. Model. 2014, 54, 582–592. [Google Scholar] [CrossRef] [PubMed]
- Nagarajan, B.; Sankaranarayanan, N.V.; Desai, U.R. Rigorous analysis of free solution glycosaminoglycan dynamics using simple, new tools. Glycobiology 2020, 30, 516–527. [Google Scholar] [CrossRef] [PubMed]
- Almond, A.; Brass, A.; Sheehan, J.K. Dynamic exchange between stabilized conformations predicted for hyaluronan tetrasaccharides: Comparison of molecular dynamics simulations with available NMR data. Glycobiology 1998, 8, 973–980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cilpa, G.; Hyvönen, M.T.; Koivuniemi, A.; Riekkola, M.L. Atomistic insight into chondroitin-6-sulfate glycosaminoglycan chain through quantum mechanics calculations and molecular dynamics simulation. J. Comput. Chem. 2010, 31, 1670–1680. [Google Scholar] [CrossRef] [PubMed]
- Sapay, N.; Cabannes, E.; Petitou, M.; Imberty, A. Molecular modeling of the interaction between heparan sulfate and cellular growth factors: Bringing pieces together. Glycobiology 2011, 21, 1181–1193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almond, A. Multiscale modeling of glycosaminoglycan structure and dynamics: Current methods and challenges. Curr. Opin. Struct. Biol. 2018, 50, 58–64. [Google Scholar] [CrossRef] [Green Version]
- Sankaranarayanan, N.V.; Nagarajan, B.; Desai, U.R. So you think computational approaches to understanding glycosaminoglycan–protein interactions are too dry and too rigid? Think again! Curr. Opin. Struct. Biol. 2018, 50, 91–100. [Google Scholar] [CrossRef]
- Guvench, O.; Hatcher, E.; Venable, R.M.; Pastor, R.W.; MacKerell, A.D., Jr. CHARMM Additive All-Atom Force Field for Glycosidic Linkages between Hexopyranoses. J. Chem. Theory Comput. 2009, 5, 2353–2370. [Google Scholar] [CrossRef] [Green Version]
- Raman, E.P.; Guvench, O.; MacKerell, A.D., Jr. CHARMM additive all-atom force field for glycosidic linkages in carbohydrates involving furanoses. J. Phys. Chem. B 2010, 114, 12981–12994. [Google Scholar] [CrossRef] [Green Version]
- Guvench, O.; Mallajosyula, S.S.; Raman, E.P.; Hatcher, E.; Vanommeslaeghe, K.; Foster, T.J.; Jamison, F.W.; MacKerell, A.D., Jr. CHARMM additive all-atom force field for carbohydrate derivatives and its utility in polysaccharide and carbohydrate–protein modeling. J. Chem. Theory Comput. 2011, 7, 3162–3180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mallajosyula, S.S.; Guvench, O.; Hatcher, E.; MacKerell, A.D., Jr. CHARMM additive all-atom force field for phosphate and sulfate linked to carbohydrates. J. Chem. Theory Comput. 2012, 8, 759–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jo, S.; Song, K.C.; Desaire, H.; MacKerell, A.D.; Im, W. Glycan reader: Automated sugar identification and simulation preparation for carbohydrates and glycoproteins. J. Comput. Chem. 2011, 32, 3135–3141. [Google Scholar] [CrossRef] [Green Version]
- Park, S.J.; Lee, J.; Patel, D.S.; Ma, H.; Lee, H.S.; Jo, S.; Im, W. Glycan Reader is improved to recognize most sugar types and chemical modifications in the Protein Data Bank. Bioinformatics 2017, 33, 3051–3057. [Google Scholar] [CrossRef] [PubMed]
- Park, S.J.; Lee, J.; Qi, Y.; Kern, N.R.; Lee, H.S.; Jo, S.; Joung, I.; Joo, K.; Lee, J.; Im, W. CHARMM-GUI Glycan Modeler for modeling and simulation of carbohydrates and glycoconjugates. Glycobiology 2019, 29, 320–331. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Yoo, J.; Aksimentiev, A. Improved Parametrization of Li+, Na+, K+, and Mg2+ Ions for All-Atom Molecular Dynamics Simulations of Nucleic Acid Systems. J. Phys. Chem. Lett. 2012, 3, 45–50. [Google Scholar] [CrossRef]
- Sterling, J.D.; Jiang, W.; Botello-Smith, W.M.; Luo, Y.L. Ion pairing and dielectric decrement in glycosaminoglycan brushes. J. Phys. Chem. B 2021, 125, 2771–2780. [Google Scholar] [CrossRef] [PubMed]
- Mark, P.; Nilsson, L. Structure and dynamics of the TIP3P, SPC, and SPC/E water models at 298 K. J. Phys. Chem. A 2001, 105, 9954–9960. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.; Fraaije, J.G. LINCS: A Linear Constraint Solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Hess, B.; Kutzner, C.; Van Der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [Green Version]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindah, E. Gromacs: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Lemkul, J. From Proteins to Perturbed Hamiltonians: A Suite of Tutorials for the GROMACS-2018 Molecular Simulation Package [Article v1.0]. Living J. Comput. Mol. Sci. 2019, 1, 5068. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Samantray, S.; Strodel, B. The effects of different glycosaminoglycans on the structure and aggregation of the amyloid-β (16–22) peptide. J. Phys. Chem. B 2021, 125, 5511–5525. [Google Scholar] [CrossRef]
- Michaud-Agrawal, N.; Denning, E.J.; Woolf, T.B.; Beckstein, O. MDAnalysis: A toolkit for the analysis of molecular dynamics simulations. J. Comput. Chem. 2011, 32, 2319–2327. [Google Scholar] [CrossRef] [Green Version]
- McGibbon, R.T.; Beauchamp, K.A.; Harrigan, M.P.; Klein, C.; Swails, J.M.; Hernández, C.X.; Schwantes, C.R.; Wang, L.P.; Lane, T.J.; Pande, V.S. MDTraj: A Modern Open Library for the Analysis of Molecular Dynamics Trajectories. Biophys. J. 2015, 109, 1528–1532. [Google Scholar] [CrossRef] [Green Version]
- Daura, X.; Gademann, K.; Jaun, B.; Seebach, D.; van Gunsteren, W.F.; Mark, A.E. Peptide Folding: When Simulation Meets Experiment. Angew. Chemie Int. Ed. 1999, 38, 236–240. [Google Scholar] [CrossRef]
- Varki, A.; Cummings, R.D.; Esko, J.D.; Stanley, P.; Hart, G.W.; Aebi, M.; Darvill, A.G.; Kinoshita, T.; Packer, N.H.; Prestegard, J.H.; et al. Essentials of Glycobiology; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2017. [Google Scholar]
- Samantray, S.; Cheung, D.L. Effect of the air—Water interface on the conformation of amyloid beta. Biointerphases 2020, 15, 061011. [Google Scholar] [CrossRef] [PubMed]
- Paul, A.; Samantray, S.; Anteghini, M.; Khaled, M.; Strodel, B. Thermodynamics and kinetics of the amyloid-β peptide revealed by Markov state models based on MD data in agreement with experiment. Chem. Sci. 2021, 12, 6652–6669. [Google Scholar] [CrossRef] [PubMed]
- Kherb, J.; Flores, S.C.; Cremer, P.S. Role of Carboxylate Side Chains in the Cation Hofmeister Series. J. Phys. Chem. B 2012, 116, 7389–7397. [Google Scholar] [CrossRef] [PubMed]
- Aziz, E.F.; Ottosson, N.; Eisebitt, S.; Eberhardt, W.; Jagoda-Cwiklik, B.; Vácha, R.; Jungwirth, P.; Winter, B. Cation-Specific Interactions with Carboxylate in Amino Acid and Acetate Aqueous Solutions: X-ray Absorption and ab initio Calculations. J. Phys. Chem. B 2008, 112, 12567–12570. [Google Scholar] [CrossRef]
- Kognole, A.; MacKerell, A. Contributions and competition of Mg2+ and K+ in folding and stabilization of the Twister Ribozyme. RNA 2020, 26, 1704–1715. [Google Scholar] [CrossRef] [PubMed]
- Duboué-Dijon, E.; Javanainen, M.; Delcroix, P.; Jungwirth, P.; Martinez-Seara, H. A practical guide to biologically relevant molecular simulations with charge scaling for electronic polarization. J. Chem. Phys. 2020, 153, 050901. [Google Scholar] [CrossRef] [PubMed]
- Wineman-Fisher, V.; Al-Hamdani, Y.; Nagy, P.R.; Tkatchenko, A.; Varma, S. Improved description of ligand polarization enhances transferability of ion–ligand interactions. J. Chem. Phys. 2020, 153, 094115. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | ||
|---|---|---|
| HA | (-GlcNAc-(1→4)-GlcUA-) | (-GlcUA-(1→3)-GlcNAc-) |
| H6S | (-GlcNAc(6S)-(1→4)-GlcUA-) | (-GlcUA-(1→4)-GlcNAc(6S)-) |
| C4S | (-GalNAc(4S)-(1→4)-GlcUA-) | (-GlcUA-(1→3)-GalNAc(4S)-) |
| C6S | (-GalNAc(6S)-(1→4)-GlcUA-) | (-GlcUA-(1→3)-GalNAc(6S)-) |
| System | 0 mM NaCl | 150 mM NaCl | 0 mM KCl | 150 mM KCl |
|---|---|---|---|---|
| HA | 5 Na, 0 Cl | 35 Na, 30 Cl | 5 K, 0 Cl | 35 K, 30 Cl |
| H6S | 10 Na, 0 Cl | 40 Na, 30 Cl | 10 K, 0 Cl | 40 K, 30 Cl |
| C4S | 10 Na, 0 Cl | 40 Na, 30 Cl | 10 K, 0 Cl | 40 K, 30 Cl |
| C6S | 10 Na, 0 Cl | 40 Na, 30 Cl | 10 K, 0 Cl | 40 K, 30 Cl |
| System | Structure | % | (nm) | (°) | (°) | |
|---|---|---|---|---|---|---|
| HA | starting | 4.5 | 83.0 | (−132.1, −146.1) | (−93.4, 76.1) | |
| cluster 1 | 34.8 | 4.2 ± 0.0 | 84.9 ± 0.1 | (−74.5 ± 0.2, −128.2 ± 0.2) | (−77.2 ± 0.2, 124.3 ± 0.3) | |
| cluster 2 | 34.6 | 4.2 ± 0.0 | 76.3 ± 0.1 | (−73.8 ± 0.2, −128.7 ± 0.2) | (−78.6 ± 0.2, 124.6 ± 0.3) | |
| cluster 3 | 25.0 | 3.8 ± 0.0 | 81.5 ± 0.2 | (−67.7 ± 0.2, −119.7 ± 0.4) | (−78.9 ± 0.3, 129.3 ± 0.2) | |
| cluster 4 | 05.6 | 3.4 ± 0.1 | 81.8 ± 0.4 | (−71.5 ± 0.5, −123.4 ± 0.8) | (−78.4 ± 0.6, 87.9 ± 1.3) | |
| H6S | starting | 4.5 | 102.00 | (−111.6, 87.2) | (45.6, 64.3) | |
| cluster 1 | 32.3 | 4.3 ± 0.0 | 109.7 ± 0.2 | (−70.9 ± 0.2, 123.9 ± 0.2) | (79.4 ± 0.4, 46.9 ± 1.0) | |
| cluster 2 | 31.4 | 4.3 ± 0.0 | 104.1 ± 0.2 | (−76.8 ± 0.2, 116.3 ± 0.2) | (80.8 ± 0.3, 40.4 ± 1.0) | |
| cluster 3 | 22.2 | 4.4 ± 0.0 | 102.7 ± 0.2 | (−73.4 ± 0.2, 119.1 ± 0.3) | (86.0 ± 0.3, −22.4 ± 0.9) | |
| cluster 4 | 14.1 | 3.8 ± 0.0 | 106.3 ± 0.3 | (−73.4 ± 0.3, 120.7 ± 0.4) | (79.6 ± 0.5, 23.1 ± 1.8) | |
| C4S | starting | 4.4 | 108.0 | (−139.4, −146.9) | (−102.8, 91.1) | |
| cluster 1 | 34.0 | 3.9 ± 0.0 | 111.1 ± 0.1 | (−67.3 ± 0.2, −121.2 ± 0.2) | (−76.4 ± 0.2, 128.4 ± 0.3) | |
| cluster 2 | 30.7 | 4.0 ± 0.0 | 101.6 ± 0.1 | (−67.6 ± 0.2, −121.6 ± 0.2) | (−78.5 ± 0.2, 125.5 ± 0.3) | |
| cluster 3 | 26.6 | 4.0 ± 0.0 | 109.0 ± 0.2 | (−67.5 ± 0.2, −121.9 ± 0.2) | (−80.2 ± 0.2, 112.7 ± 0.5) | |
| cluster 4 | 08.7 | 3.2 ± 0.0 | 105.9 ± 0.4 | (−52.3 ± 0.7, −119.0 ± 0.3) | (−78.7 ± 0.4, 114.8 ± 1.5) | |
| C6S | starting | 4.4 | 114.0 | (−120.8, −156.3) | (−106.6, 87.4) | |
| cluster 1 | 38.8 | 4.4 ± 0.0 | 111.5 ± 0.1 | (−74.3 ± 0.2, −140.6 ± 0.3) | (−72.0 ± 0.2, 118.6 ± 0.4) | |
| cluster 2 | 38.0 | 4.4 ± 0.0 | 103.3 ± 0.1 | (−74.3 ± 0.2, −140.1 ± 0.2) | (−75.4 ± 0.2, 114.0 ± 0.4) | |
| cluster 3 | 17.6 | 3.9 ± 0.0 | 106.8 ± 0.2 | (−69.8 ± 0.3, −133.4 ± 0.4) | (−75.6 ± 0.3, 111.4 ± 0.9) | |
| cluster 4 | 05.6 | 4.3 ± 0.0 | 106.3 ± 0.5 | (−73.7 ± 0.8, −78.9 ± 1.3) | (−75.7 ± 0.5, 117.0 ± 0.9) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Samantray, S.; Olubiyi, O.O.; Strodel, B. The Influences of Sulphation, Salt Type, and Salt Concentration on the Structural Heterogeneity of Glycosaminoglycans. Int. J. Mol. Sci. 2021, 22, 11529. https://doi.org/10.3390/ijms222111529
Samantray S, Olubiyi OO, Strodel B. The Influences of Sulphation, Salt Type, and Salt Concentration on the Structural Heterogeneity of Glycosaminoglycans. International Journal of Molecular Sciences. 2021; 22(21):11529. https://doi.org/10.3390/ijms222111529
Chicago/Turabian StyleSamantray, Suman, Olujide O. Olubiyi, and Birgit Strodel. 2021. "The Influences of Sulphation, Salt Type, and Salt Concentration on the Structural Heterogeneity of Glycosaminoglycans" International Journal of Molecular Sciences 22, no. 21: 11529. https://doi.org/10.3390/ijms222111529