
Nuclear and Mitochondrial Genome, Epigenome and Gut Microbiome: Emerging Molecular Biomarkers for Parkinson’s Disease

 , , and
, , and
Abstract
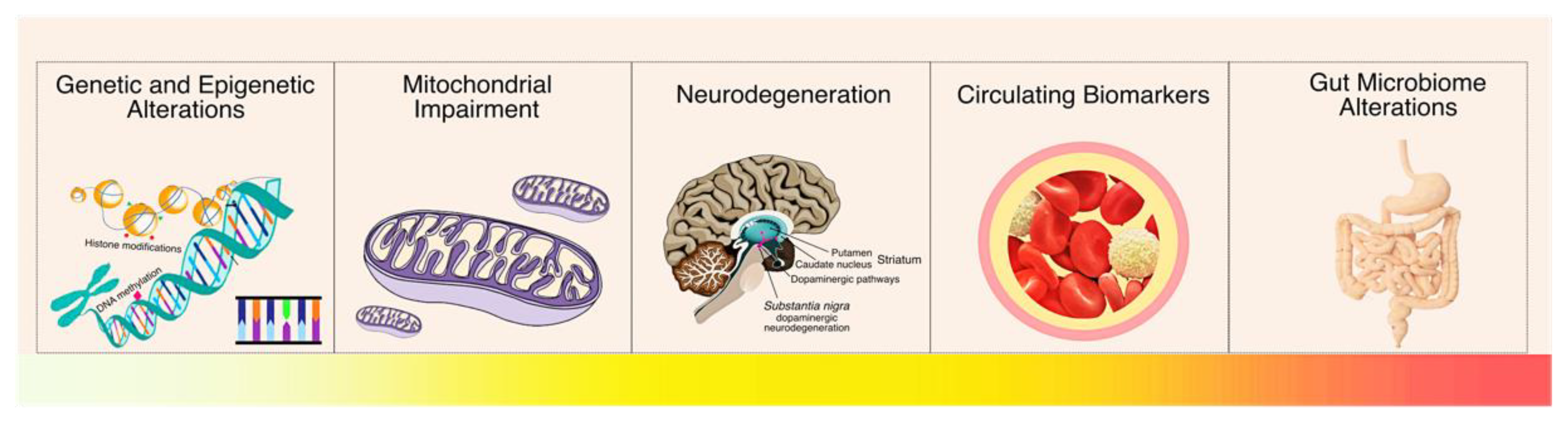
:1. Introduction
2. Parkinson’s Disease
3. Genetic Alterations in Parkinson’s Disease
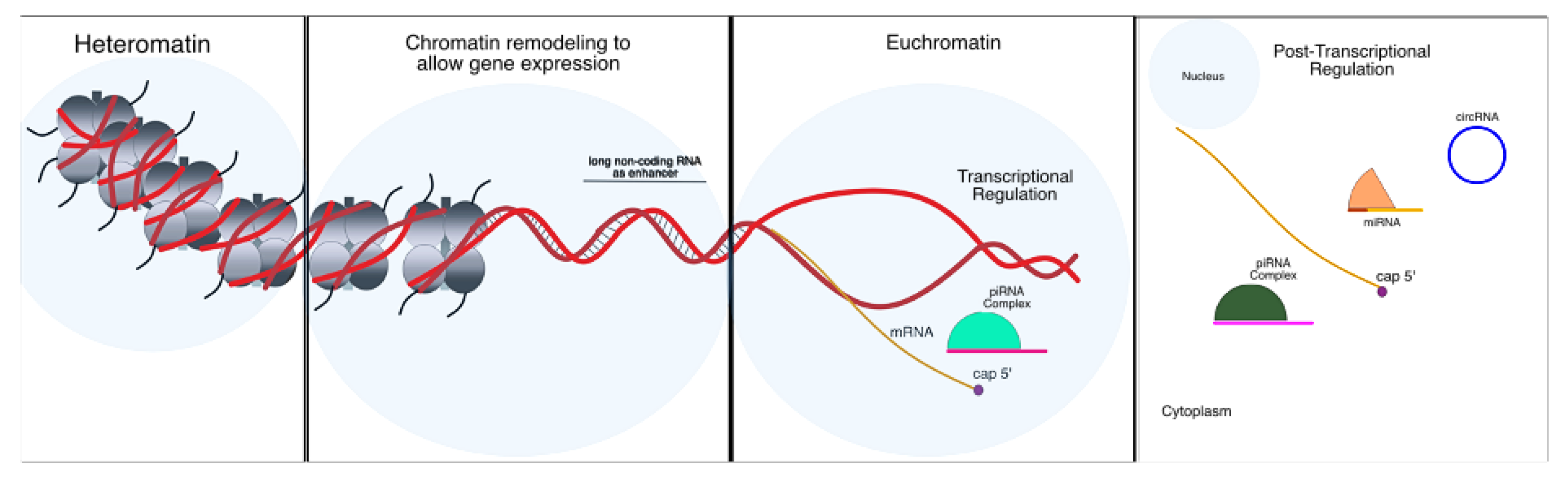
4. Epigenetic Alterations in Parkinson’s Disease
4.1. Chromatin Remodeling Mechanisms
4.2. Non-Coding RNA Regulation
4.2.1. MicroRNAs
4.2.2. Piwi-Interacting RNAs
4.2.3. Long-Noncoding RNAs
4.2.4. Circular RNAs
5. Mitochondrial Genetics and Epigenetics in Parkinson’s Disease
6. Gut Microbiome in Parkinson’s Disease
7. Future Perspectives in Parkinson’s Disease
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- GBD 2016 Neurology Collaborators. Global, regional, and national burden of neurological disorders, 1990-2016: A systematic analysis for the global burden of disease study 2016. Lancet Neurol. 2019, 18, 459–480. [Google Scholar] [CrossRef] [Green Version]
- Vinasco-Sandoval, T.; Moreira, F.C.; Vidal, A.F.; Pinto, P.; Ribeiro-dos-Santos, A.M.; Cruz, R.L.S.; Cabral, G.F.; Anaissi, A.K.M.; Lopes, K.d.P.; Ribeiro-dos-Santos, A. Global analyses of expressed piwi-interacting RNAs in gastric cancer. Int. J. Mol. Sci. 2020, 21, 7656. [Google Scholar] [CrossRef]
- Vidal, A.F.; Ribeiro-dos-Santos, A.M.; Vinasco-Sandoval, T.; Magalhães, L.; Pinto, P.; Anaissi, A.K.M.; Demachki, S.; de Assumpção, P.P.; dos Santos, S.E.B.; Ribeiro-dos-Santos, A. The comprehensive expression analysis of circular RNAs in gastric cancer and its association with field cancerization. Sci. Rep. 2017, 7, 14551. [Google Scholar] [CrossRef] [Green Version]
- Reed, X.; Bandrés-Ciga, S.; Blauwendraat, C.; Cookson, M.R. The role of monogenic genes in idiopathic Parkinson’s disease. Neurobiol. Dis. 2019, 124, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Cook Shukla, L.; Schulze, J.; Farlow, J.; Pankratz, N.D.; Wojcieszek, J.; Foroud, T. Parkinson Disease Overview. GeneReviews® 2004. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1223/ (accessed on 25 July 2019).
- Li, T.; Le, W. Biomarkers for Parkinson’s disease: How good are they? Neurosci. Bull. 2020, 36, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Erkkinen, M.G.; Kim, M.-O.; Geschwind, M.D. Clinical neurology and epidemiology of the major neurodegenerative diseases. Cold Spring Harb. Perspect. Biol. 2018, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radhakrishnan, D.; Goyal, V. Parkinson’s disease: A review. Neurol. India 2018, 66, 26–35. [Google Scholar]
- Draoui, A.; El Hiba, O.; Aimrane, A.; El Khiat, A.; Gamrani, H. Parkinson’s disease: From bench to bedside. Rev. Neurol. 2020, 176, 543–559. [Google Scholar] [CrossRef]
- Berg, D.; Postuma, R.B.; Adler, C.H.; Bloem, B.R.; Chan, P.; Dubois, B.; Gasser, T.; Goetz, C.G.; Halliday, G.; Joseph, L.; et al. MDS research criteria for prodromal Parkinson’s disease. Mov. Disord. 2015, 30, 1600–1611. [Google Scholar] [CrossRef] [PubMed]
- Postuma, R.B.; Berg, D.; Stern, M.; Poewe, W.; Olanow, C.W.; Oertel, W.; Obeso, J.; Marek, K.; Litvan, I.; Lang, A.E.; et al. MDS clinical diagnostic criteria for Parkinson’s disease. Mov. Disord. 2015, 30, 1591–1601. [Google Scholar] [CrossRef] [PubMed]
- Arnerić, S.P.; Kern, V.D.; Stephenson, D.T. Regulatory-accepted drug development tools are needed to accelerate innovative CNS disease treatments. Biochem. Pharmacol. 2018, 151, 291–306. [Google Scholar] [CrossRef]
- Lang, A.E.; Obeso, J.A. Challenges in Parkinson’s disease: Restoration of the nigrostriatal dopamine system is not enough. Lancet Neurol. 2004, 3, 309–316. [Google Scholar] [CrossRef]
- de le Boë, S.F. Opera Medica. (Editio Altera Correctior and Emendatior); Daniel Elsevir & Abraham Wolfgang: Amsterdam, The Netherlands, 1860. [Google Scholar]
- de Sauvages, F.B. Nosologia Methodica Sistens Morborum Classes: Juxtà Sydenhami Mentem & Botanicorum Ordinem. Ultima, Auctior, Emendatior; Sumptibus fratrum de Tournes: Amsterdam, The Netherlands, 1768. [Google Scholar]
- Samii, A.; Nutt, J.G.; Ransom, B.R. Parkinson’s disease. Lancet 2004, 363, 1783–1793. [Google Scholar] [CrossRef] [Green Version]
- Strafella, C.; Caputo, V.; Galota, M.R.; Zampatti, S.; Marella, G.; Mauriello, S.; Cascella, R.; Giardina, E. Application of precision medicine in neurodegenerative diseases. Front. Neurol. 2018, 9, 701. [Google Scholar] [CrossRef] [Green Version]
- Ko, J.H.; Strafella, A.P. Dopaminergic neurotransmission in the human brain: New lessons from perturbation and imaging. Neuroscientist 2012, 18, 149–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goetz, C.G. The history of Parkinson’s disease: Early clinical descriptions and neurological therapies. Cold Spring Harb. Perspect. Med. 2011, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibb, W.R.; Lees, A.J. The relevance of the Lewy body to the pathogenesis of idiopathic Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 1988, 51, 745–752. [Google Scholar] [CrossRef] [Green Version]
- Dugger, B.N.; Dickson, D.W. Pathology of neurodegenerative diseases. Cold Spring Harb. Perspect. Biol. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, M.J.; Okun, M.S. Diagnosis and treatment of Parkinson disease: A review. JAMA 2020, 323, 548. [Google Scholar] [CrossRef] [PubMed]
- Emamzadeh, F.N.; Surguchov, A. Parkinson’s disease: Biomarkers, treatment, and risk factors. Front. Neurosci. 2018, 12, 612. [Google Scholar] [CrossRef]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef] [Green Version]
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef]
- Bridi, J.C.; Hirth, F. Mechanisms of α-synuclein induced synaptopathy in Parkinson’s disease. Front. Neurosci. 2018, 12, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zolezzi, J.M.; Bastías-Candia, S.; CInestrosa, N. Molecular basis of neurodegeneration: Lessons from Alzheimer’s and Parkinson’s diseases. In Recent Advances in Neurodegeneration; Borreca, A., Ed.; IntechOpen: London, UK, 2019. [Google Scholar]
- Bellucci, A.; Zaltieri, M.; Navarria, L.; Grigoletto, J.; Missale, C.; Spano, P. From α-synuclein to synaptic dysfunctions: New insights into the pathophysiology of Parkinson’s disease. Brain Res. 2012, 1476, 183–202. [Google Scholar] [CrossRef] [PubMed]
- Perez, R.G.; Waymire, J.C.; Lin, E.; Liu, J.J.; Guo, F.; Zigmond, M.J. A role for α-synuclein in the regulation of dopamine biosynthesis. J. Neurosci. 2002, 22, 3090–3099. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Zuo, X.; Li, Y.; Zhang, C.; Zhou, M.; Zhang, Y.A.; Uéda, K.; Chan, P. Inhibition of tyrosine hydroxylase expression in α-synuclein-transfected dopaminergic neuronal cells. Neurosci. Lett. 2004, 367, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Butler, B.; Saha, K.; Rana, T.; Becker, J.P.; Sambo, D.; Davari, P.; Goodwin, S.J.; Khoshbouei, H. Dopamine transporter activity is modulated by α-synuclein. J. Biol. Chem. 2015, 290, 29542–29554. [Google Scholar] [CrossRef] [Green Version]
- Valdinocci, D.; Radford, R.; Goulding, M.; Hayashi, J.; Chung, R.; Pountney, D. Extracellular interactions of alpha-synuclein in multiple system atrophy. Int. J. Mol. Sci. 2018, 19, 4129. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.-C.; Chiu, T.-Y.; Lee, T.-Y.; Hsieh, H.-J.; Lin, C.-C.; Kao, L.-S. Soluble α-synuclein facilitates priming and fusion by releasing Ca2+ from the thapsigargin-sensitive Ca2+ pool in PC12 cells. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Logan, T.; Bendor, J.; Toupin, C.; Thorn, K.; Edwards, R.H. α-Synuclein promotes dilation of the exocytotic fusion pore. Nat. Neurosci. 2017, 20, 681–689. [Google Scholar] [CrossRef]
- Rieder, C.R.M. GBA mutations and Parkinson’s disease in Brazil. Arq. Neuro-Psiquiatr. 2019, 77, 71–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magalhaes, J.; Gegg, M.E.; Migdalska-Richards, A.; Doherty, M.K.; Whitfield, P.D.; Schapira, A.H.V. Autophagic lysosome reformation dysfunction in glucocerebrosidase deficient cells: Relevance to Parkinson disease. Hum. Mol. Genet. 2016, 25, 3432–3445. [Google Scholar] [CrossRef] [PubMed]
- Marques, A.R.A.; Mirzaian, M.; Akiyama, H.; Wisse, P.; Ferraz, M.J.; Gaspar, P.; Alfonso, P.; Irún, P.; Dahl, M.; Karlsson, S.; et al. Glucosylated cholesterol in mammalian cells and tissues: Formation and degradation by multiple cellular β-glucosidases. J. Lipid Res. 2016, 57, 451–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akiyama, H.; Kobayashi, S.; Hirabayashi, Y.; Murakami-Murofushi, K. Cholesterol glucosylation is catalyzed by transglucosylation reaction of β-glucosidase 1. Biochem. Biophys. Res. Commun. 2013, 441, 838–843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sidransky, E.; Lopez, G. The link between the GBA gene and parkinsonism. Lancet Neurol. 2012, 11, 986–998. [Google Scholar] [CrossRef] [Green Version]
- Manzoni, C. The LRRK2-macroautophagy axis and its relevance to Parkinson’s disease. Biochem. Soc. Trans. 2017, 45, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Roth, J.A.; Eichhorn, M. Down-regulation of LRRK2 in control and DAT transfected HEK cells increases manganese-induced oxidative stress and cell toxicity. Neurotoxicology 2013, 37, 100–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, O.; Seo, H.; Andrabi, S.; Guardia-Laguarta, C.; Graziotto, J.; Sundberg, M.; McLean, J.R.; Carrillo-Reid, L.; Xie, Z.; Osborn, T.; et al. Pharmacological rescue of mitochondrial deficits in iPSC-derived neural cells from patients with familial Parkinson’s disease. Sci. Transl. Med. 2012, 4. [Google Scholar] [CrossRef] [Green Version]
- Papkovskaia, T.D.; Chau, K.-Y.; Inesta-Vaquera, F.; Papkovsky, D.B.; Healy, D.G.; Nishio, K.; Staddon, J.; Duchen, M.R.; Hardy, J.; Schapira, A.H.V.; et al. G2019S leucine-rich repeat kinase 2 causes uncoupling protein-mediated mitochondrial depolarization. Hum. Mol. Genet. 2012, 21, 4201–4213. [Google Scholar] [CrossRef]
- Melrose, H.L.; Dächsel, J.C.; Behrouz, B.; Lincoln, S.J.; Yue, M.; Hinkle, K.M. Impaired dopaminergic neurotransmission and microtubule-associated protein tau alterations in human LRRK2 transgenic mice. Neurobiol. Dis. 2010, 40, 503–517. [Google Scholar] [CrossRef] [Green Version]
- Xiong, H.; Wang, D.; Chen, L.; Choo, Y.S.; Ma, H.; Tang, C. Parkin, PINK1, and DJ-1 form a ubiquitin E3 ligase complex promoting unfolded protein degradation. J. Clin. Investig. 2009, 119, 650–660. [Google Scholar] [CrossRef] [Green Version]
- Chung, K.K.; Zhang, Y.; Lim, K.L.; Tanaka, Y.; Huang, H.; Gao, J. Parkin ubiquitinates the alpha-synuclein-interacting protein, synphilin-1: Implications for Lewy-body formation in Parkinson disease. Nat. Med. 2001, 7, 1144–1150. [Google Scholar] [CrossRef]
- Wu, S.; Lei, L.; Song, Y.; Liu, M.; Lu, S.; Lou, D. Mutation of hop-1 and pink-1 attenuates vulnerability of neurotoxicity in C. elegans: The role of mitochondria-associated membrane proteins in Parkinsonism. Exp. Neurol. 2018, 309, 67–78. [Google Scholar] [CrossRef]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.K.; Harvey, K.; Gispert, S. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 2004, 304, 1158–1160. [Google Scholar] [CrossRef] [Green Version]
- Fu, K.; Ren, H.; Wang, Y.; Fei, E.; Wang, H.; Wang, G. DJ-1 inhibits TRAIL-induced apoptosis by blocking pro-caspase-8 recruitment to FADD. Oncogene 2012, 31, 1311–1322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haque, M.E.; Mount, M.P.; Safarpour, F.; Abdel-Messih, E.; Callaghan, S.; Mazerolle, C. Inactivation of pink1 gene in vivo sensitizes dopamine-producing neurons to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and can be rescued by autosomal recessive Parkinson disease genes, Parkin or DJ-1. J. Biol. Chem. 2012, 287, 23162–23170. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Song, J.; Kwon, K.; Jang, S.; Kim, C.; Baek, K. Human DJ-1 and its homologs are novel glyoxalases. Hum. Mol. Genet. 2012, 21, 3215–3225. [Google Scholar] [CrossRef] [Green Version]
- Choi, D.-H.; Hwang, O.; Lee, K.-H.; Lee, J.; Beal, M.F.; Kim, Y.-S. DJ-1 cleavage by matrix metalloproteinase 3 mediates oxidative stress-induced dopaminergic cell death. Antioxid. Redox Signal. 2011, 14, 2137–2150. [Google Scholar] [CrossRef] [Green Version]
- Guo, C.; Jeong, H.-H.; Hsieh, Y.-C.; Klein, H.-U.; Bennett, D.A.; De Jager, P.L. Tau activates transposable elements in Alzheimer’s disease. Cell Rep. 2018, 23, 2874–2880. [Google Scholar] [CrossRef] [PubMed]
- Pascale, E.; Di Battista, M.E.; Rubino, A.; Purcaro, C.; Valente, M.; Fattapposta, F. Genetic architecture of MAPT gene region in parkinson disease subtypes. Front. Cell. Neurosci. 2016, 10, 96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.-C.; Xing, A.; Tan, M.-S.; Tan, L.; Yu, J.-T. The role of MAPT in neurodegenerative diseases: Genetics, mechanisms and therapy. Mol. Neurobiol. 2016, 53, 4893–4904. [Google Scholar] [CrossRef] [PubMed]
- Pacheco, C.D.; Elrick, M.J.; Lieberman, A.P. Tau deletion exacerbates the phenotype of Niemann-Pick type C mice and implicates autophagy in pathogenesis. Hum. Mol. Genet. 2009, 18, 956–965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen Reish, H.E.; Standaert, D.G. Role of α-synuclein in inducing innate and adaptive immunity in Parkinson disease. J. Parkinson’s Dis. 2015, 5, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.-J.; Kim, C.; Lee, S.-J. Alpha-synuclein stimulation of astrocytes: Potential role for neuroinflammation and neuroprotection. Oxidative Med. Cell. Longev. 2010, 3, 283–287. [Google Scholar] [CrossRef]
- Meng, J.; Lv, Z.; Qiao, X.; Li, X.; Li, Y.; Zhang, Y. The decay of redox-stress response capacity is a substantive characteristic of aging: Revising the redox theory of aging. Redox Biol. 2017, 11, 365–374. [Google Scholar] [CrossRef]
- Pham-Huy, L.A.; He, H.; Pham-Huy, C. Free radicals, antioxidants in disease and health. Int. J. Biomed. Sci. 2008, 4, 89–96. [Google Scholar]
- Okun, E.; Griffioen, K.J.; Mattson, M.P. Toll-like receptor signaling in neural plasticity and disease. Trends Neurosci. 2011, 34, 269–281. [Google Scholar] [CrossRef] [Green Version]
- Franceschi, C.; Garagnani, P.; Vitale, G.; Capri, M.; Salvioli, S. Inflammaging and ‘garb-aging’. Trends Endocrinol. Metab. 2017, 28, 199–212. [Google Scholar] [CrossRef] [Green Version]
- Dandekar, A.; Mendez, R.; Zhang, K. Cross talk between er stress, oxidative stress, and inflammation in health and disease. In Stress Responses; Oslowski, C.M., Ed.; Springer New York: New York, NY, USA, 2015; pp. 205–214. [Google Scholar]
- Sironi, L.; Restelli, L.M.; Tolnay, M.; Neutzner, A.; Frank, S. Dysregulated interorganellar crosstalk of mitochondria in the pathogenesis of Parkinson’s disease. Cells 2020, 9, 233. [Google Scholar] [CrossRef] [Green Version]
- Puspita, L.; Chung, S.Y.; Shim, J. Oxidative stress and cellular pathologies in Parkinson’s disease. Mol. Brain 2017, 10. [Google Scholar] [CrossRef] [Green Version]
- Calì, T.; Ottolini, D.; Brini, M. Mitochondria, calcium, and endoplasmic reticulum stress in Parkinson’s disease. BioFactors 2011, 37, 228–240. [Google Scholar] [CrossRef]
- Calì, T.; Ottolini, D.; Brini, M. Mitochondrial Ca2+ and neurodegeneration. Cell Calcium 2012, 52, 73–85. [Google Scholar] [CrossRef]
- Pickrell, A.M.; Youle, R.J. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 2015, 85, 257–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, C.; Westenberger, A. Genetics of Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Winter, D.; Ashrafi, G.; Schlehe, J.; Wong, Y.L.; Selkoe, D. PINK1 and parkin target miro for phosphorylation and degradation to arrest mitochondrial motility. Cell 2011, 147, 893–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stafa, K.; Tsika, E.; Moser, R.; Musso, A.; Glauser, L.; Jones, A. Functional interaction of Parkinson’s disease-associated LRRK2 with members of the dynamin GTPase superfamily. Hum. Mol. Genet. 2014, 23, 2055–2077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, H.; Fu, K.; Wang, D.; Mu, C.; Wang, G. Oxidized DJ-1 interacts with the mitochondrial protein BCL-XL. J. Biol. Chem. 2011, 286, 35308–35317. [Google Scholar] [CrossRef] [Green Version]
- Taira, T.; Saito, Y.; Niki, T.; Iguchi-Ariga, S.M.M.; Takahashi, K.; Ariga, H. DJ-1 has a role in antioxidative stress to prevent cell death. EMBO Rep. 2004, 5, 213–218. [Google Scholar] [CrossRef] [Green Version]
- Do, J.; McKinney, C.; Sharma, P.; Sidransky, E. Glucocerebrosidase and its relevance to Parkinson disease. Mol. Neurodegener. 2019, 14, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.-R.; Chen, C.-M.; Chao, C.-Y.; Ro, L.-S.; Lyu, R.-K.; Chang, K.-H. Glucocerebrosidase gene mutation is a risk factor for early onset of Parkinson disease among taiwanese. J. Neurol. Neurosurg. Psychiatry 2007, 78, 977–979. [Google Scholar] [CrossRef] [Green Version]
- Tam, O.H.; Ostrow, L.W.; Gale Hammell, M. Diseases of the nERVous system: Retrotransposon activity in neurodegenerative disease. Mob. DNA 2019, 10, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Jacob-Hirsch, J.; Eyal, E.; Knisbacher, B.A.; Roth, J.; Cesarkas, K.; Dor, C. Whole-genome sequencing reveals principles of brain retrotransposition in neurodevelopmental disorders. Cell Res. 2018, 28, 187–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Prazak, L.; Chatterjee, N.; Grüninger, S.; Krug, L.; Theodorou, D. Activation of transposable elements during aging and neuronal decline in drosophila. Nat. Neurosci. 2013, 16, 529–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bodea, G.O.; McKelvey, E.G.Z.; Faulkner, G.J. oRetrotransposon-induced mosaicism in the neural genme. Open Biol. 2018, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Perez, J.L.; Widmann, T.J.; Adams, I.R. The impact of transposable elements on mammalian development. Development 2016, 143, 4101–4114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saleh, A.; Macia, A.; Muotri, A.R. Transposable elements, inflammation, and neurological disease. Front. Neurol. 2019, 10, 894. [Google Scholar] [CrossRef] [Green Version]
- Kassiotis, G.; Stoye, J.P. Immune responses to endogenous retroelements: Taking the bad with the good. Nat. Rev. Immunol. 2016, 16, 207–219. [Google Scholar] [CrossRef]
- Bravo, J.I.; Nozownik, S.; Danthi, P.S.; Benayoun, B.A. Transposable elements, circular RNAs and mitochondrial transcription in age-related genomic regulation. Development 2020, 147. [Google Scholar] [CrossRef]
- Huang, X.; Wong, G. An old weapon with a new function: PIWI-interacting RNAs in neurodegenerative diseases. Transl. Neurodegener. 2021, 10, 9. [Google Scholar] [CrossRef]
- Sun, W.; Samimi, H.; Gamez, M.; Zare, H.; Frost, B. Pathogenic tau-induced piRNA depletion promotes neuronal death through transposable element dysregulation in neurodegenerative tauopathies. Nat. Neurosci. 2018, 21, 1038–1048. [Google Scholar] [CrossRef]
- Nandi, S.; Chandramohan, D.; Fioriti, L.; Melnick, A.M.; Hébert, J.M.; Mason, C.E. Roles for small noncoding RNAs in silencing of retrotransposons in the mammalian brain. Proc. Natl. Acad. Sci. USA 2016, 113, 12697–12702. [Google Scholar] [CrossRef] [Green Version]
- Carlice-dos-Reis, T.; Viana, J.; Moreira, F.C.; De Cardoso, G.L.; Guerreiro, J.; Santos, S. Investigation of mutations in the HBB gene using the 1,000 genomes database. PLoS ONE 2017, 12, e0174637. [Google Scholar] [CrossRef] [Green Version]
- Cristina, T.-P.; Pablo, M.; Teresa, P.M.; Lydia, V.-D.; Irene, A.-R.; Araceli, A.-C. A genetic analysis of a spanish population with early onset Parkinson’s disease. PLoS ONE 2020, 15, e0238098. [Google Scholar] [CrossRef] [PubMed]
- Blauwendraat, C.; Makarious, M.B.; Leonard, H.L.; Bandres-Ciga, S.; Iwaki, H.; Nalls, M.A. A population scale analysis of rare SNCA variation in the UK biobank. Neurobiol. Dis. 2021, 148, 105182. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.-R.; Chang, K.-H.; Chao, C.-Y.; Lin, C.-H.; Chen, Y.-C.; Liu, T.-W. Association of SOD2 p.V16A polymorphism with Parkinson’s disease: A meta-analysis in Han Chinese. J. Formos. Med. Assoc. 2021, 120, 501–507. [Google Scholar] [CrossRef]
- Nalls, M.A.; Blauwendraat, C.; Vallerga, C.L.; Heilbron, K.; Bandres-Ciga, S.; Chang, D. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: A meta-analysis of genome-wide association studies. Lancet Neurol. 2019, 18, 1091–1102. [Google Scholar] [CrossRef]
- Chew, E.G.Y.; Tan, L.C.S.; Au, W.-L.; Prakash, K.-M.; Liu, J.; Foo, J.N. ITPKB and ZNF184 are associated with Parkinson’s disease risk in East Asians. Neurobiol. Aging 2020, 86, 201.e15–201.e17. [Google Scholar] [CrossRef]
- Moosavi, A.; Ardekani, A.M. Role of epigenetics in biology and human diseases. Iran. Biomed. J. 2016, 20, 246–258. [Google Scholar] [PubMed]
- Biswas, S.; Rao, C.M. Epigenetic tools (the writers, the readers and the erasers) and their implications in cancer therapy. Eur. J. Pharmacol. 2018, 837, 8–24. [Google Scholar] [CrossRef]
- Pereira, A.L.; Magalhães, L.; Moreira, F.C.; Reis-das-Mercês, L.; Vidal, A.F.; Ribeiro-dos-Santos, A.M. Epigenetic field cancerization in gastric cancer: MicroRNAs as promising biomarkers. J. Cancer 2019, 10, 1560–1569. [Google Scholar] [CrossRef]
- Magalhães, L.; Quintana, L.G.; Lopes, D.C.F.; Vidal, A.F.; Pereira, A.L.; Pinto, L.C.D.A.; Pinheiro, J.D.J.V.; Khayat, A.S.; Goulart, L.R.; Burbano, R.; et al. APC gene is modulated by hsa-miR-135b-5p in both diffuse and intestinal gastric cancer subtypes. BMC Cancer 2018, 18, 1055. [Google Scholar] [CrossRef] [Green Version]
- Young, J.I.; Sivasankaran, S.K.; Wang, L.; Ali, A.; Mehta, A.; Davis, D.A. Genome-wide brain DNA methylation analysis suggests epigenetic reprogramming in Parkinson disease. Neurol. Genet. 2019, 5, e342. [Google Scholar] [CrossRef] [Green Version]
- Chuang, Y.-H.; Paul, K.C.; Bronstein, J.M.; Bordelon, Y.; Horvath, S.; Ritz, B. Parkinson’s disease is associated with DNA methylation levels in human blood and saliva. Genome Med. 2017, 9, 76. [Google Scholar] [CrossRef]
- Wang, C.; Chen, L.; Yang, Y.; Zhang, M.; Wong, G. Identification of potential blood biomarkers for Parkinson’s disease by gene expression and DNA methylation data integration analysis. Clin. Epigenet. 2019, 11, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Södersten, E.; Toskas, K.; Rraklli, V.; Tiklova, K.; Björklund, Å.K.; Ringnér, M. A comprehensive map coupling histone modifications with gene regulation in adult dopaminergic and serotonergic neurons. Nat. Commun. 2018, 9, 1226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toker, L.; Tran, G.T.; Sundaresan, J.; Tysnes, O.-B.; Alves, G.; Haugarvoll, K. Genome-wide dysregulation of histone acetylation in the Parkinson’s disease brain. BioRxiv 2019. [Google Scholar] [CrossRef]
- Ha, M.; Kim, V.N. Regulation of microRNA biogenesis. Nat. Rev. Mol. Cell Biol. 2014, 15, 509–524. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [Green Version]
- Pfeffer, S.; Sewer, A.; Lagos-Quintana, M.; Sheridan, R.; Sander, C.; Grässer, F.A. Identification of microRNAs of the herpesvirus family. Nat. Methods 2005, 2, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Dueck, A.; Ziegler, C.; Eichner, A.; Berezikov, E.; Meister, G. microRNAs associated with the different human argonaute proteins. Nucleic Acids Res. 2012, 40, 9850–9862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huntzinger, E.; Izaurralde, E. Gene silencing by microRNAs: Contributions of translational repression and mRNA decay. Nat. Rev. Genet. 2011, 12, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Prodromidou, K.; Matsas, R. Species-specific miRNAs in human brain development and disease. Front. Cell. Neurosci. 2019, 13, 559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volvert, M.-L.; Prévot, P.-P.; Close, P.; Laguesse, S.; Pirotte, S.; Hemphill, J. MicroRNA targeting of CoREST controls polarization of migrating cortical neurons. Cell Rep. 2014, 7, 1168–1183. [Google Scholar] [CrossRef] [Green Version]
- Leucht, C.; Stigloher, C.; Wizenmann, A.; Klafke, R.; Folchert, A.; Bally-Cuif, L. MicroRNA-9 directs late organizer activity of the midbrain-hindbrain boundary. Nat. Neurosci. 2008, 11, 641–648. [Google Scholar] [CrossRef]
- Vatsa, N.; Kumar, V.; Singh, B.K.; Kumar, S.S.; Sharma, A.; Jana, N.R. Down-regulation of miRNA-708 promotes aberrant calcium signaling by targeting neuronatin in a mouse model of angelman syndrome. Front. Mol. Neurosci. 2019, 12, 35. [Google Scholar] [CrossRef]
- Wayman, G.A.; Davare, M.; Ando, H.; Fortin, D.; Varlamova, O.; Cheng, H.-Y.M. An activity-regulated microRNA controls dendritic plasticity by down-regulating p250GAP. Proc. Natl. Acad. Sci. USA 2008, 105, 9093–9098. [Google Scholar] [CrossRef] [Green Version]
- Rajman, M.; Schratt, G. MicroRNAs in neural development: From master regulators to fine-tuners. Development 2017, 144, 2310–2322. [Google Scholar] [CrossRef] [Green Version]
- Edbauer, D.; Neilson, J.R.; Foster, K.A.; Wang, C.-F.; Seeburg, D.P.; Batterton, M.N. Regulation of Synaptic structure and function by FMRP-associated MicroRNAs miR-125b and miR-132. Neuron 2010, 65, 373–384. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Kwon, E.J.; Tsai, L.-H. MicroRNAs in learning, memory, and neurological diseases. Learn. Mem. 2012, 19, 359–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin Jung, H.; Suh, Y. MicroRNA in aging: From discovery to biology. Curr. Genom. 2012, 13, 548–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- John, A.; Kubosumi, A.; Reddy, P.H. Mitochondrial MicroRNAs in aging and neurodegenerative diseases. Cells 2020, 9, 1345. [Google Scholar] [CrossRef]
- Ozkul, Y.; Taheri, S.; Bayram, K.K.; Sener, E.F.; Mehmetbeyoglu, E.; Öztop, D.B. A heritable profile of six miRNAs in autistic patients and mouse models. Sci. Rep. 2020, 10, 9011. [Google Scholar] [CrossRef]
- He, K.; Guo, C.; He, L.; Shi, Y. MiRNAs of peripheral blood as the biomarker of schizophrenia. Hereditas 2018, 155, 9. [Google Scholar] [CrossRef]
- Beveridge, N.J.; Cairns, M.J. MicroRNA dysregulation in schizophrenia. Neurobiol. Dis. 2012, 46, 263–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoss, A.G.; Labadorf, A.; Latourelle, J.C.; Kartha, V.K.; Hadzi, T.C.; Gusella, J.F. miR-10b-5p expression in huntington’s disease brain relates to age of onset and the extent of striatal involvement. BMC Med. Genom. 2015, 8, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, G.; Stuendl, A.; Rao, P.; Berulava, T.; Pena Centeno, T.; Kaurani, L. A combined miRNA–piRNA signature to detect Alzheimer’s disease. Transl. Psychiatry 2019, 9, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sierksma, A.; Lu, A.; Salta, E.; Vanden Eynden, E.; Callaerts-Vegh, Z.; D’Hooge, R. Deregulation of neuronal miRNAs induced by amyloid-β or TAU pathology. Mol. Neurodegener. 2018, 13, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juźwik, C.A.; SDrake, S.; Zhang, Y.; Paradis-Isler, N.; Sylvester, A.; Amar-Zifkin, A. microRNA dysregulation in neurodegenerative diseases: A systematic review. Prog. Neurobiol. 2019, 182, 101664. [Google Scholar] [CrossRef]
- Cardo, L.F.; Coto, E.; Ribacoba, R.; Menéndez, M.; Moris, G.; Suárez, E. MiRNA profile in the substantia nigra of Parkinson’s disease and healthy subjects. J. Mol. Neurosci. 2014, 54, 830–836. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Inoue, K.; Ishii, J.; Vanti, W.B.; Voronov, S.V.; Murchison, E. A MicroRNA feedback circuit in midbrain dopamine neurons. Science 2007, 317, 1220–1224. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Yang, Y.; Wang, H.; He, Y.; Li, C. MiR-29c protects against inflammation and apoptosis in Parkinson’s disease model in vivo and in vitro by targeting SP1. Clin. Exp. Pharmacol. Physiol. 2020, 47, 372–382. [Google Scholar] [CrossRef] [PubMed]
- Doxakis, E. Post-transcriptional regulation of α-synuclein expression by mir-7 and mir-153. J. Biol. Chem. 2010, 285, 12726–12734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Junn, E.; Lee, K.-W.; Jeong, B.S.; Chan, T.W.; Im, J.-Y.; Mouradian, M.M. Repression of synuclein expression and toxicity by microRNA-7. Proc. Natl. Acad. Sci. USA 2009, 6, 13052–13057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thome, A.D.; Harms, A.S.; Volpicelli-Daley, L.A.; Standaert, D.G. microRNA-155 regulates alpha-synuclein-induced inflammatory responses in models of parkinson disease. J. Neurosci. 2016, 36, 2383–2390. [Google Scholar] [CrossRef]
- Cho, H.J.; Liu, G.; Jin, S.M.; Parisiadou, L.; Xie, C.; Yu, J. MicroRNA-205 regulates the expression of Parkinson’s disease-related leucine-rich repeat kinase 2 protein. Hum. Mol. Genet. 2013, 22, 608–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gehrke, S.; Imai, Y.; Sokol, N.; Lu, B. Pathogenic LRRK2 negatively regulates microRNA-mediated translational repression. Nature 2010, 466, 637–641. [Google Scholar] [CrossRef]
- Xiong, R.; Wang, Z.; Zhao, Z.; Li, H.; Chen, W.; Zhang, B. MicroRNA-494 reduces DJ-1 expression and exacerbates neurodegeneration. Neurobiol. Aging 2014, 35, 705–714. [Google Scholar] [CrossRef]
- Oh, S.E.; Park, H.-J.; He, L.; Skibiel, C.; Junn, E.; Mouradian, M.M. The Parkinson’s disease gene product DJ-1 modulates miR-221 to promote neuronal survival against oxidative stress. Redox Biol. 2018, 19, 62–73. [Google Scholar] [CrossRef]
- Carrete, H., Jr. Parkinson’s disease and atypical parkinsonism: The importance of magnetic resonance imaging as a potential biomarker. Radiol. Bras. 2017, 50, 5–6. [Google Scholar] [CrossRef] [Green Version]
- Adusumilli, L.; Facchinello, N.; Teh, C.; Busolin, G.; Le, M.T.; Yang, H. miR-7 controls the dopaminergic/oligodendroglial fate through Wnt/β-catenin signaling regulation. Cells 2020, 9, 711. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Dongwei, Z.; Zhang, Z.; Xinhui, Q.; Kunwang, B.; Guohui, L. miR-let-7a suppresses α-synuclein-induced microglia inflammation through targeting STAT3 in Parkinson’s disease. Biochem. Biophys. Res. Commun. 2019, 519, 740–746. [Google Scholar] [CrossRef] [PubMed]
- Sadlon, A.; Takousis, P.; Alexopoulos, P.; Evangelou, E.; Prokopenko, I.; Perneczky, R. miRNAs identify shared pathways in Alzheimer’s and Parkinson’s diseases. Trends Mol. Med. 2019, 25, 662–672. [Google Scholar] [CrossRef] [PubMed]
- Aravin, A.; Gaidatzis, D.; Pfeffer, S.; Lagos-Quintana, M.; Landgraf, P.; Iovino, N. A novel class of small RNAs bind to MILI protein in mouse testes. Nature 2006, 442, 203–207. [Google Scholar] [CrossRef]
- Girard, A.; Sachidanandam, R.; Hannon, G.J.; Carmell, M.A. A germline-specific class of small RNAs binds mammalian Piwi proteins. Nature 2006, 442, 199–202. [Google Scholar] [CrossRef]
- Grivna, S.T.; Beyret, E.; Wang, Z.; Lin, H. A novel class of small RNAs in mouse spermatogenic cells. Genes Dev. 2006, 20, 1709–1714. [Google Scholar] [CrossRef] [Green Version]
- Chalbatani, G.M.; Dana, H.; Memari, F.; Gharagozlou, E.; Ashjaei, S.; Kheirandish, P. Biological function and molecular mechanism of piRNA in cancer. Pract. Lab. Med. 2019, 13, e00113. [Google Scholar] [CrossRef]
- Fonseca, C.G.; dos Santos, J.A.P.; Vidal, A.F.; Santos, S.; Ribeiro-dos-Santos, Â. piRNAs in ancer: A new approach towards translational research. Int. J. Mol. Sci. 2020, 21, 2126. [Google Scholar] [CrossRef] [Green Version]
- Czech, B.; Hannon, G.J. One loop to rule them all: The ping-pong cycle and piRNA-guided silencing. Trends Biochem. Sci. 2016, 41, 324–337. [Google Scholar] [CrossRef] [Green Version]
- Brennecke, J.; Aravin, A.A.; Stark, A.; Dus, M.; Kellis, M.; Sachidanandam, R. Discrete small RNA-generating loci as master regulators of transposon activity in drosophila. Cell 2007, 128, 1089–1103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czech, B.; Munafò, M.; Ciabrelli, F.; Eastwood, E.L.; Fabry, M.H.; Kneuss, E. piRNA-guided genome defense: From biogenesis to silencing. Annu. Rev. Genet. 2018, 52, 131–157. [Google Scholar] [CrossRef] [PubMed]
- Rojas-Ríos, P.; Simonelig, M. piRNAs and PIWI proteins: Regulators of gene expression in development and stem cells. Development 2018, 145, 161786. [Google Scholar] [CrossRef] [Green Version]
- Ross, R.J.; Weiner, M.M.; Lin, H. PIWI proteins and PIWI-interacting RNAs in the soma. Nature 2014, 505, 353–359. [Google Scholar] [CrossRef] [Green Version]
- Weick, E.M.; Miska, E.A. piRNAs: From biogenesis to function. Development 2014, 141, 3458–3471. [Google Scholar] [CrossRef] [Green Version]
- Sheng, Z.-H. The interplay of axonal energy homeostasis and mitochondrial trafficking and anchoring. Trends Cell Biol. 2017, 27, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Pantano, L.; Jodar, M.; Bak, M.; Ballescà, J.L.; Tommerup, N.; Oliva, R. The small RNA content of human sperm reveals pseudogene-derived piRNAs complementary to protein-coding genes. RNA 2015, 21, 1085–1095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajasethupathy, P.; Antonov, I.; Sheridan, R.; Frey, S.; Sander, C.; Tuschl, T. A role for neuronal piRNAs in the epigenetic control of memory-related synaptic plasticity. Cell 2012, 149, 693–707. [Google Scholar] [CrossRef] [Green Version]
- Martinez, V.D.; Enfield, K.S.S.; Rowbotham, D.A.; Lam, W.L. An atlas of gastric PIWI-interacting RNA transcriptomes and their utility for identifying signatures of gastric cancer recurrence. Gastric Cancer 2016, 19, 660–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weng, W.; Liu, N.; Toiyama, Y.; Kusunoki, M.; Nagasaka, T.; Fujiwara, T. Novel evidence for a PIWI-interacting RNA (piRNA) as an oncogenic mediator of disease progression, and a potential prognostic biomarker in colorectal cancer. Mol. Cancer 2018, 17, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.; Mai, D.; Zhang, B.; Jiang, X.; Zhang, J.; Bai, R. PIWI-interacting RNA-36712 restrains breast cancer progression and chemoresistance by interaction with SEPW1 pseudogene SEPW1P RNA. Mol. Cancer 2019, 18, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Qiu, W.; Guo, X.; Lin, X.; Yang, Q.; Zhang, W.; Zhang, Y. Transcriptome-wide piRNA profiling in human brains of Alzheimer’s disease. Neurobiol. Aging 2017, 57, 170–177. [Google Scholar] [CrossRef]
- Zuo, L.; Wang, Z.; Tan, Y.; Chen, X.; Luo, X. piRNAs and their functions in the brain. Int. J. Hum. Genet. 2016, 16, 53–60. [Google Scholar] [CrossRef] [Green Version]
- Rouget, C.; Papin, C.; Boureux, A.; Meunier, A.-C.; Franco, B.; Robine, N. Maternal mRNA deadenylation and decay by the piRNA pathway in the early drosophila embryo. Nature 2010, 467, 1128–1132. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.J.; Banerjee, S.; Zhou, H.; Jammalamadaka, A.; Arcila, M.; Manjunath, B.S. Identification of piRNAs in the central nervous system. RNA 2011, 17, 1090–1099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, Z.; Hu, H.Y.; Jiang, X.; Maierhofer, V.; Neb, E.; He, L. Widespread expression of piRNA-like molecules in somatic tissues. Nucleic Acids Res. 2011, 39, 6596–6607. [Google Scholar] [CrossRef] [PubMed]
- Jensen, P.; Myhre, C.L.; Lassen, P.S.; Metaxas, A.; Khan, A.M.; Lambertsen, K.L. TNFα affects CREB-mediated neuroprotective signaling pathways of synaptic plasticity in neurons as revealed by proteomics and phospho-proteomics. Oncotarget 2017, 8, 60223–60242. [Google Scholar] [CrossRef] [PubMed]
- Ortega-Martínez, S. A new perspective on the role of the CREB family of transcription factors in memory consolidation via adult hippocampal neurogenesis. Front. Mol. Neurosci. 2015, 8, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, S.; Yu, H.; Liu, X.; Liu, Y.; Zheng, J.; Wang, P. PIWIL1/piRNA-DQ593109 regulates the permeability of the blood-tumor barrier via the MEG3/miR-330-5p/RUNX3 axis. Mol. Ther. Nucleic Acids 2018, 10, 412–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulze, M.; Sommer, A.; Plötz, S.; Farrell, M.; Winner, B.; Grosch, J. Sporadic Parkinson’s disease derived neuronal cells show disease-specific mRNA and small RNA signatures with abundant deregulation of piRNAs. Acta Neuropathol. Commun. 2018, 6, 58. [Google Scholar] [CrossRef] [Green Version]
- Roy, J.; Sarkar, A.; Parida, S.; Ghosh, Z.; Mallick, B. Small RNA sequencing revealed dysregulated piRNAs in alzheimer’s disease and their probable role in pathogenesis. Mol. Biosyst. 2017, 13, 565–576. [Google Scholar] [CrossRef]
- Saxena, A.; Tang, D.; Carninci, P. piRNAs warrant investigation in rett syndrome: An omics perspective. Dis. Markers 2012, 33, 261–275. [Google Scholar] [CrossRef]
- Dharap, A.; Nakka, V.P.; Vemuganti, R. Altered expression of PIWI RNA in the rat brain after transient focal ischemia. Stroke 2011, 42, 1105–1109. [Google Scholar] [CrossRef]
- Kim, K.W.; Tang, N.H.; Andrusiak, M.G.; Wu, Z.; Chisholm, A.D.; Jin, Y. A neuronal piRNA pathway inhibits axon regeneration in C. elegans. Neuron 2018, 97, 511–519.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, A.; Jacobs, D.I.; Hoffman, A.E.; Zheng, T.; Zhu, Y. PIWI-interacting RNA 021285 is involved in breast tumorigenesis possibly by remodeling the cancer epigenome. Carcinogenesis 2015, 36, 1094–1102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Liu, H.; Yin, J.; Wu, W.; Zhu, D.; Amos, C.I. Genetic variants in the PIWI-piRNA pathway gene DCP1A predict melanoma disease-specific survival. Int. J. Cancer 2016, 139, 2730–2737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobs, D.I.; Qin, Q.; Lerro, M.C.; Fu, A.; Dubrow, R.; Claus, E.B. PIWI-interacting RNAs in gliomagenesis: Evidence from post-GWAS and functional analyses. Cancer Epidemiol. Biomark. Prev. 2016, 25, 1073–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, V.D.; Vucic, E.A.; Thu, K.L.; Hubaux, R.; Enfield, K.S.S.; Pikor, L.A. Unique somatic and malignant expression patterns implicate PIWI-interacting RNAs in cancer-type specific biology. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef]
- Kwon, C.; Tak, H.; Rho, M.; Chang, H.R.; Kim, Y.H.; Kim, K.T. Detection of PIWI and piRNAs in the mitochondria of mammalian cancer cells. Biochem. Biophys. Res. Commun. 2014, 446, 218–223. [Google Scholar] [CrossRef] [Green Version]
- Cavalcante, G.C.; Magalhães, L.; Ribeiro-dos-Santos, Â.; Vidal, A.F. Mitochondrial epigenetics: Non-coding RNAs as a novel layer of complexity. Int. J. Mol. Sci. 2020, 21, 1838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malik, B.; Feng, F.Y. Long noncoding RNAs in prostate cancer: Overview and clinical implications. Asian J. Androl. 2016, 18, 568–574. [Google Scholar]
- Mercer, T.R.; Mattick, J.S. Structure and function of long noncoding RNAs in epigenetic regulation. Nat. Struct. Mol. Biol. 2013, 20, 300–307. [Google Scholar] [CrossRef]
- Zhai, K.; Liu, B.; Gao, L. Long-Noncoding RNA TUG1 promotes Parkinson’s disease via modulating MiR-152-3p/PTEN pathway. Hum. Gene Ther. 2020, 31, 1274–1287. [Google Scholar] [CrossRef]
- Xu, W.; Zhang, L.; Geng, Y.; Liu, Y.; Zhang, N. Long noncoding RNA GAS5 promotes microglial inflammatory response in Parkinson’s disease by regulating NLRP3 pathway through sponging miR-223-3p. Int. Immunopharmacol. 2020, 85, 106614. [Google Scholar] [CrossRef]
- Zhang, Y.; Shao, W.; Wu, J.; Huang, S.; Yang, H.; Luo, Z. Inflammatory lncRNA AK039862 regulates paraquat-inhibited proliferation and migration of microglial and neuronal cells through the Pafah1b1/Foxa1 pathway in co-culture environments. Ecotoxicol. Environ. Saf. 2021, 208, 111424. [Google Scholar] [CrossRef]
- Chen, M.-Y.; Fan, K.; Zhao, L.-J.; Wei, J.-M.; Gao, J.-X.; Li, Z.-F. Long non-coding RNA nuclear enriched abundant transcript 1 (NEAT1) sponges microRNA-124-3p to up-regulate phosphodiesterase 4B (PDE4B) to accelerate the progression of Parkinson’s disease. Bioengineered 2021, 12, 708–719. [Google Scholar] [CrossRef]
- Sun, Q.; Zhang, Y.; Wang, S.; Yang, F.; Cai, H.; Xing, Y. NEAT1 decreasing suppresses parkinson’s disease progression via acting as miR-1301-3p sponge. J. Mol. Neurosci. 2021, 71, 369–378. [Google Scholar] [CrossRef]
- Liu, R.; Li, F.; Zhao, W. Long noncoding RNA NEAT1 knockdown inhibits MPP+-induced apoptosis, inflammation and cytotoxicity in SK-N-SH cells by regulating miR-212-5p/RAB3IP axis. Neurosci. Lett. 2020, 731, 135060. [Google Scholar] [CrossRef]
- Boros, F.A.; Maszlag-Török, R.; Vécsei, L.; Klivényi, P. Increased level of NEAT1 long non-coding RNA is detectable in peripheral blood cells of patients with Parkinson’s disease. Brain Res. 2020, 1730, 146672. [Google Scholar] [CrossRef]
- Quan, Y.; Wang, J.; Wang, S.; Zhao, J. Association of the plasma long non-coding RNA MEG3 with Parkinson’s disease. Front. Neurol. 2020, 11, 532891. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.; Guo, Y.; Wei, L.; Yu, F.; Yu, B.; Xu, A. Long noncoding RNA POU3F3 and α-synuclein in plasma L1CAM exosomes combined with β-glucocerebrosidase activity: Potential predictors of Parkinson’s disease. Neurotherapeutics 2020, 17, 1104–1119. [Google Scholar] [CrossRef] [PubMed]
- Han, C.-L.; Liu, Y.-P.; Sui, Y.-P.; Chen, N.; Du, T.-T.; Jiang, Y. Integrated transcriptome expression profiling reveals a novel lncRNA associated with L-DOPA-induced dyskinesia in a rat model of Parkinson’s disease. Aging 2020, 12, 718–739. [Google Scholar] [CrossRef] [PubMed]
- Jeck, W.R.; Sorrentino, J.A.; Wang, K.; Slevin, M.K.; Burd, C.E.; Liu, J. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA 2013, 19, 141–157. [Google Scholar] [CrossRef] [Green Version]
- D’Ambra, E.; Capauto, D.; Morlando, M. Exploring the regulatory role of circular RNAs in neurodegenerative disorders. Int. J. Mol. Sci. 2019, 20, 5477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.-L. The expanding regulatory mechanisms and cellular functions of circular RNAs. Nat. Rev. Mol. Cell Biol. 2020, 21, 475–490. [Google Scholar] [CrossRef] [PubMed]
- Wilusz, J.E. A 360° view of circular RNAs: From biogenesis to functions. WIREs RNA 2018, 9, e1478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vo, J.N.; Cieslik, M.; Zhang, Y.; Shukla, S.; Xiao, L.; Zhang, Y. The landscape of circular RNA in cancer. Cell 2019, 176, 869–881.e13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, P.; Wu, W.; Chen, S.; Zheng, Y.; Zhou, L.; Zhang, J. Expanded expression landscape and prioritization of circular RNAs in mammals. Cell Rep. 2019, 26, 3444–3460.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Fan, X.; Mao, M.; Song, X.; Wu, P.; Zhang, Y. Extensive translation of circular RNAs driven by N6-methyladenosine. Cell Res. 2017, 27, 626–641. [Google Scholar] [CrossRef] [Green Version]
- Lu, S.; Yang, X.; Wang, C.; Chen, S.; Lu, S.; Yan, W. Current status and potential role of circular RNAs in neurological disorders. J. Neurochem. 2019, 150, 237–248. [Google Scholar] [CrossRef] [Green Version]
- You, X.; Vlatkovic, I.; Babic, A.; Will, T.; Epstein, I.; Tushev, G. Neural circular RNAs are derived from synaptic genes and regulated by development and plasticity. Nat. Neurosci. 2015, 18, 603–610. [Google Scholar] [CrossRef] [Green Version]
- Qu, S.; Yang, X.; Li, X.; Wang, J.; Gao, Y.; Shang, R. Circular RNA: A new star of noncoding RNAs. Cancer Lett. 2015, 365, 141–148. [Google Scholar] [CrossRef]
- Sang, Q.; Liu, X.; Wang, L.; Qi, L.; Sun, W.; Wang, W. CircSNCA downregulation by pramipexole treatment mediates cell apoptosis and autophagy in Parkinson’s disease by targeting miR-7. Aging 2018, 10, 1281–1293. [Google Scholar] [CrossRef]
- Hansen, T.B.; Jensen, T.I.; Clausen, B.H.; Bramsen, J.B.; Finsen, B.; Damgaard, C.K. Natural RNA circles function as efficient microRNA sponges. Nature 2013, 495, 384–388. [Google Scholar] [CrossRef]
- Feng, Z.; Zhang, L.; Wang, S.; Hong, Q. Circular RNA circDLGAP4 exerts neuroprotective effects via modulating miR-134-5p/CREB pathway in Parkinson’s disease. Biochem. Biophys. Res. Commun. 2020, 522, 388–394. [Google Scholar] [CrossRef] [PubMed]
- Hanan, M.; Simchovitz, A.; Yayon, N.; Vaknine, S.; Cohen-Fultheim, R.; Karmon, M. A Parkinson’s disease circ RNA s resource reveals a link between circ SLC 8A1 and oxidative stress. EMBO Mol. Med. 2020, 12. [Google Scholar] [CrossRef]
- Sirabella, R.; Sisalli, M.J.; Costa, G.; Omura, K.; Ianniello, G.; Pinna, A. NCX1 and NCX3 as potential factors contributing to neurodegeneration and neuroinflammation in the A53T transgenic mouse model of Parkinson’s Disease. Cell Death Dis. 2018, 9, 725. [Google Scholar] [CrossRef]
- Zhou, L.; Yang, L.; Li, Y.; Mei, R.; Yu, H.; Gong, Y. MicroRNA-128 protects dopamine neurons from apoptosis and upregulates the expression of excitatory amino acid transporter 4 in parkinson’s disease by binding to AXIN1. Cell Physiol. Biochem. 2018, 51, 2275–2289. [Google Scholar] [CrossRef] [PubMed]
- Min, S.-W.; Sohn, P.D.; Cho, S.-H.; Swanson, R.A.; Gan, L. Sirtuins in neurodegenerative diseases: An update on potential mechanisms. Front. Aging Neurosci. 2013, 5, 53. [Google Scholar] [CrossRef] [Green Version]
- Shaughnessy, D.T.; McAllister, K.; Worth, L.; Haugen, A.C.; Meyer, J.N.; Domann, F.E. Mitochondria, energetics, epigenetics, and cellular responses to stress. Environ. Health Perspect. 2014, 122, 1271–1278. [Google Scholar] [CrossRef]
- Brooks, H.R. Mitochondria: Finding the power to change. Cell 2018, 175, 891–893. [Google Scholar] [CrossRef] [Green Version]
- Burke, P.J. Mitochondria, bioenergetics and apoptosis in cancer. Trends Cancer 2017, 3, 857–870. [Google Scholar] [CrossRef] [PubMed]
- Anderson, S.; Bankier, A.T.; Barrell, B.G.; de Bruijn, M.H.L.; Coulson, A.R.; Drouin, J. Sequence and organization of the human mitochondrial genome. Nature 1981, 290, 457–465. [Google Scholar] [CrossRef]
- Gammage, P.A.; Frezza, C. Mitochondrial DNA: The overlooked oncogenome? BMC Biol. 2019, 17, 53. [Google Scholar] [CrossRef] [Green Version]
- Karakaidos, P.; Rampias, T. Mitonuclear interactions in the maintenance of mitochondrial integrity. Life 2020, 10, 173. [Google Scholar] [CrossRef]
- Taanman, J.-W. The mitochondrial genome: Structure, transcription, translation and replication. Biochim. Biophys. Acta BBA Bioenerg. 1999, 1410, 103–123. [Google Scholar] [CrossRef] [Green Version]
- Alexeyev, M.; Shokolenko, I.; Wilson, G.; LeDoux, S. The maintenance of mitochondrial DNA integrity-critical analysis and update. Cold Spring Harb. Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Picard, M.; McEwen, B.S. Mitochondria impact brain function and cognition. Proc. Natl. Acad. Sci. USA 2014, 111, 7–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabral-Costa, J.V.; Kowaltowski, A.J. Neurological disorders and mitochondria. Mol. Asp. Med. 2020, 71, 100826. [Google Scholar] [CrossRef]
- Cunnane, S.C.; Trushina, E.; Morland, C.; Prigione, A.; Casadesus, G.; Andrews, Z.B. Brain energy rescue: An emerging therapeutic concept for neurodegenerative disorders of ageing. Nat. Rev. Drug Discov. 2020, 19, 609–633. [Google Scholar] [CrossRef] [PubMed]
- Langston, J.W.; Langston, E.B.; Irwin, I. MPTP-induced parkinsonism in human and non-human primates-clinical and experimental aspects. Acta Neurol. Scand. Suppl. 1984, 100, 49–54. [Google Scholar]
- Cannon, J.R.; Tapias, V.; Na, H.M.; Honick, A.S.; Drolet, R.E.; Greenamyre, J.T. A highly reproducible rotenone model of Parkinson’s disease. Neurobiol. Dis. 2009, 34, 279–290. [Google Scholar] [CrossRef] [Green Version]
- Schapira, A.H.V.; Mann, V.M.; Cooper, J.M.; Dexter, D.; Daniel, S.E.; Jenner, P. Anatomic and disease specificity of NADH CoQ1 reductase (Complex I) deficiency in Parkinson’s disease. J. Neurochem. 1990, 55, 2142–2145. [Google Scholar] [CrossRef] [PubMed]
- Blin, O.; Desnuelle, C.; Rascol, O.; Borg, M.; Paul, H.P.S.; Azulay, J.P. Mitochondrial respiratory failure in skeletal muscle from patients with Parkinson’s disease and multiple system atrophy. J. Neurol. Sci. 1994, 125, 95–101. [Google Scholar] [CrossRef]
- Haas, R.H.; Nasirian, F.; Nakano, K.; Ward, D.; Pay, M.; Hill, R. Low platelet mitochondrial complex I and complex II/III activity in early untreated parkinson’s disease: Abnormalities of electron transport complexes in PD. Ann. Neurol. 1995, 37, 714–722. [Google Scholar] [CrossRef] [PubMed]
- Gu, M.; Cooper, J.M.; Taanman, J.W.; Schapira, A.H.V. Mitochondrial DNA transmission of the mitochondrial defect in Parkinson’s disease. Ann. Neurol. 1998, 44, 177–186. [Google Scholar] [CrossRef]
- Bose, A.; Beal, M.F. Mitochondrial dysfunction in Parkinson’s disease. J. Neurochem. 2016, 139, 216–231. [Google Scholar] [CrossRef]
- Taylor, R.W.; Turnbull, D.M. Mitochondrial DNA mutations in human disease. Nat. Rev. Genet. 2005, 6, 389–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schon, E.A.; DiMauro, S.; Hirano, M. Human mitochondrial DNA: Roles of inherited and somatic mutations. Nat. Rev. Genet. 2012, 13, 878–890. [Google Scholar] [CrossRef] [PubMed]
- Antonyová, V.; Kejík, Z.; Brogyányi, T.; Kaplánek, R.; Pajková, M.; Talianová, V. Role of mtDNA disturbances in the pathogenesis of Alzheimer’s and Parkinson’s disease. DNA Repair 2020, 91–92, 102871. [Google Scholar] [CrossRef]
- Richter, G.; Sonnenschein, A.; Grünewald, T.; Reichmann, H.; Janetzky, B. Novel mitochondrial DNA mutations in Parkinson’s disease. J. Neural. Transm. 2002, 109, 721–729. [Google Scholar] [CrossRef]
- Shoffner, J.M.; Brown, M.D.; Torroni, A.; Lott, M.T.; Cabell, M.F.; Mirra, S.S. Mitochondrial DNA variants observed in Alzheimer disease and Parkinson disease patients. Genomics 1993, 17, 171–184. [Google Scholar] [CrossRef]
- Huerta, C.; Castro, M.G.; Coto, E.; Blázquez, M.; Ribacoba, R.; Guisasola, L.M. Mitochondrial DNA polymorphisms and risk of Parkinson’s disease in Spanish population. J. Neurol. Sci. 2005, 236, 49–54. [Google Scholar] [CrossRef]
- Egensperger, R.; Kösel, S.; Schnopp, N.M.; Mehraein, P.; Graeber, M.B. Association of the mitochondrial tRNAA4336G mutation with Alzheimer’s and Parkinson’s diseases. Neuropathol. Appl. Neurobiol. 1997, 23, 315–321. [Google Scholar] [CrossRef] [PubMed]
- Nido, G.S.; Dölle, C.; Flønes, I.; Tuppen, H.A.; Alves, G.; Tysnes, O.-B. Ultradeep mapping of neuronal mitochondrial deletions in Parkinson’s disease. Neurobiol. Aging 2018, 63, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Coxhead, J.; Kurzawa-Akanbi, M.; Hussain, R.; Pyle, A.; Chinnery, P.; Hudson, G. Somatic mtDNA variation is an important component of Parkinson’s disease. Neurobiol. Aging 2016, 38, 217.e1–217.e6. [Google Scholar] [CrossRef] [Green Version]
- Ross, O.A.; McCormack, R.; Maxwell, L.D.; Duguid, R.A.; Quinn, D.J.; Barnett, Y.A. mt4216C variant in linkage with the mtDNA TJ cluster may confer a susceptibility to mitochondrial dysfunction resulting in an increased risk of Parkinson’s disease in the Irish. Exp. Gerontol. 2003, 38, 397–405. [Google Scholar] [CrossRef]
- van der Walt, J.M.; Nicodemus, K.K.; Martin, E.R.; Scott, W.K.; Nance, M.A.; Watts, R.L. Mitochondrial Polymorphisms Significantly Reduce the Risk of Parkinson Disease. Am. J. Hum. Genet. 2003, 72, 804–811. [Google Scholar] [CrossRef] [Green Version]
- Hudson, G.; Nalls, M.; Evans, J.R.; Breen, D.P.; Winder-Rhodes, S.; Morrison, K.E. Two-stage association study and meta-analysis of mitochondrial DNA variants in Parkinson disease. Neurology 2013, 80, 2042–2048. [Google Scholar] [CrossRef] [PubMed]
- Rosner, S.; Giladi, N.; Orr-Urtreger, A. Advances in the genetics of Parkinson’s disease. Acta Pharmacol. Sin. 2008, 29, 21–34. [Google Scholar] [CrossRef] [Green Version]
- Singleton, A.; Hardy, J. The evolution of genetics: Alzheimer’s and Parkinson’s Diseases. Neuron 2016, 90, 1154–1163. [Google Scholar] [CrossRef] [Green Version]
- Grünewald, A.; Kumar, K.R.; Sue, C.M. New insights into the complex role of mitochondria in Parkinson’s disease. Prog. Neurobiol. 2019, 177, 73–93. [Google Scholar] [CrossRef]
- Ashrafi, G.; Schwarz, T.L. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2013, 20, 31–42. [Google Scholar] [CrossRef] [Green Version]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef]
- Valente, E.M.; Bentivoglio, A.R.; Dixon, P.H.; Ferraris, A.; Ialongo, T.; Frontali, M. Localization of a novel locus for autosomal recessive early-onset parkinsonism, park6, on human chromosome 1p35-p36. Am. J. Hum. Genet. 2001, 68, 895–900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Duijn, C.M.; Dekker, M.C.J.; Bonifati, V.; Galjaard, R.J.; Houwing-Duistermaat, J.J.; Snijders, P.J.L.M. PARK7, a novel locus for autosomal recessive early-onset parkinsonism, on chromosome 1p36. Am. J. Hum. Genet. 2001, 69, 629–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, Q.; Jeong, Y.Y. Mitophagy in Alzheimer’s disease and other age-related neurodegenerative diseases. Cells 2020, 9, 150. [Google Scholar] [CrossRef] [Green Version]
- Iacobazzi, V.; Castegna, A.; Infantino, V.; Andria, G. Mitochondrial DNA methylation as a next-generation biomarker and diagnostic tool. Mol. Genet. Metab. 2013, 110, 25–34. [Google Scholar] [CrossRef]
- D’Aquila, P.; Bellizzi, D.; Passarino, G. Mitochondria in health, aging and diseases: The epigenetic perspective. Biogerontology 2015, 16, 569–585. [Google Scholar] [CrossRef] [PubMed]
- Devall, M.; Smith, R.G.; Jeffries, A.; Hannon, E.; Davies, M.N.; Schalkwyk, L. Regional differences in mitochondrial DNA methylation in human post-mortem brain tissue. Clin. Epigenet. 2017, 9, 47. [Google Scholar] [CrossRef] [Green Version]
- Patil, V.; Cuenin, C.; Chung, F.; Aguilera, J.R.R.; Fernandez-Jimenez, N.; Romero-Garmendia, I. Human mitochondrial DNA is extensively methylated in a non-CpG context. Nucleic Acids Res. 2019, 47, 10072–10085. [Google Scholar] [CrossRef] [Green Version]
- Mercer, T.R.; Neph, S.; Dinger, M.E.; Crawford, J.; Smith, M.A.; Shearwood, A.-M.J. The human mitochondrial transcriptome. Cell 2011, 146, 645–658. [Google Scholar] [CrossRef] [Green Version]
- Gusic, M.; Prokisch, H. ncRNAs: New players in mitochondrial health and disease? Front. Genet. 2020, 11, 95. [Google Scholar] [CrossRef] [Green Version]
- Blanch, M.; Mosquera, J.L.; Ansoleaga, B.; Ferrer, I.; Barrachina, M. Altered mitochondrial DNA methylation pattern in Alzheimer disease–related pathology and in Parkinson disease. Am. J. Pathol. 2016, 186, 385–397. [Google Scholar] [CrossRef] [Green Version]
- Chuang, Y.-H.; Lu, A.T.; Paul, K.C.; Folle, A.D.; Bronstein, J.M.; Bordelon, Y. Longitudinal epigenome-wide methylation study of cognitive decline and motor progression in Parkinson’s disease. JPD 2019, 9, 389–400. [Google Scholar] [CrossRef]
- Lyu, Y.; Bai, L.; Qin, C. Long noncoding RNAs in neurodevelopment and Parkinson’s disease. Anim. Models. Exp. Med. 2019, 2, 239–251. [Google Scholar] [CrossRef] [Green Version]
- Cho, I.; Blaser, M.J. The human microbiome: At the interface of health and disease. Nat. Rev. Genet. 2012, 13, 260–270. [Google Scholar] [CrossRef] [Green Version]
- Lloyd-Price, J.; Abu-Ali, G.; Huttenhower, C. The healthy human microbiome. Genome Med. 2016, 8, 51. [Google Scholar] [CrossRef] [Green Version]
- Sasmita, A.O. Modification of the gut microbiome to combat neurodegeneration. Rev. Neurosci. 2019, 30, 795–805. [Google Scholar] [CrossRef]
- Rhee, S.H.; Pothoulakis, C.; Mayer, E.A. Principles and clinical implications of the brain–gut–enteric microbiota axis. Nat. Rev. Gastroenterol. Hepatol. 2009, 6, 306–314. [Google Scholar] [CrossRef] [Green Version]
- Braak, H.; Tredici, K.D.; Rüb, U.; de Vos, R.A.I.; Jansen Steur, E.N.H.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Svensson, E.; Horváth-Puhó, E.; Thomsen, R.W.; Djurhuus, J.C.; Pedersen, L.; Borghammer, P. Vagotomy and subsequent risk of Parkinson’s disease: Vagotomy and risk of PD. Ann. Neurol. 2015, 78, 522–529. [Google Scholar] [CrossRef]
- Sampson, T.R.; Debelius, J.W.; Thron, T.; Janssen, S.; Shastri, G.G.; Ilhan, Z.E. Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson’s disease. Cell 2016, 167, 1469–1480.e12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattarai, Y.; Si, J.; Pu, M.; Ross, O.A.; McLean, P.J.; Till, L. Role of gut microbiota in regulating gastrointestinal dysfunction and motor symptoms in a mouse model of Parkinson’s disease. Gut Microbes 2021, 13. [Google Scholar] [CrossRef] [PubMed]
- Dodiya, H.B.; Forsyth, C.B.; Voigt, R.M.; Engen, P.A.; Patel, J.; Shaikh, M. Chronic stress-induced gut dysfunction exacerbates Parkinson’s disease phenotype and pathology in a rotenone-induced mouse model of Parkinson’s disease. Neurobiol. Dis. 2020, 135, 104352. [Google Scholar] [CrossRef] [PubMed]
- Gorecki, A.M.; Preskey, L.; Bakeberg, M.C.; Kenna, J.E.; Gildenhuys, C.; MacDougall, G. Altered gut microbiome in Parkinson’s disease and the influence of lipopolysaccharide in a human α-synuclein over-expressing mouse model. Front. Neurosci. 2019, 13, 839. [Google Scholar] [CrossRef] [Green Version]
- Aho, V.T.E.; Pereira, P.A.B.; Voutilainen, S.; Paulin, L.; Pekkonen, E.; Auvinen, P. Gut microbiota in Parkinson’s disease: Temporal stability and relations to disease progression. EBioMedicine 2019, 44, 691–707. [Google Scholar] [CrossRef] [Green Version]
- Cilia, R.; Piatti, M.; Cereda, E.; Bolliri, C.; Caronni, S.; Ferri, V. Does gut microbiota influence the course of Parkinson’s disease? A 3-year prospective exploratory study in de novo patients. JPD 2021, 11, 159–170. [Google Scholar] [CrossRef]
- Unger, M.M.; Spiegel, J.; Dillmann, K.-U.; Grundmann, D.; Philippeit, H.; Bürmann, J. Short chain fatty acids and gut microbiota differ between patients with Parkinson’s disease and age-matched controls. Parkinsonism Relat. Disord. 2016, 32, 66–72. [Google Scholar] [CrossRef]
- Lin, C.-H.; Chen, C.-C.; Chiang, H.-L.; Liou, J.-M.; Chang, C.-M.; Lu, T.-P. Altered gut microbiota and inflammatory cytokine responses in patients with Parkinson’s disease. J. Neuroinflamm. 2019, 16, 129. [Google Scholar] [CrossRef]
- Scheperjans, F.; Aho, V.; Pereira, P.A.B.; Koskinen, K.; Paulin, L.; Pekkonen, E. Gut microbiota are related to Parkinson’s disease and clinical phenotype. Mov. Disord. 2015, 30, 350–358. [Google Scholar] [CrossRef]
- Hegelmaier, T.; Lebbing, M.; Duscha, A.; Tomaske, L.; Tönges, L.; Holm, J.B. Interventional influence of the intestinal microbiome through dietary intervention and bowel cleansing might improve motor symptoms in Parkinson’s disease. Cells 2020, 9, 376. [Google Scholar] [CrossRef] [Green Version]
- Hertel, J.; Harms, A.C.; Heinken, A.; Baldini, F.; Thinnes, C.C.; Glaab, E. Integrated analyses of microbiome and longitudinal metabolome data reveal microbial-host interactions on sulfur metabolism in Parkinson’s disease. Cell Rep. 2019, 29, 1767–1777.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldini, F.; Hertel, J.; Sandt, E.; Thinnes, C.C.; Neuberger-Castillo, L.; Pavelka, L. Parkinson’s disease-associated alterations of the gut microbiome predict disease-relevant changes in metabolic functions. BMC Biol. 2020, 18, 62. [Google Scholar] [CrossRef] [PubMed]
- Morrison, D.J.; Preston, T. Formation of short chain fatty acids by the gut microbiota and their impact on human metabolism. Gut Microbes 2016, 7, 189–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, Y.; Shan, C.; Zhuang, S.; Zhuang, Q.; Ghosh, A.; Zhu, K. Gut microbiota-derived propionate mediates the neuroprotective effect of osteocalcin in a mouse model of Parkinson’s disease. Microbiome 2021, 9, 34. [Google Scholar] [CrossRef]
- Nishiwaki, H.; Hamaguchi, T.; Ito, M.; Ishida, T.; Maeda, T.; Kashihara, K. Short-chain fatty acid-producing gut microbiota is decreased in Parkinson’s disease but not in rapid-eye-movement sleep behavior disorder. MSystems 2020, 5, e00797-20. [Google Scholar] [CrossRef]
- Cirstea, M.S.; Yu, A.C.; Golz, E.; Sundvick, K.; Kliger, D.; Radisavljevic, N. Microbiota composition and metabolism are associated with gut function in Parkinson’s disease. Mov. Disord. 2020, 35, 1208–1217. [Google Scholar] [CrossRef]
- Rowland, I.; Gibson, G.; Heinken, A.; Scott, K.; Swann, J.; Thiele, I. Gut microbiota functions: Metabolism of nutrients and other food components. Eur. J. Nutr. 2018, 57, 1–24. [Google Scholar] [CrossRef] [Green Version]
- Ho, L.; Zhao, D.; Ono, K.; Ruan, K.; Mogno, I.; Tsuji, M. Heterogeneity in gut microbiota drive polyphenol metabolism that influences α-synuclein misfolding and toxicity. J. Nutr. Biochem. 2019, 64, 170–181. [Google Scholar] [CrossRef]
- Haikal, C.; Chen, Q.-Q.; Li, J.-Y. Microbiome changes: An indicator of Parkinson’s disease? Transl. Neurodegener. 2019, 8, 38. [Google Scholar] [CrossRef] [Green Version]
- Vascellari, S.; Palmas, V.; Melis, M.; Pisanu, S.; Cusano, R.; Uva, P. Gut microbiota and metabolome alterations associated with Parkinson’s disease. MSystems 2020, 5, e00561-20. [Google Scholar] [CrossRef]
- Losurdo, G.; D’Abramo, F.S.; Indellicati, G.; Lillo, C.; Ierardi, E.; Di Leo, A. The influence of small intestinal bacterial overgrowth in digestive and extra-intestinal disorders. Int. J. Mol. Sci. 2020, 21, 3531. [Google Scholar] [CrossRef]
- Tan, A.H.; Mahadeva, S.; Thalha, A.M.; Gibson, P.R.; Kiew, C.K.; Yeat, C.M. Small intestinal bacterial overgrowth in Parkinson’s disease. Park. Relat. Disord. 2014, 20, 535–540. [Google Scholar] [CrossRef]
- Hewel, C.; Kaiser, J.; Wierczeiko, A.; Linke, J.; Reinhardt, C.; Endres, K. Common miRNA patterns of alzheimer’s disease and Parkinson’s disease and their putative impact on commensal gut microbiota. Front. Neurosci. 2019, 13, 113. [Google Scholar] [CrossRef] [Green Version]
- Fong, W.; Li, Q.; Yu, J. Gut microbiota modulation: A novel strategy for prevention and treatment of colorectal cancer. Oncogene 2020, 39, 4925–4943. [Google Scholar] [CrossRef]
- Vendrik, K.E.W.; Ooijevaar, R.E.; de Jong, P.R.C.; Laman, J.D.; van Oosten, B.W.; van Hilten, J.J. Fecal microbiota transplantation in neurological disorders. Front. Cell Infect. Microbiol. 2020, 10, 98. [Google Scholar] [CrossRef] [Green Version]
- Koutzoumis, D.N.; Vergara, M.; Pino, J.; Buddendorff, J.; Khoshbouei, H.; Mandel, R.J. Alterations of the gut microbiota with antibiotics protects dopamine neuron loss and improve motor deficits in a pharmacological rodent model of Parkinson’s disease. Exp. Neurol. 2020, 325, 113159. [Google Scholar] [CrossRef] [PubMed]
- Uyar, G.Ö.; Yildiran, H. A nutritional approach to microbiota in Parkinson’s disease. Biosci. Microbiota Food Health 2019, 38, 115–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.; Xu, H.; Luo, Q.; He, J.; Li, M.; Chen, H.; Tang, W.; Nie, Y.; Zhou, Y. Fecal microbiota transplantation to treat Parkinsonʼs disease with constipation: A case report. Medicine 2019, 98, e16163. [Google Scholar] [CrossRef] [PubMed]
- Hazan, S. Rapid improvement in Alzheimer’s disease symptoms following fecal microbiota transplantation: A case report. J. Int. Med. Res. 2020, 48. [Google Scholar] [CrossRef] [PubMed]
- Scheperjans, F.; Derkinderen, P.; Borghammer, P. The gut and Parkinson’s disease: Hype or hope? JPD 2018, 8, S31–S39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borghammer, P.; Van Den Berge, N. Brain-first versus gut-first parkinson’s disease: A hypothesis. JPD 2019, 9, S281–S295. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Gene | Protein | PD Type | Main Function | Reference |
|---|---|---|---|---|
| SNCA | α-Synuclein | Monogenic and Sporadic PD | Increase local Ca2+ release to enhance ATP-induced exocytosis; Regulation of synaptic vesicle trafficking and neurotransmitter release; Modulation of Dopamine transporter (DAT) | [26] [33] [34] [31] |
| GBA | Glucocerebrosidase | Sporadic PD | Degradation of complex lipids; Cholesterol metabolism | [35] [36] [37] [38] [39] |
| LRRK2 | Dardarin | Monogenic and Sporadic PD | Cellular response to Dopamine Mitochondrial organization, location Regulation of autophagy Mitochondrial depolarization | [40] [41] [42] [43] [44] |
| PARK2 | Parkin | Familial and Sporadic PD | Mediates ubiquitination to remove and/or detox abnormal folded or damaged proteins | [45] [46] |
| PARK6 | Pink1 | Familial and Sporadic PD | Regulation of damaged mitochondrial clearance by mitophagy | [47] [45] [48] |
| PARK7 | DJ-1 | Sporadic PD | Mitophagy Response to ROS Regulation of neural apoptosis | [49] [50] [51] [52] |
| MAPT | TAU | Sporadic PD | Astrocyte activation; Axonal transport of mitochondria Microglial cellular activation Cellular response against ROS Regulation of Autophagy Regulation of Synaptic Plasticity | [53] [54] [55] [56] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fonseca Cabral, G.; Schaan, A.P.; Cavalcante, G.C.; Sena-dos-Santos, C.; de Souza, T.P.; Souza Port’s, N.M.; dos Santos Pinheiro, J.A.; Ribeiro-dos-Santos, Â.; Vidal, A.F. Nuclear and Mitochondrial Genome, Epigenome and Gut Microbiome: Emerging Molecular Biomarkers for Parkinson’s Disease. Int. J. Mol. Sci. 2021, 22, 9839. https://doi.org/10.3390/ijms22189839
Fonseca Cabral G, Schaan AP, Cavalcante GC, Sena-dos-Santos C, de Souza TP, Souza Port’s NM, dos Santos Pinheiro JA, Ribeiro-dos-Santos Â, Vidal AF. Nuclear and Mitochondrial Genome, Epigenome and Gut Microbiome: Emerging Molecular Biomarkers for Parkinson’s Disease. International Journal of Molecular Sciences. 2021; 22(18):9839. https://doi.org/10.3390/ijms22189839
Chicago/Turabian StyleFonseca Cabral, Gleyce, Ana Paula Schaan, Giovanna C. Cavalcante, Camille Sena-dos-Santos, Tatiane Piedade de Souza, Natacha M. Souza Port’s, Jhully Azevedo dos Santos Pinheiro, Ândrea Ribeiro-dos-Santos, and Amanda F. Vidal. 2021. "Nuclear and Mitochondrial Genome, Epigenome and Gut Microbiome: Emerging Molecular Biomarkers for Parkinson’s Disease" International Journal of Molecular Sciences 22, no. 18: 9839. https://doi.org/10.3390/ijms22189839