
Molecular Basis, Diagnostic Challenges and Therapeutic Approaches of Alport Syndrome: A Primer for Clinicians

 ,
,
Abstract
:1. Introduction
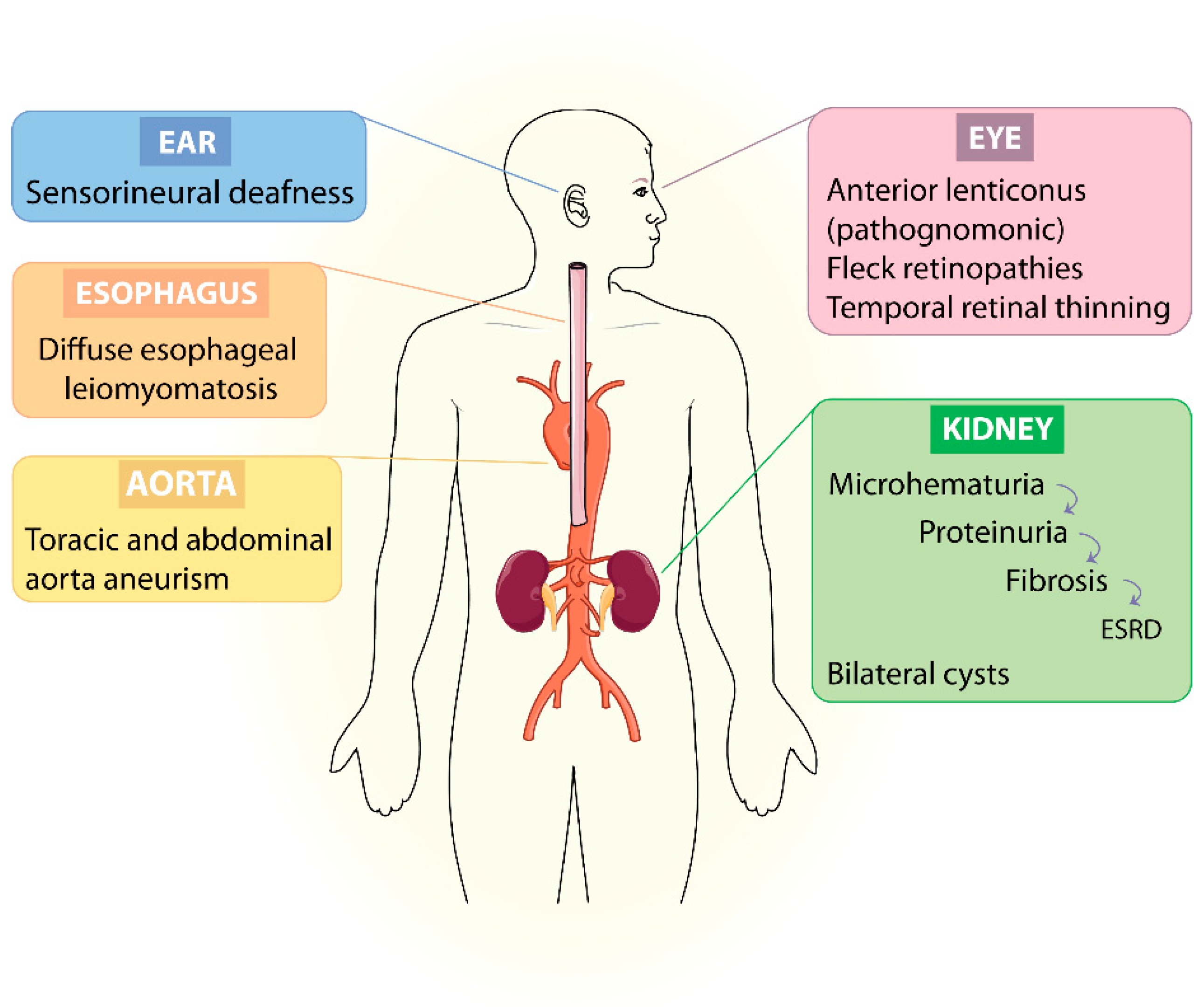
1.1. Clinical Manifestations
1.2. Clinical Presentation
1.3. Prevalence
2. Molecular Basis of the Disease
2.1. In Vitro and In Vivo Modeling of Alport Syndrome
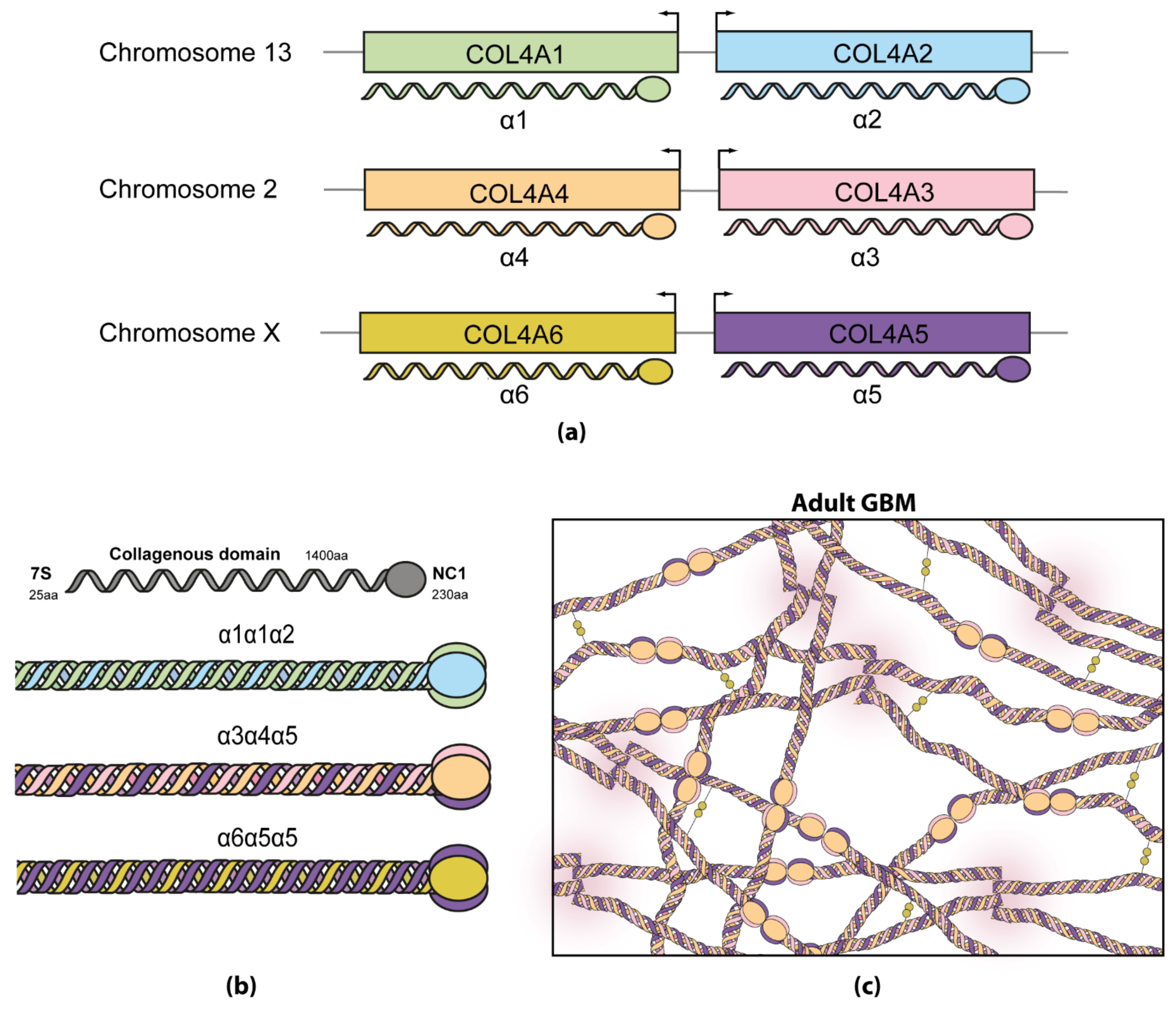
2.2. Collagen in Alport Syndrome
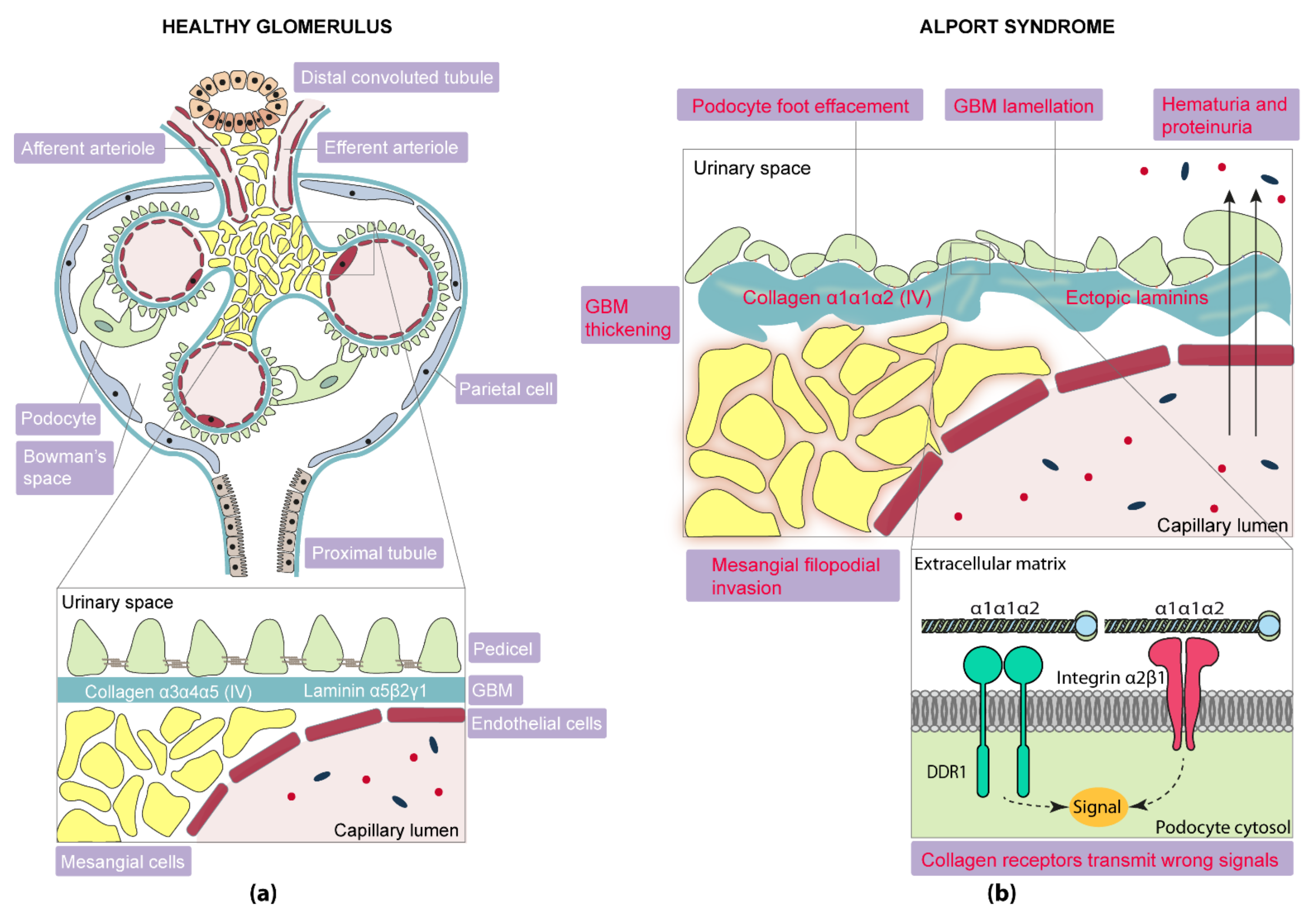
2.3. Other Glomerular Alterations in Alport Syndrome
2.3.1. Laminins
2.3.2. Receptors
Integrins
Collagen Receptors
CC Chemokine Receptor 2
2.3.3. Mesangial Filopodial Invasion
2.3.4. Permeability
2.3.5. Podocyte Detachment
2.3.6. Fibrosis
3. Diagnosis
3.1. Clinical Diagnosis
- Hematuria and proteinuria could be caused by many diseases that affect the glomerulus and the blood filtration process.
- Familial information could be useful, although not in all cases. In ARAS, cases can skip generations, and it is estimated that 10–15% of the cases of men with XLAS are due to de novo mutations [8].
- Extrarenal manifestations are hardly ever present, only in the most severe cases.
- The most common histological sign of AS is FSGS, which can be caused by other genetic or non-genetic diseases. However, it was recently described that mutations in COL4A genes explain 38% of cases of familial FSGS and 3% of sporadic FSGS [75]. Skin biopsy could also be useful in cases of XLAS, although it is currently an obsolete technique [54]. Histological techniques have been used for decades for the diagnosis of AS and innovations are ongoing. A new immunostaining technique is able to differentiate AS with incidental IgA deposits from IgA nephropathy [76].
3.2. Genetic Diagnosis
3.3. Prognosis
4. Treatment
4.1. Clinical Trials for Alport Syndrome
- Bardoxolone is an anti-inflammatory agent that acts by activating the transcription factor Nrf2 (erythroid 2-related factor 2) and inhibiting the NF-κB (kappa-light-chain-enhancer of activated B cells) pathway [91]. The safety and efficacy of bardoxolone was evaluated in the CARDINAL clinical trial (NCT03019185). A total of 187 adult and pediatric participants at various stages of the disease, with and without previous ACEi/ARB treatment, were enrolled in this study. Long-term safety is now being evaluated in a phase 3 EAGLE clinical trial (NCT03749447) that includes 480 participants.
- In a Col4a3 Alport mouse model (129 Sv/J), paricalcitol demonstrated renal protective and antifibrotic effects. Paricalcitol was assessed along with an ACE inhibitor and the results show a synergistic effect capable of delaying ESRD onset [92]. This drug is being tested in an observational clinical trial (NCT02378805).
- The HERA clinical trial (NCT02855268) is now recruiting for a phase 2 interventional study of lademirsen (previously known as RG-012), an inhibitor of miR-21. In vivo experiments have shown how the silencing of this miRNA reduces the inflammation and fibrosis of AS [93].
- Atrasentan is a selective endothelin A receptor antagonist that reduces albuminuria without causing fluid retention, as other members of its family do [94]. It has been assessed in the SONAR clinical trial (NCT01858532) in diabetic patients and the final results show that the risk of kidney events decreases, protecting renal function [95]. Currently, the AFFINITY clinical trial (NCT04573920) is recruiting for testing atrasentan in proteinuric glomerular diseases, including AS.
- Spirinolactone is an aldosterone antagonist that could help treat AS in those cases in which ACE inhibitors lose effectivity. In a Col4a3 Alport animal model (129 Sv/J), a concomitant treatment of ACE and spironolactone reduced proteinuria levels and fibrosis. However, the premature death of some mice could be a side effect of the treatment [96]. Adverse effects of this combination have been shown also in humans [97]; therefore, it must be administered under strict supervision. Spirinolactone effects in humans are being tested in an observational clinical trial (NCT02378805).
- HMG-CoA reductase inhibitors or statins are well known for their action in regulating cholesterol levels. Nonetheless, anti-inflammatory and anti-fibrotic effects have been associated with them, which are clearly of interest in AS. An example is cerivastatin, which is able to reduce proteinuria and fibrosis, prolonging the lifespan of the Alport mice model (129 Sv/J) [98]. The results of the use of statins to treat AS are being evaluated in an observational study (NCT02378805).
- Sparsentan is a dual-acting drug, angiotensin II type 1 (AT1) receptor and endothelin A receptor (ETAR) blocker. The clinical trial EPPIK (NCT05003986) is still recruiting to test sparsentan for the treatment of various proteinuric glomerular diseases, including AS in a pediatric population. Sparsentan has given good results treating FSGS in a phase 2 DUET clinical trial (NCT01613118) [99] and is currently in phase 3 (NCT03493685) [100].
4.2. Pre-Clinical Trials for Alport Syndrome
5. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Stokman, M.F.; Renkema, K.Y.; Giles, R.H.; Schaefer, F.; Knoers, N.V.A.M.; Van Eerde, A.M. The expanding phenotypic spectra of kidney diseases: Insights from genetic studies. Nat. Rev. Nephrol. 2016, 12, 472–483. [Google Scholar] [CrossRef] [PubMed]
- Sá, M.J.N.; Fieremans, N.; De Brouwer, A.P.M.; Sousa, R.; Costa, F.T.; Brito, M.J.; Carvalho, F.; Rodrigues, M.; de Sousa, F.T.; Felgueiras, J.; et al. Deletion of the 5′exons of COL4A6 is not needed for the development of diffuse leiomyomatosis in patients with Alport syndrome. J. Med. Genet. 2013, 50, 745–753. [Google Scholar] [CrossRef] [PubMed]
- Kruegel, J.; Rubel, D.; Gross, O. Alport syndrome—Insights from basic and clinical research. Nat. Rev. Nephrol. 2013, 9, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Byrne, M.C.; Budisavljevic, M.N.; Fan, Z.; Self, S.E.; Ploth, D.W. Renal transplant in patients with Alport’s syndrome. Am. J. Kidney Dis. 2002, 39, 769–775. [Google Scholar] [CrossRef] [PubMed]
- Mallett, A.; Tang, W.; Clayton, P.A.; Stevenson, S.; McDonald, S.P.; Hawley, C.M.; Badve, S.V.; Boudville, N.; Brown, F.G.; Campbell, S.B.; et al. End-stage kidney disease due to Alport syndrome: Outcomes in 296 consecutive Australia and New Zealand dialysis and transplant registry cases. Nephrol. Dial. Transplant. 2014, 29, 2277–2286. [Google Scholar] [CrossRef] [Green Version]
- Gulati, A.; Sevillano, A.M.; Praga, M.; Gutierrez, E.; Alba, I.; Dahl, N.K.; Besse, W.; Choi, J.; Somlo, S. Collagen IV Gene Mutations in Adults With Bilateral Renal Cysts and CKD. Kidney Int. Rep. 2020, 5, 103–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sevillano, A.M.; Gutierrez, E.; Morales, E.; Hernandez, E.; Molina, M.; Gonzalez, E.; Praga, M. Multiple kidney cysts in thin basement membrane disease with proteinuria and kidney function impairment. Clin. Kidney J. 2014, 7, 251–256. [Google Scholar] [CrossRef] [Green Version]
- Kashtan, C.E. Collagen IV-Related Nephropathies (Alport Syndrome and Thin Basement Membrane Nephropathy). In GeneReviews™; University of Washington: Seattle, WA, USA, 2001; pp. 1–18. [Google Scholar]
- Boeckhaus, J.; Strenzke, N.; Storz, C.; Gross, O. Characterization of sensorineural hearing loss in children with alport syndrome. Life 2020, 10, 360. [Google Scholar] [CrossRef]
- Chen, Y.; Colville, D.; Ierino, F.; Symons, A.; Savige, J. Temporal retinal thinning and the diagnosis of Alport syndrome and Thin basement membrane nephropathy. Ophthalmic Genet. 2018, 39, 208–214. [Google Scholar] [CrossRef]
- Savige, J.; Sheth, S.; Leys, A.; Nicholson, A.; Mack, H.G.; Colville, D. Ocular features in Alport syndrome: Pathogenesis and clinical significance. Clin. J. Am. Soc. Nephrol. 2015, 10, 703–709. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Mochizuki, T.; Smeets, H.; Antignac, C.; Laurila, P.; De Paepe, A.; Tryggvason, K.; Reeders, S.T. Deletion of the paired α5 (IV) and α6 (1V) collagen genes in inherited smooth muscle tumors. Science 1993, 261, 1167–1169. [Google Scholar] [CrossRef]
- Nozu, K.; Minamikawa, S.; Yamada, S.; Oka, M.; Yanagita, M.; Morisada, N.; Fujinaga, S.; Nagano, C.; Gotoh, Y.; Takahashi, E.; et al. Characterization of contiguous gene deletions in COL4A6 and COL4A5 in Alport syndrome-diffuse leiomyomatosis. J. Hum. Genet. 2017, 62, 733–735. [Google Scholar] [CrossRef]
- Kashtan, C.E.; Segal, Y.; Flinter, F.; Makanjuola, D.; Gan, J.-S.; Watnick, T. Aortic abnormalities in males with Alport syndrome. Nephrol. Dial. Transplant. 2010, 25, 3554–3560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, J.; Abt, P.; Cheng, K.; Aurigemma, G.; Rosenthal, L. Type A Dissection in a Patient with Alport Syndrome. Circ. Cardiovasc. Imaging 2020, 13, e010701. [Google Scholar] [CrossRef] [PubMed]
- Jais, J.P.; Knebelmann, B.; Giatras, I.; de Marchi, M.; Rizzoni, G.; Renieri, A.; Weber, M.; Gross, O.; Netzer, K.-O.; Flinter, F.; et al. X-linked Alport Syndrome: Natural History in 195 Families and Genotype_Phenotype Correlations in males. J. Am. Soc. Nephrol. 2000, 11, 649–657. [Google Scholar] [CrossRef] [PubMed]
- Jais, J.P.; Knebelmann, B.; Giatras, I.; De Marchi, M.; Rizzoni, G.; Renieri, A.; Weber, M.; Gross, O.; Netzer, K.O.; Flinter, F.; et al. X-linked Alport syndrome: Natural history and genotype-phenotype correlations in girls and women belonging to 195 families: A “European Community Alport Syndrome Concerted Action” study. J. Am. Soc. Nephrol. 2003, 14, 2603–2610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bekheirnia, M.R.; Reed, B.; Gregory, M.C.; McFann, K.; Shamshirsaz, A.A.; Masoumi, A.; Schrier, R.W. Genotype-phenotype correlation in X-linked Alport syndrome. J. Am. Soc. Nephrol. 2010, 21, 876–883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savige, J.; Colville, D.; Rheault, M.; Gear, S.; Lennon, R.; Lagas, S.; Finlay, M.; Flinter, F. Alport syndrome in women and girls. Clin. J. Am. Soc. Nephrol. 2016, 11, 1713–1720. [Google Scholar] [CrossRef] [Green Version]
- Storey, H.; Savige, J.; Sivakumar, V.; Abbs, S.; Flinter, F.A. COL4A3/COL4A4 mutations and features in individuals with autosomal recessive alport syndrome. J. Am. Soc. Nephrol. 2013, 24, 1945–1954. [Google Scholar] [CrossRef] [Green Version]
- Pescucci, C.; Mari, F.; Longo, I.; Vogiatzi, P.; Caselli, R.; Scala, E.; Abaterusso, C.; Gusmano, R.; Seri, M.; Miglietti, N.; et al. Autosomal-dominant Alport syndrome: Natural history of a disease due to COL4A3 or COL4A4 gene. Kidney Int. 2004, 65, 1598–1603. [Google Scholar] [CrossRef] [Green Version]
- Fallerini, C.; Baldassarri, M.; Trevisson, E.; Morbidoni, V.; La Manna, A.; Lazzarin, R.; Pasini, A.; Barbano, G.; Pinciaroli, A.R.; Garosi, G.; et al. Alport syndrome: Impact of digenic inheritance in patients management. Clin. Genet. 2017, 92, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Fallerini, C.; Dosa, L.; Tita, R.; Prete, D.D.; Feriozzi, S.; Gai, G.; Clementi, M.; La Manna, A.; Miglietti, N.; Mancini, R.; et al. Unbiased next generation sequencing analysis confirms the existence of autosomal dominant Alport syndrome in a relevant fraction of cases. Clin. Genet. 2014, 86, 252–257. [Google Scholar] [CrossRef] [PubMed]
- Furlano, M.; Martínez, V.; Pybus, M.; Arce, Y.; Crespi, J.; Venegas, M.d.P.; Bullich, G.; Domingo, A.; Ayasreh, N.; Benito, S.; et al. Clinical and Genetic Features of Autosomal Dominant Alport Syndrome: A Case Series. Am. J. Kidney Dis. 2021, 78, 560–570. [Google Scholar] [CrossRef] [PubMed]
- Altun, I.; Saygılı, S.; Canpolat, N.; Özlük, Y.; Hürdoğan, Ö.; Yeşil, G.; Çalışkan, S.; Sever, L. Strong mesangial IgA staining—Does it always refer to IgA nephropathy in a patient with proteinuria and hematuria? Answers. Pediatr. Nephrol. 2021, 36, 2043–2045. [Google Scholar] [CrossRef]
- Mencarelli, M.A.; Heidet, L.; Storey, H.; Van Geel, M.; Knebelmann, B.; Fallerini, C.; Miglietti, N.; Antonucci, M.F.; Cetta, F.; Sayer, J.A.; et al. Evidence of digenic inheritance in alport syndrome. J. Med. Genet. 2015, 52, 163–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daga, S.; Fallerini, C.; Furini, S.; Pecoraro, C.; Scolari, F.; Ariani, F.; Bruttini, M.; Mencarelli, M.A.; Mari, F.; Renieri, A.; et al. Non-collagen genes role in digenic alport syndrome. BMC Nephrol. 2019, 20, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barua, M.; Paterson, A.D. Population-based studies reveal an additive role of type IV collagen variants in hematuria and albuminuria. Pediatr. Nephrol. 2021, 1–10. [Google Scholar] [CrossRef]
- Frese, J.; Kettwig, M.; Zappel, H.; Hofer, J.; Gröne, H.J.; Nagel, M.; Sunder-Plassmann, G.; Kain, R.; Neuweiler, J.; Gross, O. Kidney injury by variants in the COL4A5 gene aggravated by polymorphisms in slit diaphragm genes causes focal segmental glomerulosclerosis. Int. J. Mol. Sci. 2019, 20, 519. [Google Scholar] [CrossRef] [Green Version]
- Voskarides, K.; Papagregoriou, G.; Hadjipanagi, D.; Petrou, I.; Savva, I.; Elia, A.; Athanasiou, Y.; Pastelli, A.; Kkolou, M.; Hadjigavriel, M.; et al. COL4A5 and LAMA5 variants co-inherited in familial hematuria: Digenic inheritance or genetic modifier effect? BMC Nephrol. 2018, 19, 114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savige, J.; Ariani, F.; Mari, F.; Bruttini, M.; Renieri, A.; Gross, O.; Deltas, C.; Flinter, F.; Ding, J.; Gale, D.P.; et al. Expert consensus guidelines for the genetic diagnosis of Alport syndrome. Pediatr. Nephrol. 2019, 34, 1175–1189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Odiatis, C.; Savva, I.; Pieri, M.; Ioannou, P.; Petrou, P.; Papagregoriou, G.; Antoniadou, K.; Makrides, N.; Stefanou, C.; Ljubanović, D.G.; et al. A glycine substitution in the collagenous domain of Col4a3 in mice recapitulates late onset Alport syndrome. Matrix Biol. Plus 2021, 9, 100053. [Google Scholar] [CrossRef] [PubMed]
- Warady, B.A.; Agarwal, R.; Bangalore, S.; Chapman, A.; Levin, A.; Stenvinkel, P.; Toto, R.D.; Chertow, G.M. Alport Syndrome Classification and Management. Kidney Med. 2020, 2, 639–649. [Google Scholar] [CrossRef] [PubMed]
- Groopman, E.E.; Marasa, M.; Cameron-Christie, S.; Petrovski, S.; Aggarwal, V.S.; Milo-Rasouly, H.; Li, Y.; Zhang, J.; Nestor, J.; Krithivasan, P.; et al. Diagnostic Utility of Exome Sequencing for Kidney Disease. N. Engl. J. Med. 2019, 380, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Cosgrove, D.; Kalluri, R.; Miner, J.H.; Segal, Y.; Borza, D.B. Choosing a mouse model to study the molecular pathobiology of Alport glomerulonephritis. Kidney Int. 2007, 71, 615–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falcone, S.; Wisby, L.; Nicol, T.; Blease, A.; Starbuck, B.; Parker, A.; Sanderson, J.; Brown, S.D.M.; Scudamore, C.L.; Pusey, C.D.; et al. Modification of an aggressive model of Alport Syndrome reveals early differences in disease pathogenesis due to genetic background. Sci. Rep. 2019, 9, 20398. [Google Scholar] [CrossRef] [PubMed]
- Takemon, Y.; Wright, V.; Davenport, B.; Gatti, D.M.; Sheehan, S.M.; Letson, K.; Savage, H.S.; Lennon, R.; Korstanje, R. Uncovering Modifier Genes of X-Linked Alport Syndrome Using a Novel Multiparent Mouse Model. J. Am. Soc. Nephrol. 2021, 32, 1961–1973. [Google Scholar] [CrossRef] [PubMed]
- Dufek, B.; Meehan, D.T.; Delimont, D.; Cheung, L.; Gratton, M.A.; Phillips, G.; Song, W.; Liu, S.; Cosgrove, D. Endothelin A receptor activation on mesangial cells initiates Alport glomerular disease. Kidney Int. 2016, 90, 300–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.J.; David, J.M.; Wilbon, S.S.; Santos, J.V.; Patel, D.M.; Ahmad, A.; Mitrofanova, A.; Liu, X.; Mallela, S.K.; Ducasa, G.M.; et al. Discoidin domain receptor 1 activation links extracellular matrix to podocyte lipotoxicity in Alport syndrome. EBioMedicine 2021, 63, 103162. [Google Scholar] [CrossRef]
- Cosgrove, D.; Meehan, D.T.; Grunkemeyer, J.A.; Kornak, J.M.; Sayers, R.; Hunter, W.J.; Samuelson, G.C. Collagen COL4A3 knockout: A mouse model for autosomal Alport syndrome. Genes Dev. 1996, 10, 2981–2992. [Google Scholar] [CrossRef] [Green Version]
- Cosgrove, D.; Samuelson, G.; Meehan, D.T.; Miller, C.; McGee, J.; Walsh, E.J.; Siegel, M. Ultrastructural, physiological, and molecular defects in the inner ear of a gene-knockout mouse model for autosomal Alport syndrome. Hear. Res. 1998, 121, 84–98. [Google Scholar] [CrossRef]
- Miner, J.H.; Sanes, J.R. Molecular and functional defects in kidneys of mice lacking collagen α3(IV): Implications for Alport syndrome. J. Cell Biol. 1996, 135, 1403–1413. [Google Scholar] [CrossRef] [PubMed]
- Andrews, K.L.; Betsuyaku, T.; Rogers, S.S.; Michael Shipley, J.; Senior, R.M.; Miner, J.H. Gelatinase B (MMP-9) is not essential in the normal kidney and does not influence progression of renal disease in a mouse model of alport syndrome. Am. J. Pathol. 2000, 157, 303–311. [Google Scholar] [CrossRef] [Green Version]
- Korstanje, R.; Caputo, C.R.; Doty, R.A.; Cook, S.A.; Bronson, R.T.; Davisson, M.T.; Miner, J.H. A mouse Col4a4 mutation causing Alport glomerulosclerosis with abnormal collagen α3α4α5(IV) trimers. Kidney Int. 2014, 85, 1461–1468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnold, C.N.; Xia, Y.; Lin, P.; Ross, C.; Schwander, M.; Smart, N.G.; Müller, U.; Beutler, B. Rapid identification of a disease allele in mouse through whole genome sequencing and bulk segregation analysis. Genetics 2011, 187, 633–641. [Google Scholar] [CrossRef] [Green Version]
- Rheault, M.N.; Kren, S.M.; Thielen, B.K.; Mesa, H.A.; Crosson, J.T.; Thomas, W.; Sado, Y.; Kashtan, C.E.; Segal, Y. Mouse model of X-linked Alport syndrome. J. Am. Soc. Nephrol. 2004, 15, 1466–1474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gyoneva, L.; Segal, Y.; Dorfman, K.D.; Barocas, V.H. Mechanical response of wild-type and Alport murine lens capsules during osmotic swelling. Exp. Eye Res. 2013, 113, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Hashikami, K.; Asahina, M.; Nozu, K.; Iijima, K.; Nagata, M.; Takeyama, M. Establishment of X-linked Alport syndrome model mice with a Col4a5 R471X mutation. Biochem. Biophys. Rep. 2019, 17, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Bult, C.J.; Kadin, J.A.; Richardson, J.E.; Blake, J.A.; Eppig, J.T. The mouse genome database: Enhancements and updates. Nucleic Acids Res. 2009, 38, D586–D592. [Google Scholar] [CrossRef] [Green Version]
- Naylor, R.W.; Morais, M.R.P.T.; Lennon, R. Complexities of the glomerular basement membrane. Nat. Rev. Nephrol. 2021, 17, 112–127. [Google Scholar] [CrossRef]
- Quinlan, C.; Rheault, M.N. Genetic Basis of Type IV Collagen Disorders of the Kidney. Clin. J. Am. Soc. Nephrol. 2021, 16, 1101–1109. [Google Scholar] [CrossRef] [PubMed]
- Suh, J.H.; Miner, J.H. The glomerular basement membrane as a barrier to albumin. Nat. Rev. Nephrol. 2013, 9, 470–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Ge, G. Complexity of type IV collagens: From network assembly to function. Biol. Chem. 2019, 400, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Savige, J.; Storey, H.; Watson, E.; Hertz, J.M.; Deltas, C.; Renieri, A.; Mari, F.; Hilbert, P.; Plevova, P.; Byers, P.; et al. Consensus statement on standards and guidelines for the molecular diagnostics of Alport syndrome: Refining the ACMG criteria. Eur. J. Hum. Genet. 2021, 29, 1186–1197. [Google Scholar] [CrossRef] [PubMed]
- Vanacore, R.; Ham, A.J.L.; Voehler, M.; Sanders, C.R.; Conrads, T.P.; Veenstra, T.D.; Sharpless, B.; Dawson, P.E.; Hudson, B.G. A Sulfilimine Bond Identified in Collagen IV. Science 2009, 325, 1230–1234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedchenko, V.; Boudko, S.P.; Barber, M.; Mikhailova, T.; Saus, J.; Harmange, J.-C.; Hudson, B.G. Collagen IVα345 dysfunction in glomerular basement membrane diseases. III. A functional framework for α345 hexamer assembly. J. Biol. Chem. 2021, 296, 100592. [Google Scholar] [CrossRef] [PubMed]
- Abrahamson, D.R. Role of the Podocyte (and Glomerular Endothelium) in Building the GBM. Semin. Nephrol. 2012, 32, 342–349. [Google Scholar] [CrossRef] [Green Version]
- St. John, P.L.; Abrahamson, D.R. Glomerular endothelial cells and podocytes jointly synthesize laminin-1 and -11 chains. Kidney Int. 2001, 60, 1037–1046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kashyan, C.E.; Kim, Y.; Lees, G.E.; Thorner, P.S.; Virtanen, I.; Miner, J.H. Abnormal Glomerular Basement Membrane Laminins in Murine, Canine, and Human Alport Syndrome: Aberrant Laminin α2 Deposition Is Species Independent. J. Am. Soc. Nephrol. 2001, 12, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Abrahamson, D.R.; Prettyman, A.C.; Robert, B.; St. John, P.L. Laminin-1 reexpression in Alport mouse glomerular basement membranes. Kidney Int. 2003, 63, 826–834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zallocchi, M.; Johnson, B.M.; Meehan, D.T.; Delimont, D.; Cosgrove, D. α1β1 Integrin/rac1-dependent mesangial invasion of glomerular capillaries in alport syndrome. Am. J. Pathol. 2013, 183, 1269–1280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steenhard, B.M.; Vanacore, R.; Friedman, D.; Zelenchuk, A.; Stroganova, L.; Isom, K.; John, S.P.L.; Hudson, B.G.; Abrahamson, D.R. Upregulated Expression of Integrin α1 in Mesangial Cells and Integrin α3 and Vimentin in Podocytes of Col4a3-Null (Alport) Mice. PLoS ONE 2012, 7, e50745. [Google Scholar] [CrossRef] [Green Version]
- Cosgrove, D.; Meehan, D.T.; Delimont, D.; Pozzi, A.; Chen, X.; Rodgers, K.D.; Tempero, R.M.; Zallocchi, M.; Rao, V.H. Integrin α1β1 regulates matrix metalloproteinases via p38 mitogen-activated protein kinase in mesangial cells: Implications for alport syndrome. Am. J. Pathol. 2008, 172, 761–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hahm, K.; Lukashev, M.E.; Luo, Y.; Yang, W.J.; Dolinski, B.M.; Weinreb, P.H.; Simon, K.J.; Li, C.W.; Leone, D.R.; Lobb, R.R.; et al. αvβ6 Integrin Regulates Renal Fibrosis and Inflammation in Alport Mouse. Am. J. Pathol. 2007, 170, 110–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suleiman, H.; Zhang, L.; Roth, R.; Heuser, J.E.; Miner, J.H.; Shaw, A.S.; Dani, A. Nanoscale protein architecture of the kidney glomerular basement membrane. eLife 2013, 2, e01149. [Google Scholar] [CrossRef] [PubMed]
- Gross, O.; Girgert, R.; Beirowski, B.; Kretzler, M.; Kang, H.G.; Kruegel, J.; Miosge, N.; Busse, A.C.; Segerer, S.; Vogel, W.F.; et al. Loss of collagen-receptor DDR1 delays renal fibrosis in hereditary type IV collagen disease. Matrix Biol. 2010, 29, 346–356. [Google Scholar] [CrossRef] [PubMed]
- Richter, H.; Satz, A.L.; Bedoucha, M.; Buettelmann, B.; Petersen, A.C.; Harmeier, A.; Hermosilla, R.; Hochstrasser, R.; Burger, D.; Gsell, B.; et al. DNA-Encoded Library-Derived DDR1 Inhibitor Prevents Fibrosis and Renal Function Loss in a Genetic Mouse Model of Alport Syndrome. ACS Chem. Biol. 2018, 14, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Rubel, D.; Frese, J.; Martin, M.; Leibnitz, A.; Girgert, R.; Miosge, N.; Eckes, B.; Müller, G.A.; Gross, O. Collagen receptors integrin alpha2beta1 and discoidin domain receptor 1 regulate maturation of the glomerular basement membrane and loss of integrin alpha2beta1 delays kidney fibrosis in COL4A3 knockout mice. Matrix Biol. 2014, 34, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Sannomiya, Y.; Kaseda, S.; Kamura, M.; Yamamoto, H.; Yamada, H.; Inamoto, M.; Kuwazuru, J.; Niino, S.; Shuto, T.; Suico, M.A.; et al. The role of discoidin domain receptor 2 in the renal dysfunction of alport syndrome mouse model. Ren. Fail. 2021, 43, 510–519. [Google Scholar] [CrossRef] [PubMed]
- Rao, V.H.; Meehan, D.T.; Delimont, D.; Nakajima, M.; Wada, T.; Gratton, M.A.; Cosgrove, D. Role for macrophage metalloelastase in glomerular basement membrane damage associated with Alport syndrome. Am. J. Pathol. 2006, 169, 32–46. [Google Scholar] [CrossRef] [Green Version]
- Delimont, D.; Dufek, B.M.; Meehan, D.T.; Zallocchi, M.; Gratton, M.A.; Phillips, G.; Cosgrove, D. Laminin α2-mediated focal adhesion kinase activation triggers Alport glomerular pathogenesis. PLoS ONE 2014, 9, e99083. [Google Scholar] [CrossRef] [Green Version]
- Abrahamson, D.R.; Isom, K.; Roach, E.; Stroganova, L.; Zelenchuk, A.; Miner, J.H.; St. John, P.L. Laminin compensation in collagen α3(IV) knockout (Alport) glomeruli contributes to permeability defects. J. Am. Soc. Nephrol. 2007, 18, 2465–2472. [Google Scholar] [CrossRef]
- Ding, F.; Wickman, L.; Wang, S.Q.; Zhang, Y.; Wang, F.; Afshinnia, F.; Hodgin, J.; Ding, J.; Wiggins, R.C. Accelerated podocyte detachment and progressive podocyte loss from glomeruli with age in Alport Syndrome. Kidney Int. 2017, 92, 1515–1525. [Google Scholar] [CrossRef] [PubMed]
- Clauss, S.; Gross, O.; Kulkarni, O.; Avila-Ferrufino, A.; Radomska, E.; Segerer, S.; Eulberg, D.; Klussmann, S.; Anders, H.J. Ccl2/Mcp-I blockade reduces glomerular and interstitial macrophages but does not ameliorate renal pathology in co//agen4A3-deficient mice with autosomal recessive alport nephropathy. J. Pathol. 2009, 218, 40–47. [Google Scholar] [CrossRef]
- Gast, C.; Pengelly, R.J.; Lyon, M.; Bunyan, D.J.; Seaby, E.G.; Graham, N.; Venkat-Raman, G.; Ennis, S. Collagen (COL4A) mutations are the most frequent mutations underlying adult focal segmental glomerulosclerosis. Nephrol. Dial. Transplant. 2016, 31, 961–970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishiko, S.; Tanaka, A.; Takeda, A.; Hara, M.; Hamano, N.; Koizumi, M.; Ueno, T.; Hayashi, H.; Kondo, A.; Nagai, S.; et al. Utility of glomerular Gd-IgA1 staining for indistinguishable cases of IgA nephropathy or Alport syndrome. Clin. Exp. Nephrol. 2021, 25, 779–787. [Google Scholar] [CrossRef] [PubMed]
- Morinière, V.; Dahan, K.; Hilbert, P.; Lison, M.; Lebbah, S.; Topa, A.; Bole-Feysot, C.; Pruvost, S.; Nitschke, P.; Plaisier, E.; et al. Improving mutation screening in familial hematuric nephropathies through next generation sequencing. J. Am. Soc. Nephrol. 2014, 25, 2740–2751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Zhang, Y.; Wang, F.; Nair, V.; Ding, F.; Xiao, H.; Yao, Y.; Kretzler, M.; Ju, W.; Ding, J. Urinary epidermal growth factor as a prognostic marker for the progression of Alport syndrome in children. Pediatr. Nephrol. 2018, 33, 1731–1739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Temme, J.; Kramer, A.; Jager, K.J.; Lange, K.; Peters, F.; Müller, G.A.; Kramar, R.; Heaf, J.G.; Finne, P.; Palsson, R.; et al. Outcomes of male patients with Alport syndrome undergoing renal replacement therapy. Clin. J. Am. Soc. Nephrol. 2012, 7, 1969–1976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savva, I.; Pierides, A.; Deltas, C. RAAS inhibition and the course of Alport syndrome. Pharmacol. Res. 2016, 107, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Liu, H.; Liu, D.; Liu, Y.; Li, H.; Tan, X.; Liu, F.; Peng, Y.; Zhang, H. Effects of RAAS Inhibitors in Patients with Kidney Disease. Curr. Hypertens. Rep. 2017, 19, 72. [Google Scholar] [CrossRef] [PubMed]
- Yamamura, T.; Horinouchi, T.; Nagano, C.; Omori, T.; Sakakibara, N.; Aoto, Y.; Ishiko, S.; Nakanishi, K.; Shima, Y.; Nagase, H.; et al. Genotype-phenotype correlations influence the response to angiotensin-targeting drugs in Japanese patients with male X-linked Alport syndrome. Kidney Int. 2020, 98, 1605–1614. [Google Scholar] [CrossRef] [PubMed]
- Temme, J.; Peters, F.; Lange, K.; Pirson, Y.; Heidet, L.; Torra, R.; Grunfeld, J.P.; Weber, M.; Licht, C.; Müller, G.A.; et al. Incidence of renal failure and nephroprotection by RAAS inhibition in heterozygous carriers of X-chromosomal and autosomal recessive Alport mutations. Kidney Int. 2012, 81, 779–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Böckhaus, J.; Wang, F.; Wang, S.; Rubel, D.; Gross, O.; Ding, J. Genotype–phenotype correlations and nephroprotective effects of RAAS inhibition in patients with autosomal recessive Alport syndrome. Pediatr. Nephrol. 2021, 36, 2719–2730. [Google Scholar] [CrossRef] [PubMed]
- Gross, O.; Licht, C.; Anders, H.J.; Hoppe, B.; Beck, B.; Tönshoff, B.; Höcker, B.; Wygoda, S.; Ehrich, J.H.H.; Pape, L.; et al. Early angiotensin-converting enzyme inhibition in Alport syndrome delays renal failure and improves life expectancy. Kidney Int. 2012, 81, 494–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarad, G.; Knutsen, R.H.; Mecham, R.P.; Miner, J.H. Albumin contributes to kidney disease progression in alport syndrome. Am. J. Physiol. Ren. Physiol. 2016, 311, F120–F130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gross, O.; Tönshoff, B.; Weber, L.T.; Pape, L.; Latta, K.; Fehrenbach, H.; Lange-Sperandio, B.; Zappel, H.; Hoyer, P.; Staude, H.; et al. A multicenter, randomized, placebo-controlled, double-blind phase 3 trial with open-arm comparison indicates safety and efficacy of nephroprotective therapy with ramipril in children with Alport’s syndrome. Kidney Int. 2020, 97, 1275–1286. [Google Scholar] [CrossRef] [Green Version]
- Gross, O.; Beirowski, B.; Koepke, M.L.; Kuck, J.; Reiner, M.; Addicks, K.; Smyth, N.; Schulze-Lohoff, E.; Weber, M. Preemptive ramipril therapy delays renal failure and reduces renal fibrosis in COL4A3-knockout mice with Alport syndrome. Kidney Int. 2003, 63, 438–446. [Google Scholar] [CrossRef] [Green Version]
- Kashtan, C.E.; Gross, O. Clinical practice recommendations for the diagnosis and management of Alport syndrome in children, adolescents, and young adults–an update for 2020. Pediatr. Nephrol. 2021, 36, 711–719. [Google Scholar] [CrossRef] [PubMed]
- MN, R.; WE, S. Long-term ACE inhibition in Alport syndrome: Are the benefits worth the risks? Kidney Int. 2020, 97, 1104–1106. [Google Scholar] [CrossRef]
- Stenvinkel, P.; Chertow, G.M.; Devarajan, P.; Levin, A.; Andreoli, S.P.; Bangalore, S.; Warady, B.A. Chronic Inflammation in Chronic Kidney Disease Progression: Role of Nrf2. Kidney Int. Rep. 2021, 6, 1775–1787. [Google Scholar] [CrossRef]
- Rubel, D.; Stock, J.; Ciner, A.; Hiller, H.; Girgert, R.; Müller, G.A.; Gross, O. Antifibrotic, nephroprotective effects of paricalcitol versus calcitriol on top of ACE-inhibitor therapy in the COL4A3 knockout mouse model for progressive renal fibrosis. Nephrol. Dial. Transplant. 2014, 29, 1012–1019. [Google Scholar] [CrossRef] [Green Version]
- Gomez, I.G.; MacKenna, D.A.; Johnson, B.G.; Kaimal, V.; Roach, A.M.; Ren, S.; Nakagawa, N.; Xin, C.; Newitt, R.; Pandya, S.; et al. Anti-microRNA-21 oligonucleotides prevent Alport nephropathy progression by stimulating metabolic pathways. J. Clin. Investig. 2015, 125, 141–156. [Google Scholar] [CrossRef] [PubMed]
- De Zeeuw, D.; Coll, B.; Andress, D.; Brennan, J.J.; Tang, H.; Houser, M.; Correa-Rotter, R.; Kohan, D.; Heerspink, H.J.L.; Makino, H.; et al. The endothelin antagonist atrasentan lowers residual albuminuria in patients with type 2 diabetic nephropathy. J. Am. Soc. Nephrol. 2014, 25, 1083–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heerspink, H.J.L.; Parving, H.H.; Andress, D.L.; Bakris, G.; Correa-Rotter, R.; Hou, F.F.; Kitzman, D.W.; Kohan, D.; Makino, H.; McMurray, J.J.V.; et al. Atrasentan and renal events in patients with type 2 diabetes and chronic kidney disease (SONAR): A double-blind, randomised, placebo-controlled trial. Lancet 2019, 393, 1937–1947. [Google Scholar] [CrossRef]
- Rubel, D.; Zhang, Y.; Sowa, N.; Girgert, R.; Gross, O. Organoprotective Effects of Spironolactone on Top of Ramipril Therapy in a Mouse Model for Alport Syndrome. J. Clin. Med. 2021, 10, 2958. [Google Scholar] [CrossRef] [PubMed]
- Juurlink, D.N.; Mamdani, M.M.; Lee, D.S.; Kopp, A.; Austin, P.C.; Laupacis, A.; Redelmeier, D.A. Rates of Hyperkalemia after Publication of the Randomized Aldactone Evaluation Study. N. Engl. J. Med. 2004, 351, 543–551. [Google Scholar] [CrossRef] [PubMed]
- Koepke, M.L.; Weber, M.; Schulze-Lohoff, E.; Beirowski, B.; Segerer, S.; Gross, O. Nephroprotective effect of the HMG-CoA-reductase inhibitor cerivastatin in a mouse model of progressive renal fibrosis in Alport syndrome. Nephrol. Dial. Transplant. 2007, 22, 1062–1069. [Google Scholar] [CrossRef] [Green Version]
- Trachtman, H.; Nelson, P.; Adler, S.; Campbell, K.N.; Chaudhuri, A.; Derebail, V.K.; Gambaro, G.; Gesualdo, L.; Gipson, D.S.; Hogan, J.; et al. DUET: A Phase 2 Study Evaluating the Efficacy and Safety of Sparsentan in Patients with FSGS. J. Am. Soc. Nephrol. 2018, 29, 2745–2754. [Google Scholar] [CrossRef] [Green Version]
- Komers, R.; Diva, U.; Inrig, J.K.; Loewen, A.; Trachtman, H.; Rote, W.E. Study Design of the Phase 3 Sparsentan Versus Irbesartan (DUPLEX) Study in Patients With Focal Segmental Glomerulosclerosis. Kidney Int. Rep. 2020, 5, 494–502. [Google Scholar] [CrossRef]
- Weinstock, B.A.; Feldman, D.L.; Fornoni, A.; Gross, O.; Kashtan, C.E.; Lagas, S.; Lennon, R.; Miner, J.H.; Rheault, M.N.; Simon, J.F.; et al. Clinical trial recommendations for potential Alport syndrome therapies. Kidney Int. 2020, 97, 1109–1116. [Google Scholar] [CrossRef]
- Omachi, K.; Kaseda, S.; Yokota, T.; Kamura, M.; Teramoto, K.; Kuwazuru, J.; Kojima, H.; Nohara, H.; Koyama, K.; Ohtsuki, S.; et al. Metformin ameliorates the severity of experimental Alport syndrome. Sci. Rep. 2021, 11, 7053. [Google Scholar] [CrossRef] [PubMed]
- Suh, S.H.; Choi, H.S.; Kim, C.S.; Kim, I.J.; Ma, S.K.; Scholey, J.W.; Kim, S.W.; Bae, E.H. Olmesartan attenuates kidney fibrosis in a murine model of alport syndrome by suppressing tubular expression of TGFβ. Int. J. Mol. Sci. 2019, 20, 3843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suh, S.H.; Mathew, A.P.; Choi, H.S.; Vasukutty, A.; Kim, C.S.; Kim, I.J.; Ma, S.K.; Kim, S.W.; Park, I.-K.; Bae, E.H. Kidney-accumulating olmesartan-loaded nanomicelles ameliorate the organ damage in a murine model of Alport syndrome. Int. J. Pharm. 2021, 600, 120497. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Strain | Gene | Genetics | References |
|---|---|---|---|
| 129-Col4a3tm1Dec/J | COL4A3 | Col4a3tm1Dec/Col4a3tm1Dec | [40] |
| 129X1/SvJ-Col4a3tm1Dec | COL4A3 | Col4a3tm1Dec/Col4a3tm1Dec | [41] |
| 129S1/Sv * 129X1/SvJ | COL4A3 | Col4a3tm1Jhm/Col4a3tm1Jhm | [42] |
| 129S1/Sv * 129S6/SvEvTac * 129X1/SvJ | COL4A3 | Col4a3tm1Jhm/Col4a3tm1Jhm Mmp9tm1Tvu/Mmp9tm1Tvu | [43] |
| 129X1/SvJ * C57BL/6 | COL4A3 | Col4a3tm1Dec/Col4a3tm1Dec | [40] |
| 129S1.NON(NZO)-Col4a4bwk/PgnJ | COL4A4 | Col4a4bwk/Col4a4bwk | [44] |
| C57BL/6J-Col4a4m1Btlr | COL4A4 | Col4a4m1Btlr/Col4a4m1Btlr | [45] |
| D2.NON(NZO)-Col4a4bwk/GrsrJ | COL4A4 | Col4a4bwk/Col4a4bwk | [44] |
| C3H/HeH * C57BL/6J | COLA4A | Col4a4m1H/Col4a4m1H | [36] |
| NON;NZO-Col4a4bwk/J | COL4A4 | Col4a4bwk/Col4a4bwk | [44] |
| B6.Cg-Col4a5tm1Yseg | COL4A5 | Col4a5tm1Yseg/Col4a5+ | [46,47] |
| B6.Cg-Col4a5tm1Yseg | COL4A5 | Col4a5tm1Yseg/Y | [46,47] |
| C57BL/6J-Col4a5em1Keha | COL4A5 | Col4a5em1Keha/Y | [48] |
| Identifier | Study | Status | Interventions | Characteristics | Population | Sponsor |
|---|---|---|---|---|---|---|
| NCT01485978 | Efficacy and Safety Study to Delay Renal Failure in Children with Alport Syndrome | Completed | Drug: ramipril Drug: placebo to ramipril | Phase 3 | From 24 months to 18 years | Institut fuer anwe dungsorientierte Forschung und klinische Studien GmbH University Medical Center Goettingen German Federal Ministry of Education and Research |
| NCT03019185 | A Phase 2/3 Trial of the Efficacy and Safety of Bardoxolone Methyl in Patients With Alport Syndrome -CARDINAL | Completed | Drug: placebo oral capsule Drug: bardoxolone methyl | Phase 2 Phase 3 | From 12 years to 60 years | Reata Pharmaceuticals, Inc. |
| NCT03749447 | An Extended Access Program for Bardoxolone Methyl in Patients with CKD (EAGLE) | Recruiting | Drug: bardoxolone methyl | Phase 3 | Of 12 years and older | Reata Pharmaceuticals, Inc. |
| NCT02378805 | European Alport Therapy Registry European Initiative Towards Delaying Renal Failure in Alport Syndrome | Recruiting | Drug: ACE inhibitor Drug: AT1 inhibitor Drug: HMG coenzyme inhibitor (statin) Drug: spironolactone Drug: paricalcitol | Observational | Child, adult, older adult | University Hospital Goettingen Society for Pediatric Nephrology (Germany) Deutsche Gesellschaft für Nephrologie Alport Selbsthilfe e.V. Association pour l’Information et la Recherche sur les Maladies Rénales Génétiques (AIRG) KfH Foundation Preventive Medicine |
| NCT02855268 | Study of Lademirsen (SAR339375) in Patients with Alport Syndrome | Recruiting | Drug: lademirsen (SAR339375) Drug: placebo | Phase 2 | From 18 years to 55 years | Genzyme, a Sanofi Company Sanofi |
| NCT04573920 | Atrasentan in Patients with Proteinuric Glomerular Diseases | Recruiting | Drug: atrasentan | Phase 2 | Of 18 years and older | Chinook Therapeutics U.S., Inc. Chinook Therapeutics, Inc. |
| NCT05003986 | Study of Sparsentan Treatment in Pediatrics with Proteinuric Glomerular Diseases | Recruiting | Drug: sparsentan | Phase 2 | From 1 year to 17 years | Travere Therapeutics, Inc. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martínez-Pulleiro, R.; García-Murias, M.; Fidalgo-Díaz, M.; García-González, M.Á. Molecular Basis, Diagnostic Challenges and Therapeutic Approaches of Alport Syndrome: A Primer for Clinicians. Int. J. Mol. Sci. 2021, 22, 11063. https://doi.org/10.3390/ijms222011063
Martínez-Pulleiro R, García-Murias M, Fidalgo-Díaz M, García-González MÁ. Molecular Basis, Diagnostic Challenges and Therapeutic Approaches of Alport Syndrome: A Primer for Clinicians. International Journal of Molecular Sciences. 2021; 22(20):11063. https://doi.org/10.3390/ijms222011063
Chicago/Turabian StyleMartínez-Pulleiro, Raquel, María García-Murias, Manuel Fidalgo-Díaz, and Miguel Ángel García-González. 2021. "Molecular Basis, Diagnostic Challenges and Therapeutic Approaches of Alport Syndrome: A Primer for Clinicians" International Journal of Molecular Sciences 22, no. 20: 11063. https://doi.org/10.3390/ijms222011063