
Classification and Functional Analysis between Cancer and Normal Tissues Using Explainable Pathway Deep Learning through RNA-Sequencing Gene Expression


Abstract
:1. Introduction
2. Results
2.1. Selecting the Pathway Database for the PathDeep Approach
2.2. PathDeep Performance Compared with That of Other Methods
2.3. Sample Visualization Using Gene Expression and Pathway Index
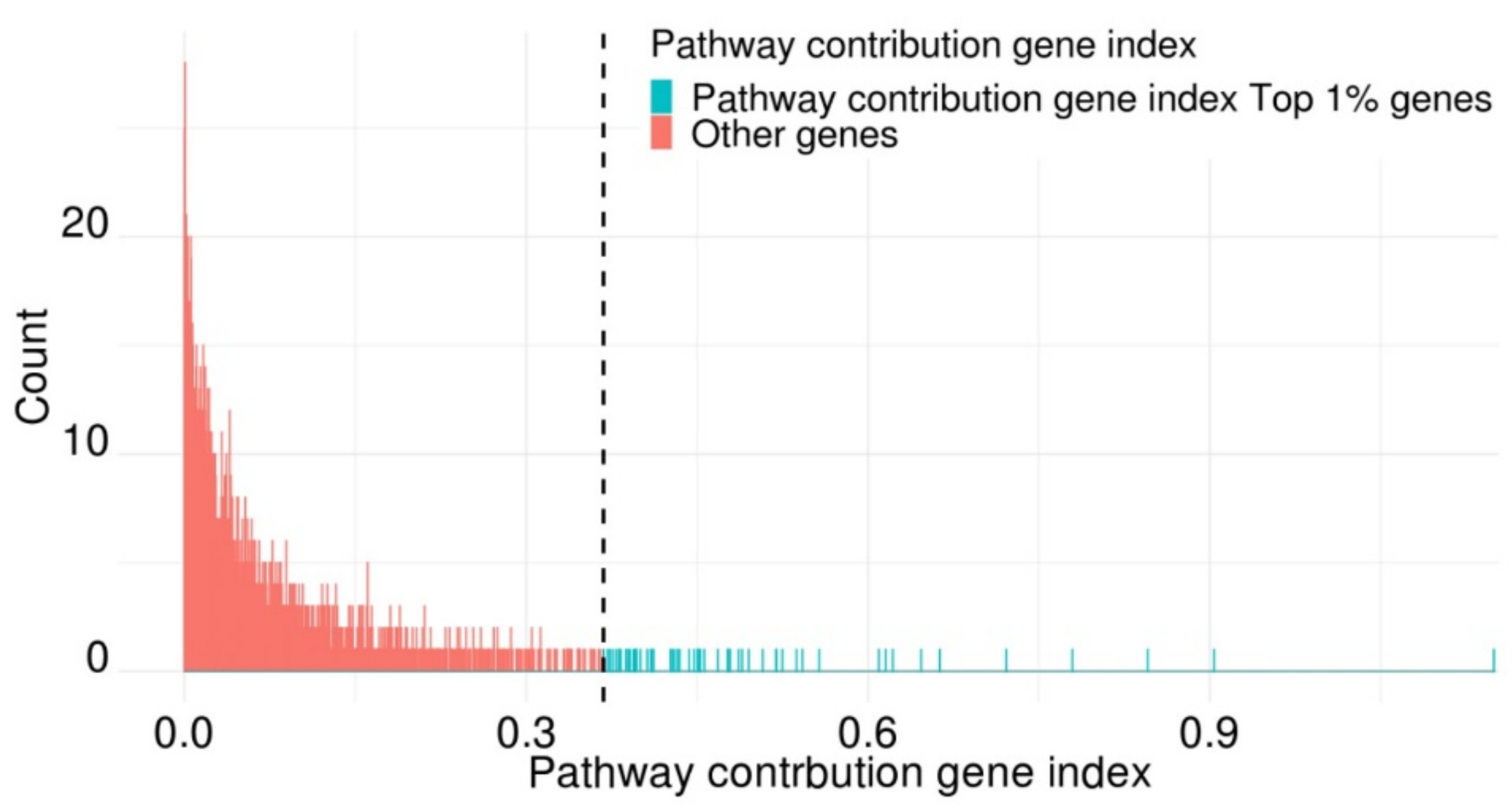
2.4. Functional Analysis Using Highly Contributing Pathway Genes
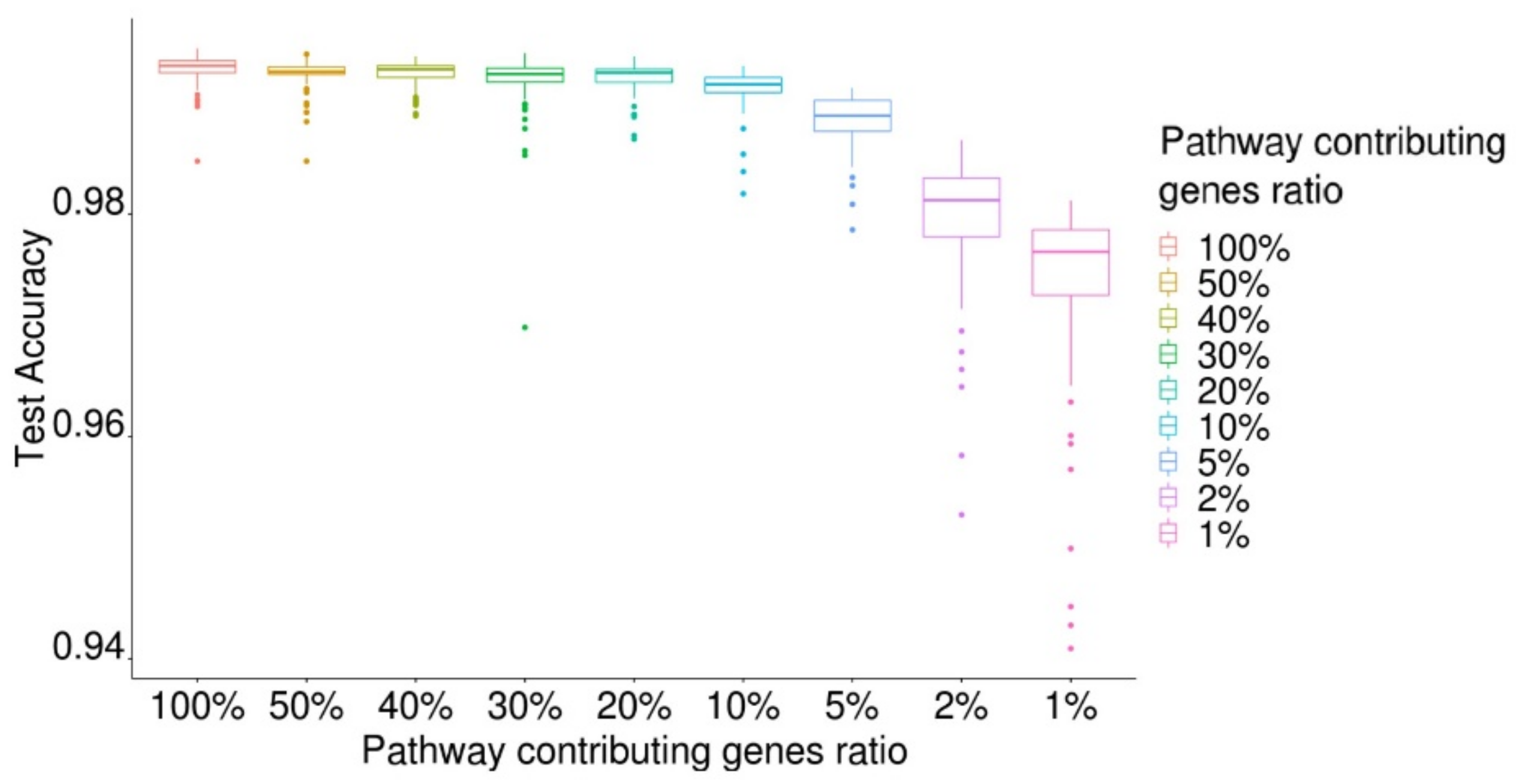
2.5. Classification of Cancer and Normal Tissues Using Key Genes with a High Pathway-Contributing Index
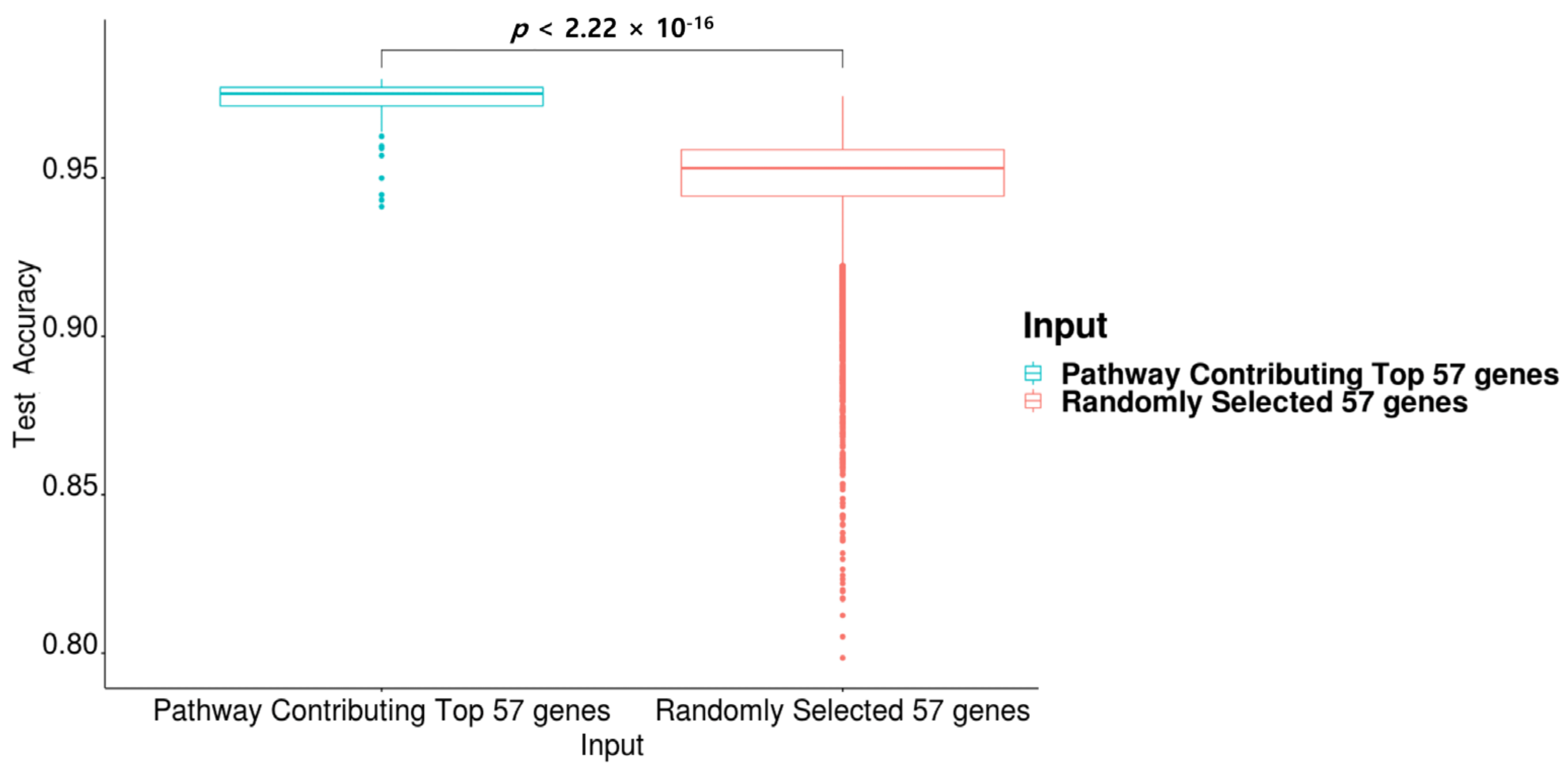
2.6. Pathway Contribution Gene Indexes of the Top 57 Genes in PathDeep versus Those of a Randomly Selected 57 Genes in PathDeep
2.7. Identification of Essential Pathways for Cancer Prediction and Evaluation of Model Performance of the Essential Pathways
3. Discussion
4. Materials and Methods
4.1. Gene Expression Data
4.2. MSigDB with TCGA TARGET GTEx
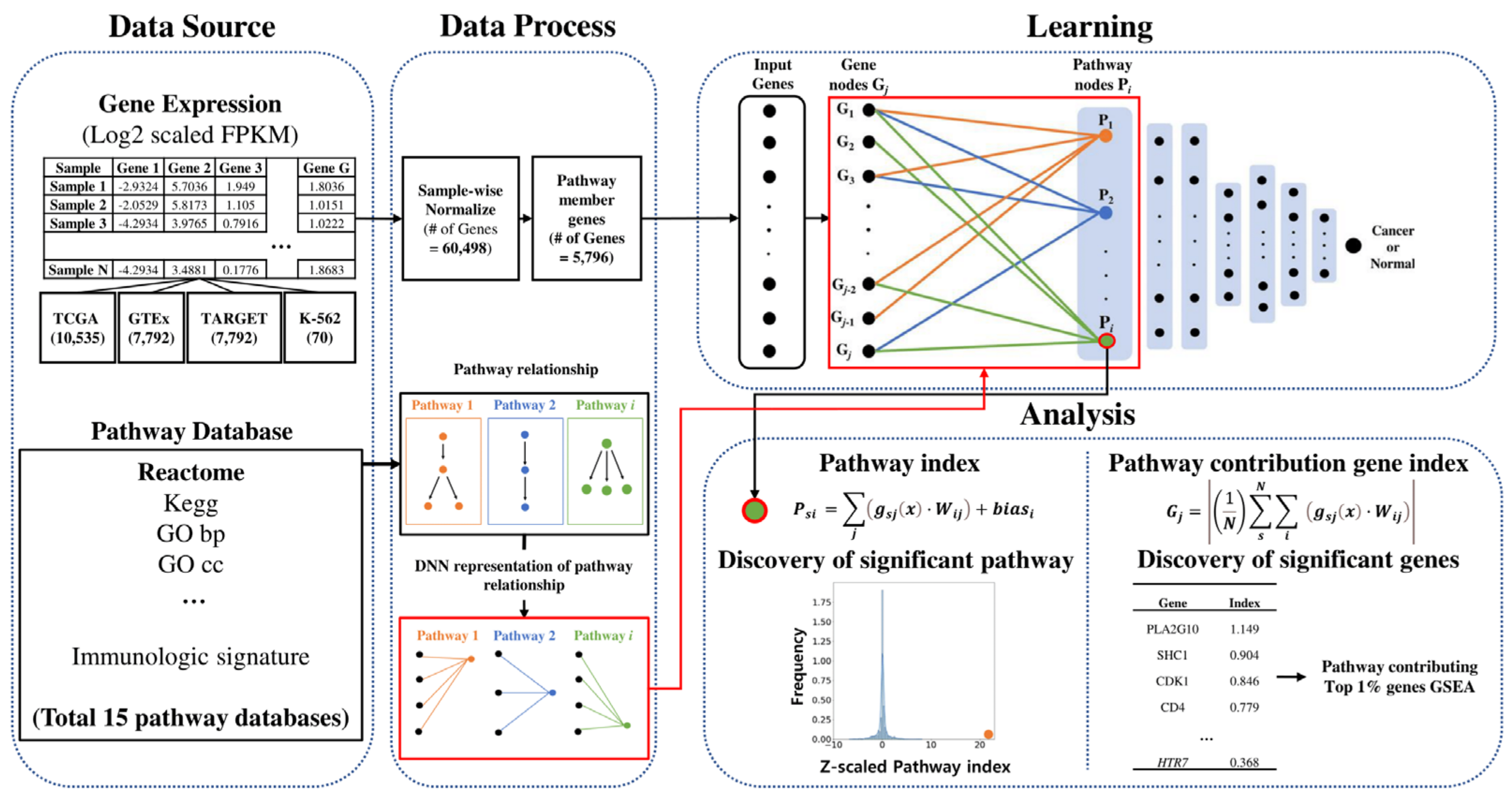
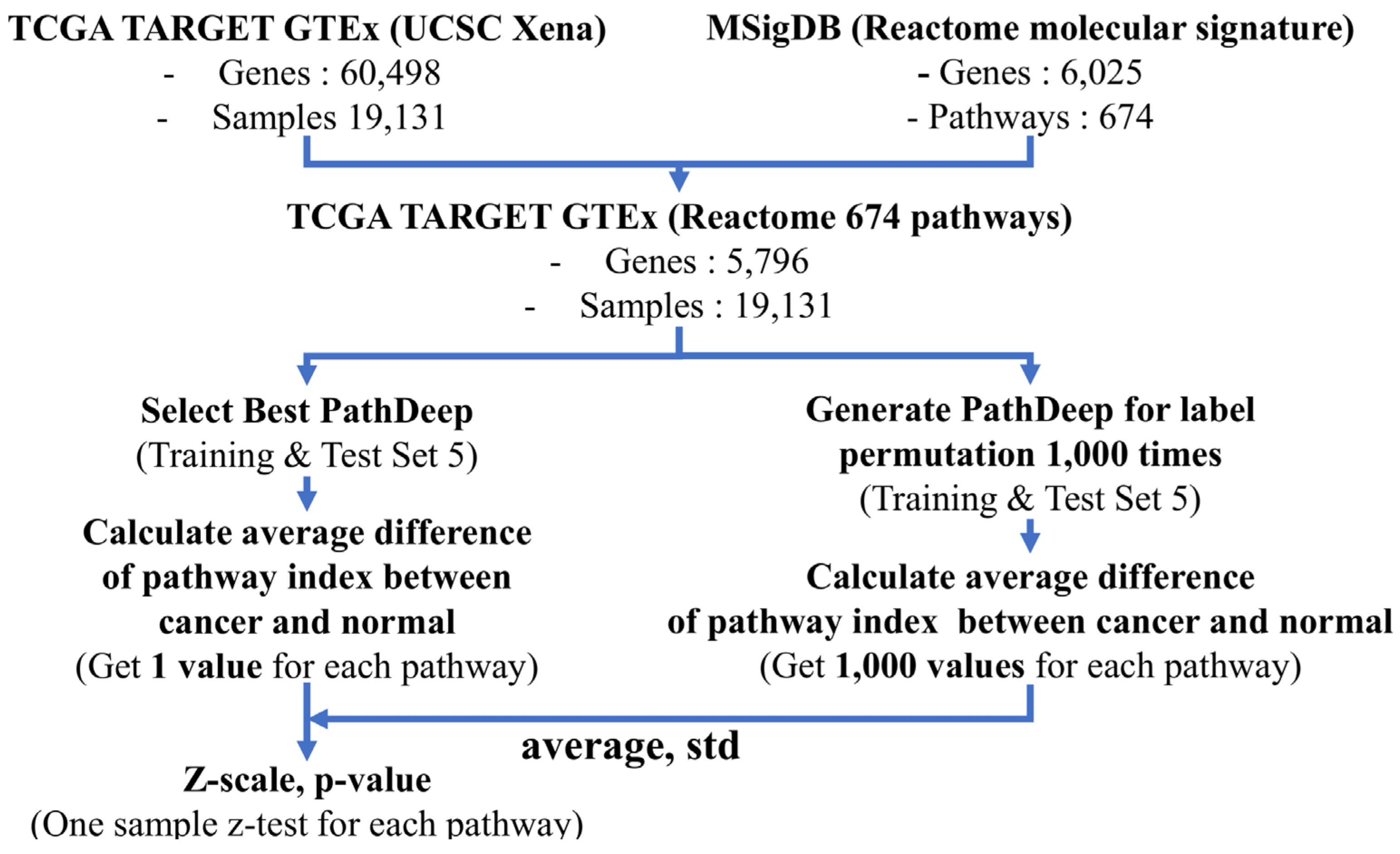
4.3. Data Processing and Workflow
4.4. PathDeep Model Structure
4.5. Selecting an Appropriate Pathway Database for PathDeep
4.6. Visualizing Pathway Index
4.7. Calculating Pathway Index and Pathway Contribution Gene Index
4.8. Functional Enrichment Test of the Top 1% Pathway-Contributing Genes
4.9. Identifying Cancer-Related Pathways Using Label Permutation
5. Patent
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Ouahabi, A.; Taleb-Ahmed, A. Deep Learning for Real-Time Semantic Segmentation: Application in Ultrasound Imaging. Pattern Recognit. Lett. 2021, 144, 27–34. [Google Scholar] [CrossRef]
- Shen, L.; Margolies, L.R.; Rothstein, J.H.; Fluder, E.; McBride, R.; Sieh, W. Deep Learning to Improve Breast Cancer Detection on Screening Mammography. Sci. Rep. 2019, 9, 12495. [Google Scholar] [CrossRef] [PubMed]
- Noorbakhsh, J.; Farahmand, S.; Foroughi pour, A.; Namburi, S.; Caruana, D.; Rimm, D.; Soltanieh-ha, M.; Zarringhalam, K.; Chuang, J.H. Deep Learning-Based Cross-Classifications Reveal Conserved Spatial Behaviors within Tumor Histological Images. Nat. Commun. 2020, 11, 6367. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A Revolutionary Tool for Transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Khalifa, N.E.M.; Taha, M.H.N.; Ezzat Ali, D.; Slowik, A.; Hassanien, A.E. Artificial Intelligence Technique for Gene Expression by Tumor RNA-Seq Data: A Novel Optimized Deep Learning Approach. IEEE Access 2020, 8, 22874–22883. [Google Scholar] [CrossRef]
- Hinton, G.E. Learning Multiple Layers of Representation. Trends Cogn. Sci. 2007, 11, 428–434. [Google Scholar] [CrossRef]
- Gao, F.; Wang, W.; Tan, M.; Zhu, L.; Zhang, Y.; Fessler, E.; Vermeulen, L.; Wang, X. DeepCC: A Novel Deep Learning-Based Framework for Cancer Molecular Subtype Classification. Oncogenesis 2019, 8, 44. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene Set Enrichment Analysis: A Knowledge-Based Approach for Interpreting Genome-Wide Expression Profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Cancer Genome Atlas Program—National Cancer Institute. Available online: https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga (accessed on 12 October 2021).
- Edgar, R. Gene Expression Omnibus: NCBI Gene Expression and Hybridization Array Data Repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [Green Version]
- Floyd, E.T.; James, M.D.; Thompson, E.B. Sequential Comparative Hybridizations Analyzed by Computerized Image Processing Can Identify and Quantitate Regulated RNAs. DNA 1983, 2, 309–327. [Google Scholar] [CrossRef]
- Ogata, H.; Goto, S.; Sato, K.; Fujibuchi, W.; Bono, H.; Kanehisa, M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 1999, 27, 29–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-García, G.; Jerez, J.M.; Franco, L.; Veredas, F.J. Transfer Learning with Convolutional Neural Networks for Cancer Survival Prediction Using Gene-Expression Data. PLoS ONE 2020, 15, e0230536. [Google Scholar] [CrossRef]
- O’Shea, K.; Nash, R. An Introduction to Convolutional Neural Networks. arXiv 2015, arXiv:1511.08458. [Google Scholar]
- Cortes, C.; Vapnik, V. Support-Vector Networks. Mach. Learn. 1995, 20, 273–297. [Google Scholar] [CrossRef]
- Ho, T.K. Random Decision Forests. In Proceedings of the 3rd International Conference on Document Analysis and Recognition, Montreal, QC, Canada, 14–16 August 1995; IEEE Computer Society Press: Montreal, QC, Canada, 1995; Volume 1, pp. 278–282. [Google Scholar]
- Deng, L.; Cai, Y.; Zhang, W.; Yang, W.; Gao, B.; Liu, H. Pathway-Guided Deep Neural Network toward Interpretable and Predictive Modeling of Drug Sensitivity. J. Chem. Inf. Model. 2020, 60, 4497–4505. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Soares, J.; Greninger, P.; Edelman, E.J.; Lightfoot, H.; Forbes, S.; Bindal, N.; Beare, D.; Smith, J.A.; Thompson, I.R.; et al. Genomics of Drug Sensitivity in Cancer (GDSC): A Resource for Therapeutic Biomarker Discovery in Cancer Cells. Nucleic Acids Res. 2012, 41, D955–D961. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, M.; von Mering, C.; Campillos, M.; Jensen, L.J.; Bork, P. STITCH: Interaction Networks of Chemicals and Proteins. Nucleic Acids Res. 2007, 36, D684–D688. [Google Scholar] [CrossRef]
- Gaudelet, T.; Malod-Dognin, N.; Sánchez-Valle, J.; Pancaldi, V.; Valencia, A.; Pržulj, N. Unveiling New Disease, Pathway, and Gene Associations via Multi-Scale Neural Network. PLoS ONE 2020, 15, e0231059. [Google Scholar] [CrossRef]
- Hao, J.; Kim, Y.; Kim, T.-K.; Kang, M. PASNet: Pathway-Associated Sparse Deep Neural Network for Prognosis Prediction from High-Throughput Data. BMC Bioinform. 2018, 19, 510. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Guestrin, C. XGBoost: A Scalable Tree Boosting System. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, San Francisco, CA, USA, 13–17 August 2016; ACM: San Francisco, CA, USA, 2016; pp. 785–794. [Google Scholar]
- Friedman, J.H. Greedy Function Approximation: A Gradient Boosting Machine. Ann. Stat. 2001, 29, 1189–1232. [Google Scholar] [CrossRef]
- Bar-Shavit, R.; Maoz, M.; Kancharla, A.; Nag, J.; Agranovich, D.; Grisaru-Granovsky, S.; Uziely, B. G Protein-Coupled Receptors in Cancer. Int. J. Mol. Sci. 2016, 17, 1320. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.; Sun, L.; Jiao, Y.; Lee, L.T.O. The Role of G Protein-Coupled Receptor Kinases in Cancer. Int. J. Biol. Sci. 2018, 14, 189–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, H.; Taketomi, Y.; Murakami, M. Metabolic Regulation by Secreted Phospholipase A2. Inflamm. Regener. 2016, 36, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pucer, A.; Brglez, V.; Payré, C.; Pungerčar, J.; Lambeau, G.; Petan, T. Group X Secreted Phospholipase A2 Induces Lipid Droplet Formation and Prolongs Breast Cancer Cell Survival. Mol. Cancer 2013, 12, 111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, K.F.; Sajinovic, M.; Hein, J.; Nixdorf, S.; Galettis, P.; Liauw, W.; de Souza, P.; Dong, Q.; Graham, G.G.; Russell, P.J. Emerging Roles for Phospholipase A2 Enzymes in Cancer. Biochimie 2010, 92, 601–610. [Google Scholar] [CrossRef]
- Lee, J.-S.; Oh, S.-J.; Choi, H.-J.; Kang, J.H.; Lee, S.-H.; Ha, J.S.; Woo, S.M.; Jang, H.; Lee, H.; Kim, S.-Y. ATP Production Relies on Fatty Acid Oxidation Rather than Glycolysis in Pancreatic Ductal Adenocarcinoma. Cancers 2020, 12, 2477. [Google Scholar] [CrossRef]
- Wang, F.; Li, Y.; Zhang, Z.; Wang, J.; Wang, J. SHCBP1 Regulates Apoptosis in Lung Cancer Cells through Phosphatase and Tensin Homolog. Oncol. Lett. 2019, 18, 1888–1894. [Google Scholar] [CrossRef] [Green Version]
- Alam, S.M.; Rajendran, M.; Ouyang, S.; Veeramani, S.; Zhang, L.; Lin, M.-F. A Novel Role of Shc Adaptor Proteins in Steroid Hormone-Regulated Cancers. Endocr. Relat. Cancer 2009, 16, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Youngson, R.M. Collins Dictionary of Human Biology; Collins: Glasgow, Scotland, 2006. [Google Scholar]
- Bartek, J.; Lukas, J. Pathways Governing G1/S Transition and Their Response to DNA Damage. FEBS Lett. 2001, 490, 117–122. [Google Scholar] [CrossRef] [Green Version]
- Swinnen, J.V.; Dehairs, J.; Talebi, A. Membrane Lipid Remodeling Takes Center Stage in Growth Factor Receptor-Driven Cancer Development. Cell Metab. 2019, 30, 407–408. [Google Scholar] [CrossRef] [PubMed]
- Goldman, M.J.; Craft, B.; Hastie, M.; Repečka, K.; McDade, F.; Kamath, A.; Banerjee, A.; Luo, Y.; Rogers, D.; Brooks, A.N.; et al. Visualizing and Interpreting Cancer Genomics Data via the Xena Platform. Nat. Biotechnol. 2020, 38, 675–678. [Google Scholar] [CrossRef]
- Van der Maaten, L.; Hinton, G. Visualizing Data Using T-SNE. J. Mach. Learn. Res. 2008, 9, 2579–2605. [Google Scholar]
- Warde-Farley, D.; Donaldson, S.L.; Comes, O.; Zuberi, K.; Badrawi, R.; Chao, P.; Franz, M.; Grouios, C.; Kazi, F.; Lopes, C.T.; et al. The GeneMANIA Prediction Server: Biological Network Integration for Gene Prioritization and Predicting Gene Function. Nucleic Acids Res. 2010, 38, W214–W220. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathway Database Sources | Accuracy | p Value |
|---|---|---|
| c2 reactome | 0.994 | 5.19 × 10−2 |
| c6 oncogenic signatures | 0.993 | 1.12 × 10−1 |
| c5 GO cc | 0.993 | 1.57 × 10−1 |
| c2 kegg | 0.992 | 2.40 × 10−1 |
| c3 tft | 0.991 | 2.49 × 10−1 |
| c3 mir | 0.989 | 2.66 × 10−1 |
| c2 cp biocarta | 0.992 | 2.73 × 10−1 |
| c5 GO bp | 0.993 | 2.98 × 10−1 |
| c4 cm | 0.990 | 3.25 × 10−1 |
| c4 cgn | 0.992 | 3.41 × 10−1 |
| c2 cp | 0.993 | 3.77 × 10−1 |
| c5 GO mf | 0.992 | 3.89 × 10−1 |
| c2 cgp | 0.993 | 4.16 × 10−1 |
| c7 immunologic signatures | 0.977 | 4.50 × 10−1 |
| c1 positional | 0.991 | 4.58 × 10−1 |
| Pathway (c2 Reactome) | FDR (q-Value) |
|---|---|
| GPCR DOWNSTREAM SIGNALING | 0 |
| GPCR LIGAND BINDING | 4.54 × 10−100 |
| NEURONAL SYSTEM | 9.86 × 10−73 |
| SLC MEDIATED TRANSMEMBRANE TRANSPORT | 1.11 × 10−64 |
| SIGNALING BY EGFR IN CANCER | 4.13 × 10−55 |
| AXON GUIDANCE | 6.81 × 10−53 |
| PEPTIDE LIGAND BINDING RECEPTORS | 1.31 × 10−29 |
| DIABETES PATHWAYS | 9.06 × 10−20 |
| GASTRIN CREB SIGNALING PATHWAY VIA PKC AND MAPK | 4.20 × 10−17 |
| SIGNALING BY FGFR MUTANTS | 9.46 × 10−17 |
| COLLAGEN FORMATION | 3.74 × 10−16 |
| FATTY ACYL COA BIOSYNTHESIS | 3.42 × 10−15 |
| CIRCADIAN REPRESSION OF EXPRESSION BY REV ERBA | 8.17 × 10−12 |
| NFKB IS ACTIVATED AND SIGNALS SURVIVAL | 1.73 × 10−11 |
| ACETYLCHOLINE BINDING AND DOWNSTREAM EVENTS | 6.24 × 10−11 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, S.; Huang, E.; Ahn, T. Classification and Functional Analysis between Cancer and Normal Tissues Using Explainable Pathway Deep Learning through RNA-Sequencing Gene Expression. Int. J. Mol. Sci. 2021, 22, 11531. https://doi.org/10.3390/ijms222111531
Park S, Huang E, Ahn T. Classification and Functional Analysis between Cancer and Normal Tissues Using Explainable Pathway Deep Learning through RNA-Sequencing Gene Expression. International Journal of Molecular Sciences. 2021; 22(21):11531. https://doi.org/10.3390/ijms222111531
Chicago/Turabian StylePark, Sangick, Eunchong Huang, and Taejin Ahn. 2021. "Classification and Functional Analysis between Cancer and Normal Tissues Using Explainable Pathway Deep Learning through RNA-Sequencing Gene Expression" International Journal of Molecular Sciences 22, no. 21: 11531. https://doi.org/10.3390/ijms222111531