
From Menopause to Neurodegeneration—Molecular Basis and Potential Therapy


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Sex Differences in Major Neurodegenerative Diseases
2.1. Alzheimer’s Disease (AD)
2.2. Parkinson’s Disease (PD)
2.3. Dementia with Lewy Bodies (DLB)
3. Role of Hormones and Trophic Factors in Neurodegenerative Diseases
3.1. Effects of Estrogen on Neuroplasticity
3.2. Immunomodulatory Effects of Estrogen
3.3. The Role of Estrogen in AD
3.4. The Role of Estrogen in PD
3.5. The Effects of Progesterone on Neurodegenerative Diseases
3.6. The Role of IGF-1 in Neurodegenerative Diseases
4. Menopause and Estrogen, Progesterone, and IGF-1 Signals
4.1. Genomic Action of Estrogen
4.2. Non-Genomic Pathway Signals of Estrogen
4.3. IGF-1 and IGF-1R Signals in AD
4.4. Progesterone Signals and Neuroprotection
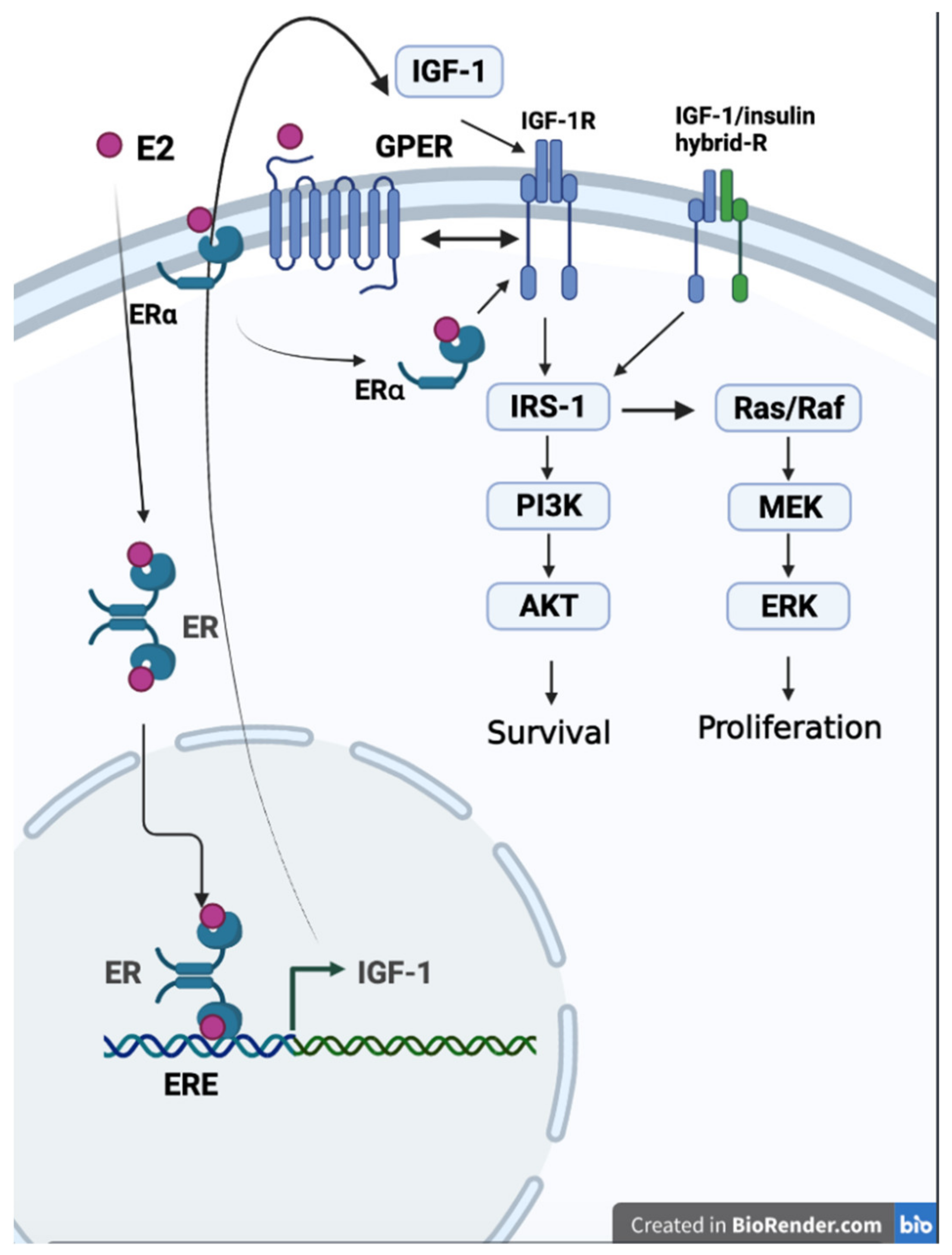
4.5. Estrogen, IGF-1, and IGF-1R Signals
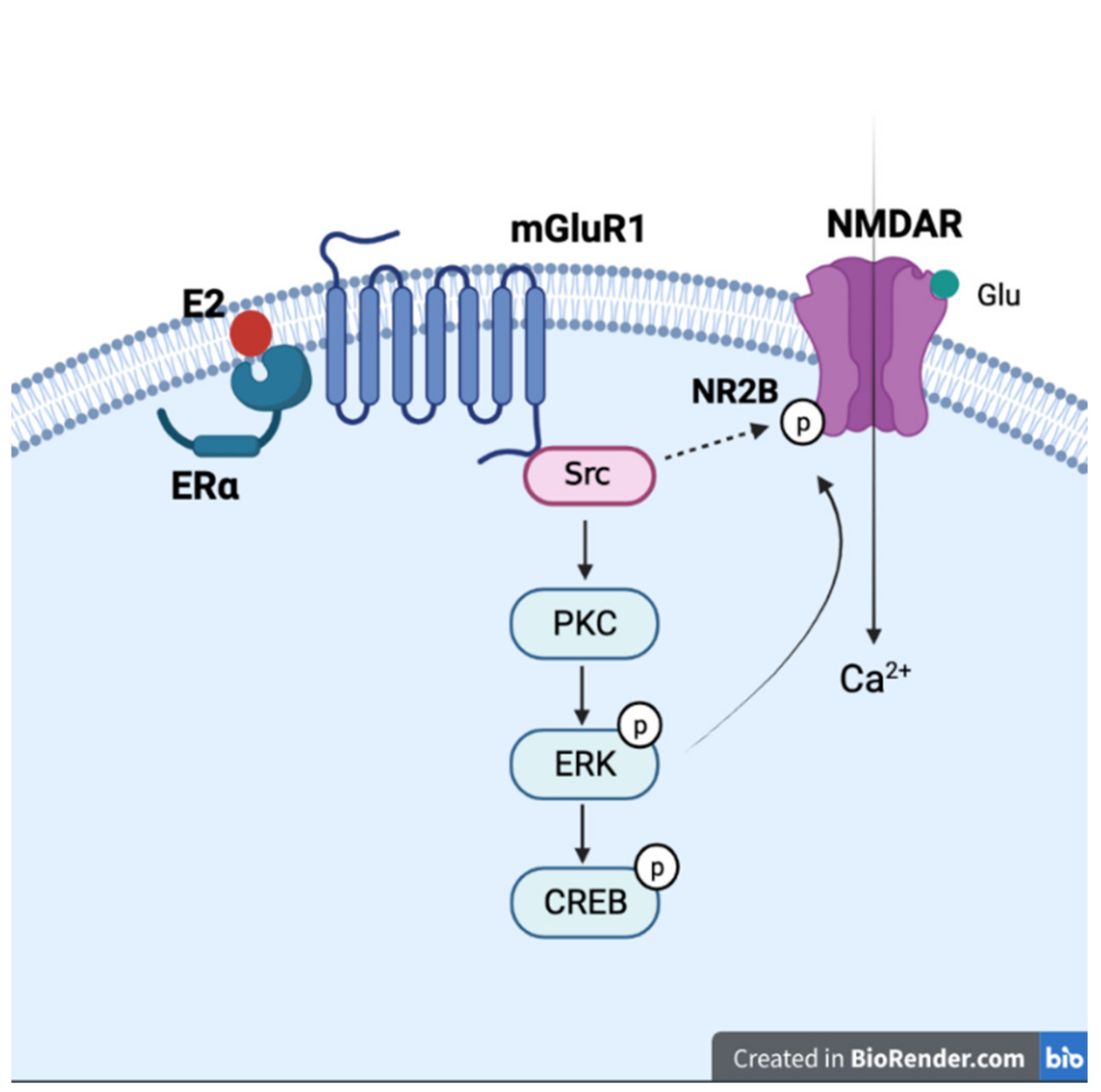
5. Estrogen and NMDA Receptor Signals in Neurodegenerative Diseases
6. Other Potential Therapeutic Strategies
6.1. Effects of Phytoestrogens on AD
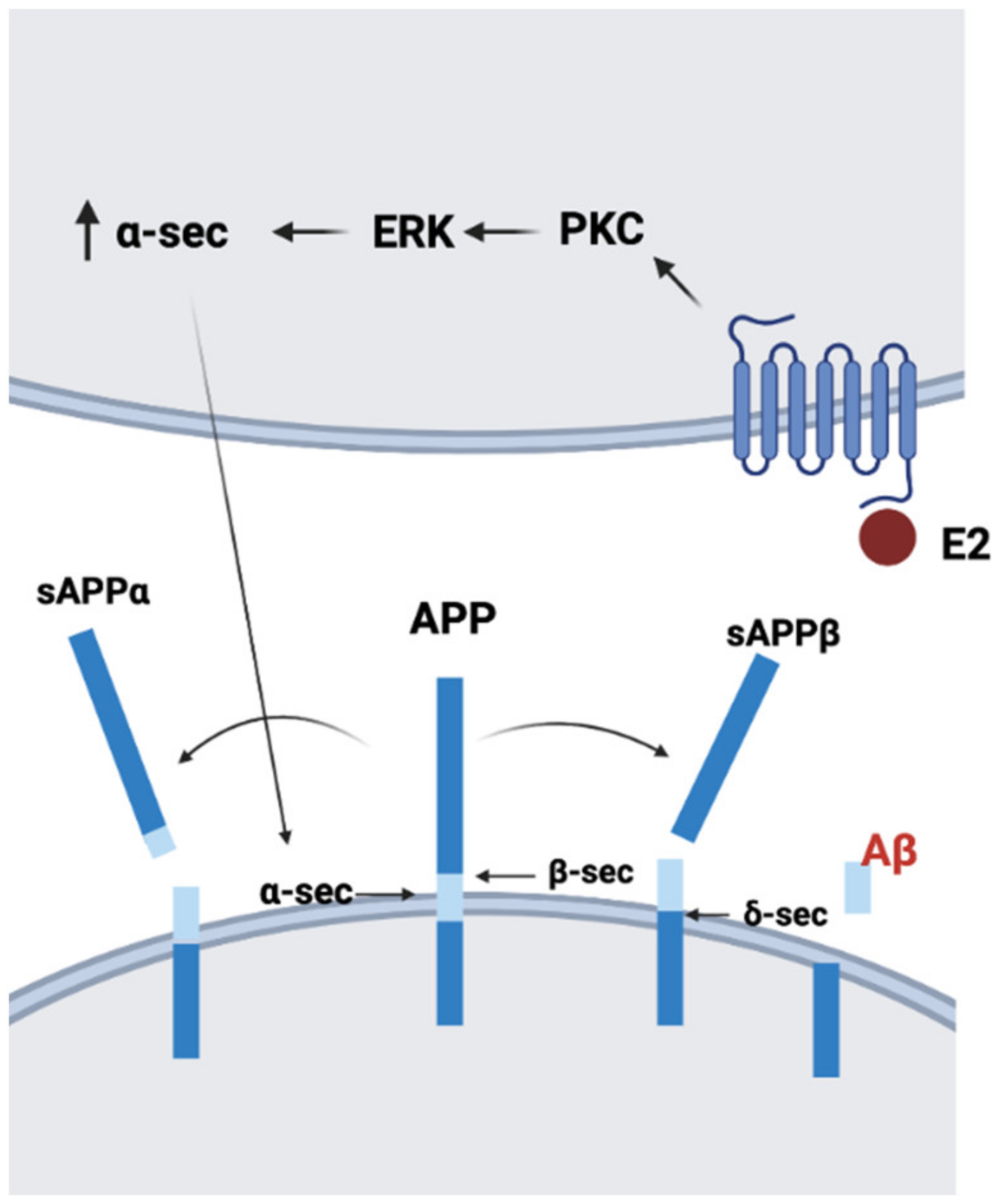
6.2. Genomic and Non-Genomic Aspects of AD
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Garre-Olmo, J. Epidemiology of Alzheimer’s disease and other dementias. Rev. Neurol. 2018, 66, 377–386. [Google Scholar]
- GBD 2016 Parkinson’s Disease Collaborators. Global, regional, and national burden of Parkinson’s disease, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2018, 17, 939–953. [Google Scholar] [CrossRef] [Green Version]
- Kane, J.P.M.; Surendranathan, A.; Bentley, A.; Barker, S.A.H.; Taylor, J.P.; Thomas, A.J.; Allan, L.M.; McNally, R.J.; James, P.W.; McKeith, I.G.; et al. Clinical prevalence of Lewy body dementia. Alzheimers Res. Ther. 2018, 10, 19. [Google Scholar] [CrossRef] [Green Version]
- West, M.J.; Coleman, P.D.; Flood, D.G.; Troncoso, J.C. Differences in the pattern of hippocampal neuronal loss in normal ageing and Alzheimer’s disease. Lancet 1994, 344, 769–772. [Google Scholar] [CrossRef]
- Schulz, J.B. Neuronal pathology in Parkinson’s disease. Cell Tissue Res. 2005, 320, 211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erskine, D.; Thomas, A.J.; Attems, J.; Taylor, J.P.; McKeith, I.G.; Morris, C.M.; Khundakar, A.A. Specific patterns of neuronal loss in the pulvinar nucleus in dementia with lewy bodies. Mov. Disord. 2017, 32, 414–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurtishi, A.; Rosen, B.; Patil, K.S.; Alves, G.W.; Moller, S.G. Cellular Proteostasis in Neurodegeneration. Mol. Neurobiol. 2019, 56, 3676–3689. [Google Scholar] [CrossRef]
- Atwood, C.S.; Bowen, R.L. The endocrine dyscrasia that accompanies menopause and andropause induces aberrant cell cycle signaling that triggers re-entry of post-mitotic neurons into the cell cycle, neurodysfunction, neurodegeneration and cognitive disease. Horm. Behav. 2015, 76, 63–80. [Google Scholar] [CrossRef] [Green Version]
- Mielke, M.M. Sex and Gender Differences in Alzheimer’s Disease Dementia. Psychiatr. Times 2018, 35, 14–17. [Google Scholar]
- Jurado-Coronel, J.C.; Cabezas, R.; Avila Rodriguez, M.F.; Echeverria, V.; Garcia-Segura, L.M.; Barreto, G.E. Sex differences in Parkinson’s disease: Features on clinical symptoms, treatment outcome, sexual hormones and genetics. Front. Neuroendocrinol. 2018, 50, 18–30. [Google Scholar] [CrossRef]
- Lin, C.H.; Chen, P.K.; Wang, S.H.; Lane, H.Y. Effect of Sodium Benzoate on Cognitive Function Among Patients With Behavioral and Psychological Symptoms of Dementia: Secondary Analysis of a Randomized Clinical Trial. JAMA Netw. Open 2021, 4, e216156. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.X.; Jacobs, D.; Stern, Y.; Marder, K.; Schofield, P.; Gurland, B.; Andrews, H.; Mayeux, R. Effect of oestrogen during menopause on risk and age at onset of Alzheimer’s disease. Lancet 1996, 348, 429–432. [Google Scholar] [CrossRef]
- Kawas, C.; Resnick, S.; Morrison, A.; Brookmeyer, R.; Corrada, M.; Zonderman, A.; Bacal, C.; Lingle, D.D.; Metter, E. A prospective study of estrogen replacement therapy and the risk of developing Alzheimer’s disease: The Baltimore Longitudinal Study of Aging. Neurology 1997, 48, 1517–1521. [Google Scholar] [CrossRef]
- Gould, E.; Woolley, C.S.; Frankfurt, M.; McEwen, B.S. Gonadal steroids regulate dendritic spine density in hippocampal pyramidal cells in adulthood. J. Neurosci. 1990, 10, 1286–1291. [Google Scholar] [CrossRef] [Green Version]
- Hara, Y.; Park, C.S.; Janssen, W.G.; Roberts, M.T.; Morrison, J.H.; Rapp, P.R. Synaptic correlates of memory and menopause in the hippocampal dentate gyrus in rhesus monkeys. Neurobiol. Aging 2012, 33, 421.e17–421.e28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hara, Y.; Yuk, F.; Puri, R.; Janssen, W.G.; Rapp, P.R.; Morrison, J.H. Estrogen Restores Multisynaptic Boutons in the Dorsolateral Prefrontal Cortex while Promoting Working Memory in Aged Rhesus Monkeys. J. Neurosci. 2016, 36, 901–910. [Google Scholar] [CrossRef] [Green Version]
- Zagni, E.; Simoni, L.; Colombo, D. Sex and Gender Differences in Central Nervous System-Related Disorders. Neurosci. J. 2016, 2016, 2827090. [Google Scholar] [CrossRef] [Green Version]
- Ott, A.; Stolk, R.P.; van Harskamp, F.; Pols, H.A.; Hofman, A.; Breteler, M.M. Diabetes mellitus and the risk of dementia: The Rotterdam Study. Neurology 1999, 53, 1937–1942. [Google Scholar] [CrossRef]
- Paykel, E.S.; Brayne, C.; Huppert, F.A.; Gill, C.; Barkley, C.; Gehlhaar, E.; Beardsall, L.; Girling, D.M.; Pollitt, P.; O’Connor, D. Incidence of dementia in a population older than 75 years in the United Kingdom. Arch. Gen. Psychiatry 1994, 51, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Beam, C.R.; Kaneshiro, C.; Jang, J.Y.; Reynolds, C.A.; Pedersen, N.L.; Gatz, M. Differences Between Women and Men in Incidence Rates of Dementia and Alzheimer’s Disease. J. Alzheimers Dis. 2018, 64, 1077–1083. [Google Scholar] [CrossRef]
- Corrada, M.M.; Brookmeyer, R.; Paganini-Hill, A.; Berlau, D.; Kawas, C.H. Dementia incidence continues to increase with age in the oldest old: The 90+ study. Ann. Neurol. 2010, 67, 114–121. [Google Scholar] [CrossRef] [Green Version]
- Mouton, A.; Blanc, F.; Gros, A.; Manera, V.; Fabre, R.; Sauleau, E.; Gomez-Luporsi, I.; Tifratene, K.; Friedman, L.; Thummler, S.; et al. Sex ratio in dementia with Lewy bodies balanced between Alzheimer’s disease and Parkinson’s disease dementia: A cross-sectional study. Alzheimers Res. Ther. 2018, 10, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinforiani, E.; Citterio, A.; Zucchella, C.; Bono, G.; Corbetta, S.; Merlo, P.; Mauri, M. Impact of gender differences on the outcome of Alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 2010, 30, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Barnes, L.L.; Wilson, R.S.; Bienias, J.L.; Schneider, J.A.; Evans, D.A.; Bennett, D.A. Sex differences in the clinical manifestations of Alzheimer disease pathology. Arch. Gen. Psychiatry 2005, 62, 685–691. [Google Scholar] [CrossRef] [PubMed]
- Mattsson, N.; Lonneborg, A.; Boccardi, M.; Blennow, K.; Hansson, O.; Geneva Task Force for the Roadmap of Alzheimer’s Biomarkers. Clinical validity of cerebrospinal fluid Abeta42, tau, and phospho-tau as biomarkers for Alzheimer’s disease in the context of a structured 5-phase development framework. Neurobiol. Aging 2017, 52, 196–213. [Google Scholar] [CrossRef]
- Johnson, K.A.; Schultz, A.; Betensky, R.A.; Becker, J.A.; Sepulcre, J.; Rentz, D.; Mormino, E.; Chhatwal, J.; Amariglio, R.; Papp, K.; et al. Tau positron emission tomographic imaging in aging and early Alzheimer disease. Ann. Neurol. 2016, 79, 110–119. [Google Scholar] [CrossRef] [Green Version]
- Jack, C.R., Jr.; Wiste, H.J.; Weigand, S.D.; Therneau, T.M.; Knopman, D.S.; Lowe, V.; Vemuri, P.; Mielke, M.M.; Roberts, R.O.; Machulda, M.M.; et al. Age-specific and sex-specific prevalence of cerebral beta-amyloidosis, tauopathy, and neurodegeneration in cognitively unimpaired individuals aged 50–95 years: A cross-sectional study. Lancet Neurol. 2017, 16, 435–444. [Google Scholar] [CrossRef]
- Buckley, R.F.; Mormino, E.C.; Chhatwal, J.; Schultz, A.P.; Rabin, J.S.; Rentz, D.M.; Acar, D.; Properzi, M.J.; Dumurgier, J.; Jacobs, H.; et al. Associations between baseline amyloid, sex, and APOE on subsequent tau accumulation in cerebrospinal fluid. Neurobiol. Aging 2019, 78, 178–185. [Google Scholar] [CrossRef] [Green Version]
- Knapskog, A.B.; Eldholm, R.S.; Braekhus, A.; Engedal, K.; Saltvedt, I. Factors that influence the levels of cerebrospinal fluid biomarkers in memory clinic patients. BMC Geriatr. 2017, 17, 210. [Google Scholar] [CrossRef] [Green Version]
- Pringsheim, T.; Jette, N.; Frolkis, A.; Steeves, T.D. The prevalence of Parkinson’s disease: A systematic review and meta-analysis. Mov. Disord. 2014, 29, 1583–1590. [Google Scholar] [CrossRef]
- Baldereschi, M.; Di Carlo, A.; Rocca, W.A.; Vanni, P.; Maggi, S.; Perissinotto, E.; Grigoletto, F.; Amaducci, L.; Inzitari, D. Parkinson’s disease and parkinsonism in a longitudinal study: Two-fold higher incidence in men. ILSA Working Group. Italian Longitudinal Study on Aging. Neurology 2000, 55, 1358–1363. [Google Scholar] [CrossRef]
- Swerdlow, R.H.; Parker, W.D.; Currie, L.J.; Bennett, J.P.; Harrison, M.B.; Trugman, J.M.; Wooten, G.F. Gender ratio differences between Parkinson’s disease patients and their affected relatives. Parkinsonism Relat. Disord. 2001, 7, 129–133. [Google Scholar] [CrossRef]
- Van Den Eeden, S.K.; Tanner, C.M.; Bernstein, A.L.; Fross, R.D.; Leimpeter, A.; Bloch, D.A.; Nelson, L.M. Incidence of Parkinson’s disease: Variation by age, gender, and race/ethnicity. Am. J. Epidemiol. 2003, 157, 1015–1022. [Google Scholar] [CrossRef]
- Taylor, K.S.; Cook, J.A.; Counsell, C.E. Heterogeneity in male to female risk for Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 2007, 78, 905–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wooten, G.F.; Currie, L.J.; Bovbjerg, V.E.; Lee, J.K.; Patrie, J. Are men at greater risk for Parkinson’s disease than women? J. Neurol. Neurosurg. Psychiatry 2004, 75, 637–639. [Google Scholar] [CrossRef]
- Meoni, S.; Macerollo, A.; Moro, E. Sex differences in movement disorders. Nat. Rev. Neurol. 2020, 16, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Currie, L.J.; Harrison, M.B.; Trugman, J.M.; Bennett, J.P.; Wooten, G.F. Postmenopausal estrogen use affects risk for Parkinson disease. Arch. Neurol. 2004, 61, 886–888. [Google Scholar] [CrossRef] [PubMed]
- Ragonese, P.; D’Amelio, M.; Callari, G.; Salemi, G.; Morgante, L.; Savettieri, G. Age at menopause predicts age at onset of Parkinson’s disease. Mov. Disord. 2006, 21, 2211–2214. [Google Scholar] [CrossRef]
- Ragonese, P.; D’Amelio, M.; Savettieri, G. Implications for estrogens in Parkinson’s disease: An epidemiological approach. Ann. N. Y. Acad. Sci. 2006, 1089, 373–382. [Google Scholar] [CrossRef]
- Marder, K.; Tang, M.X.; Alfaro, B.; Mejia, H.; Cote, L.; Jacobs, D.; Stern, Y.; Sano, M.; Mayeux, R. Postmenopausal estrogen use and Parkinson’s disease with and without dementia. Neurology 1998, 50, 1141–1143. [Google Scholar] [CrossRef]
- Nicoletti, A.; Nicoletti, G.; Arabia, G.; Annesi, G.; De Mari, M.; Lamberti, P.; Grasso, L.; Marconi, R.; Epifanio, A.; Morgante, L.; et al. Reproductive factors and Parkinson’s disease: A multicenter case-control study. Mov. Disord. 2011, 26, 2563–2566. [Google Scholar] [CrossRef]
- Song, Y.J.; Li, S.R.; Li, X.W.; Chen, X.; Wei, Z.X.; Liu, Q.S.; Cheng, Y. The Effect of Estrogen Replacement Therapy on Alzheimer’s Disease and Parkinson’s Disease in Postmenopausal Women: A Meta-Analysis. Front. Neurosci. 2020, 14, 157. [Google Scholar] [CrossRef]
- Vann Jones, S.A.; O’Brien, J.T. The prevalence and incidence of dementia with Lewy bodies: A systematic review of population and clinical studies. Psychol. Med. 2014, 44, 673–683. [Google Scholar] [CrossRef]
- Fereshtehnejad, S.M.; Religa, D.; Westman, E.; Aarsland, D.; Lokk, J.; Eriksdotter, M. Demography, diagnostics, and medication in dementia with Lewy bodies and Parkinson’s disease with dementia: Data from the Swedish Dementia Quality Registry (SveDem). Neuropsychiatr. Dis. Treat. 2013, 9, 927–935. [Google Scholar] [CrossRef] [Green Version]
- Yue, W.; Wang, X.D.; Shi, Z.; Wang, Y.; Liu, S.; Liu, S.; Zhang, Y.; Zhang, Y.; Lu, H.; Su, W.; et al. The prevalence of dementia with Lewy bodies in a rural area of China. Parkinsonism Relat. Disord. 2016, 29, 72–77. [Google Scholar] [CrossRef]
- Goodman, R.A.; Lochner, K.A.; Thambisetty, M.; Wingo, T.S.; Posner, S.F.; Ling, S.M. Prevalence of dementia subtypes in United States Medicare fee-for-service beneficiaries, 2011–2013. Alzheimers Dement. 2017, 13, 28–37. [Google Scholar] [CrossRef] [Green Version]
- Savica, R.; Grossardt, B.R.; Bower, J.H.; Boeve, B.F.; Ahlskog, J.E.; Rocca, W.A. Incidence of dementia with Lewy bodies and Parkinson disease dementia. JAMA Neurol. 2013, 70, 1396–1402. [Google Scholar] [CrossRef]
- Breitve, M.H.; Hynninen, M.J.; Bronnick, K.; Chwiszczuk, L.J.; Auestad, B.H.; Aarsland, D.; Rongve, A. A longitudinal study of anxiety and cognitive decline in dementia with Lewy bodies and Alzheimer’s disease. Alzheimers Res. Ther. 2016, 8, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farina, E.; Baglio, F.; Caffarra, P.; Magnani, G.; Scarpini, E.; Appollonio, I.; Bascelli, C.; Cheldi, A.; Nemni, R.; Franceschi, M.; et al. Frequency and clinical features of Lewy body dementia in Italian memory clinics. Acta Biomed. 2009, 80, 57–64. [Google Scholar] [PubMed]
- Van de Beek, M.; Babapour Mofrad, R.; van Steenoven, I.; Vanderstichele, H.; Scheltens, P.; Teunissen, C.E.; Lemstra, A.W.; van der Flier, W.M. Sex-specific associations with cerebrospinal fluid biomarkers in dementia with Lewy bodies. Alzheimers Res. Ther. 2020, 12, 44. [Google Scholar] [CrossRef] [PubMed]
- Hestiantoro, A.; Wiwie, M.; Shadrina, A.; Ibrahim, N.; Purba, J.S. FSH to estradiol ratio can be used as screening method for mild cognitive impairment in postmenopausal women. Climacteric 2017, 20, 577–582. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.; Gao, G.; Huang, W. A study on co-localization of FSH and its receptor in rat hippocampus. J. Mol. Histol. 2008, 39, 49–55. [Google Scholar] [CrossRef]
- Kandasamy, M.; Radhakrishnan, R.K.; Poornimai Abirami, G.P.; Roshan, S.A.; Yesudhas, A.; Balamuthu, K.; Prahalathan, C.; Shanmugaapriya, S.; Moorthy, A.; Essa, M.M.; et al. Possible Existence of the Hypothalamic-Pituitary-Hippocampal (HPH) Axis: A Reciprocal Relationship Between Hippocampal Specific Neuroestradiol Synthesis and Neuroblastosis in Ageing Brains with Special Reference to Menopause and Neurocognitive Disorders. Neurochem. Res. 2019, 44, 1781–1795. [Google Scholar] [CrossRef] [PubMed]
- Grimm, A.; Schmitt, K.; Lang, U.E.; Mensah-Nyagan, A.G.; Eckert, A. Improvement of neuronal bioenergetics by neurosteroids: Implications for age-related neurodegenerative disorders. Biochim. Biophys. Acta 2014, 1842, 2427–2438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendell, A.L.; MacLusky, N.J. Neurosteroid Metabolites of Gonadal Steroid Hormones in Neuroprotection: Implications for Sex Differences in Neurodegenerative Disease. Front. Mol. Neurosci. 2018, 11, 359. [Google Scholar] [CrossRef] [Green Version]
- Santoro, N. The menopausal transition. Am. J. Med. 2005, 118 (Suppl. 12B), 8–13. [Google Scholar] [CrossRef] [PubMed]
- Santoro, N.; Lasley, B.; McConnell, D.; Allsworth, J.; Crawford, S.; Gold, E.B.; Finkelstein, J.S.; Greendale, G.A.; Kelsey, J.; Korenman, S.; et al. Body size and ethnicity are associated with menstrual cycle alterations in women in the early menopausal transition: The Study of Women’s Health across the Nation (SWAN) Daily Hormone Study. J. Clin. Endocrinol. Metab. 2004, 89, 2622–2631. [Google Scholar] [CrossRef] [Green Version]
- Guennoun, R. Progesterone in the Brain: Hormone, Neurosteroid and Neuroprotectant. Int. J. Mol. Sci. 2020, 21, 5271. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.M.; Zhao, H.Y.; Ning, G.; Chen, Y.; Zhang, L.Z.; Sun, L.H.; Zhao, Y.J.; Xu, M.Y.; Chen, J.L. IGF-1 as an early marker for low bone mass or osteoporosis in premenopausal and postmenopausal women. J. Bone Miner. Metab. 2008, 26, 159–164. [Google Scholar] [CrossRef]
- Nasu, M.; Sugimoto, T.; Chihara, M.; Hiraumi, M.; Kurimoto, F.; Chihara, K. Effect of natural menopause on serum levels of IGF-I and IGF-binding proteins: Relationship with bone mineral density and lipid metabolism in perimenopausal women. Eur. J. Endocrinol. 1997, 136, 608–616. [Google Scholar] [CrossRef] [PubMed]
- Cao, P.; Maximov, A.; Sudhof, T.C. Activity-dependent IGF-1 exocytosis is controlled by the Ca(2+)-sensor synaptotagmin-10. Cell 2011, 145, 300–311. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Sareddy, G.R.; Wang, J.; Zhang, Q.; Tang, F.L.; Pratap, U.P.; Tekmal, R.R.; Vadlamudi, R.K.; Brann, D.W. Neuron-Derived Estrogen Is Critical for Astrocyte Activation and Neuroprotection of the Ischemic Brain. J. Neurosci. 2020, 40, 7355–7374. [Google Scholar] [CrossRef] [PubMed]
- Kuo, J.; Hamid, N.; Bondar, G.; Prossnitz, E.R.; Micevych, P. Membrane estrogen receptors stimulate intracellular calcium release and progesterone synthesis in hypothalamic astrocytes. J. Neurosci. 2010, 30, 12950–12957. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, Y.; Hojo, Y.; Kojima, H.; Ikeda, M.; Hotta, K.; Sato, R.; Ooishi, Y.; Yoshiya, M.; Chung, B.C.; Yamazaki, T.; et al. Estradiol rapidly modulates synaptic plasticity of hippocampal neurons: Involvement of kinase networks. Brain Res. 2015, 1621, 147–161. [Google Scholar] [CrossRef]
- Spencer-Segal, J.L.; Tsuda, M.C.; Mattei, L.; Waters, E.M.; Romeo, R.D.; Milner, T.A.; McEwen, B.S.; Ogawa, S. Estradiol acts via estrogen receptors alpha and beta on pathways important for synaptic plasticity in the mouse hippocampal formation. Neuroscience 2012, 202, 131–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phan, A.; Lancaster, K.E.; Armstrong, J.N.; MacLusky, N.J.; Choleris, E. Rapid effects of estrogen receptor alpha and beta selective agonists on learning and dendritic spines in female mice. Endocrinology 2011, 152, 1492–1502. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Day, M.; Muniz, L.C.; Bitran, D.; Arias, R.; Revilla-Sanchez, R.; Grauer, S.; Zhang, G.; Kelley, C.; Pulito, V.; et al. Activation of estrogen receptor-beta regulates hippocampal synaptic plasticity and improves memory. Nat. Neurosci. 2008, 11, 334–343. [Google Scholar] [CrossRef]
- Boulware, M.I.; Heisler, J.D.; Frick, K.M. The memory-enhancing effects of hippocampal estrogen receptor activation involve metabotropic glutamate receptor signaling. J. Neurosci. 2013, 33, 15184–15194. [Google Scholar] [CrossRef]
- Wang, W.; Le, A.A.; Hou, B.; Lauterborn, J.C.; Cox, C.D.; Levin, E.R.; Lynch, G.; Gall, C.M. Memory-Related Synaptic Plasticity Is Sexually Dimorphic in Rodent Hippocampus. J. Neurosci. 2018, 38, 7935–7951. [Google Scholar] [CrossRef] [Green Version]
- Briz, V.; Liu, Y.; Zhu, G.; Bi, X.; Baudry, M. A novel form of synaptic plasticity in field CA3 of hippocampus requires GPER1 activation and BDNF release. J. Cell Biol. 2015, 210, 1225–1237. [Google Scholar] [CrossRef]
- Khadilkar, S.V.; Patil, V.A. Sex Hormones and Cognition: Where Do We Stand? J. Obstet. Gynaecol. India 2019, 69, 303–312. [Google Scholar] [CrossRef]
- Jacobs, E.G.; Weiss, B.K.; Makris, N.; Whitfield-Gabrieli, S.; Buka, S.L.; Klibanski, A.; Goldstein, J.M. Impact of Sex and Menopausal Status on Episodic Memory Circuitry in Early Midlife. J. Neurosci. 2016, 36, 10163–10173. [Google Scholar] [CrossRef] [Green Version]
- Rentz, D.M.; Weiss, B.K.; Jacobs, E.G.; Cherkerzian, S.; Klibanski, A.; Remington, A.; Aizley, H.; Goldstein, J.M. Sex differences in episodic memory in early midlife: Impact of reproductive aging. Menopause 2017, 24, 400–408. [Google Scholar] [CrossRef] [Green Version]
- Gilsanz, P.; Lee, C.; Corrada, M.M.; Kawas, C.H.; Quesenberry, C.P., Jr.; Whitmer, R.A. Reproductive period and risk of dementia in a diverse cohort of health care members. Neurology 2019, 92, e2005–e2014. [Google Scholar] [CrossRef] [PubMed]
- McRoberts, J.A.; Li, J.; Ennes, H.S.; Mayer, E.A. Sex-dependent differences in the activity and modulation of N-methyl-d-aspartic acid receptors in rat dorsal root ganglia neurons. Neuroscience 2007, 148, 1015–1020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, C.C.; McMahon, L.L. Estradiol-induced increase in the magnitude of long-term potentiation is prevented by blocking NR2B-containing receptors. J. Neurosci. 2006, 26, 8517–8522. [Google Scholar] [CrossRef] [Green Version]
- Kramar, E.A.; Chen, L.Y.; Brandon, N.J.; Rex, C.S.; Liu, F.; Gall, C.M.; Lynch, G. Cytoskeletal changes underlie estrogen’s acute effects on synaptic transmission and plasticity. J. Neurosci. 2009, 29, 12982–12993. [Google Scholar] [CrossRef] [Green Version]
- Koss, W.A.; Haertel, J.M.; Philippi, S.M.; Frick, K.M. Sex Differences in the Rapid Cell Signaling Mechanisms Underlying the Memory-Enhancing Effects of 17beta-Estradiol. eNeuro 2018, 5. [Google Scholar] [CrossRef] [Green Version]
- Nicholson, K.; MacLusky, N.J.; Leranth, C. Synaptic effects of estrogen. Vitam. Horm. 2020, 114, 167–210. [Google Scholar] [PubMed]
- Imtiaz, B.; Tuppurainen, M.; Rikkonen, T.; Kivipelto, M.; Soininen, H.; Kroger, H.; Tolppanen, A.M. Postmenopausal hormone therapy and Alzheimer disease: A prospective cohort study. Neurology 2017, 88, 1062–1068. [Google Scholar] [CrossRef] [Green Version]
- Waring, S.C.; Rocca, W.A.; Petersen, R.C.; O’Brien, P.C.; Tangalos, E.G.; Kokmen, E. Postmenopausal estrogen replacement therapy and risk of AD: A population-based study. Neurology 1999, 52, 965–970. [Google Scholar] [CrossRef] [Green Version]
- Manson, J.E.; Aragaki, A.K.; Rossouw, J.E.; Anderson, G.L.; Prentice, R.L.; LaCroix, A.Z.; Chlebowski, R.T.; Howard, B.V.; Thomson, C.A.; Margolis, K.L.; et al. Menopausal Hormone Therapy and Long-term All-Cause and Cause-Specific Mortality: The Women’s Health Initiative Randomized Trials. JAMA 2017, 318, 927–938. [Google Scholar] [CrossRef]
- Marongiu, R. Accelerated Ovarian Failure as a Unique Model to Study Peri-Menopause Influence on Alzheimer’s Disease. Front. Aging Neurosci. 2019, 11, 242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.J.; Soto, M.; Branigan, G.L.; Rodgers, K.; Brinton, R.D. Association between menopausal hormone therapy and risk of neurodegenerative diseases: Implications for precision hormone therapy. Alzheimers Dement. 2021, 7, e12174. [Google Scholar]
- Veenman, L. Raloxifene as Treatment for Various Types of Brain Injuries and Neurodegenerative Diseases: A Good Start. Int. J. Mol. Sci. 2020, 21, 7586. [Google Scholar] [CrossRef] [PubMed]
- Blanquart, E.; Laffont, S.; Guery, J.C. Sex hormone regulation of innate lymphoid cells. Biomed. J. 2021, 44, 144–156. [Google Scholar] [CrossRef] [PubMed]
- Amjadi, F.; Salehi, E.; Mehdizadeh, M.; Aflatoonian, R. Role of the innate immunity in female reproductive tract. Adv. Biomed. Res. 2014, 3, 1. [Google Scholar]
- Medina-Estrada, I.; Alva-Murillo, N.; Lopez-Meza, J.E.; Ochoa-Zarzosa, A. Immunomodulatory Effects of 17beta-Estradiol on Epithelial Cells during Bacterial Infections. J. Immunol. Res. 2018, 2018, 6098961. [Google Scholar] [CrossRef]
- Li, S.; Herrera, G.G.; Tam, K.K.; Lizarraga, J.S.; Beedle, M.T.; Winuthayanon, W. Estrogen Action in the Epithelial Cells of the Mouse Vagina Regulates Neutrophil Infiltration and Vaginal Tissue Integrity. Sci. Rep. 2018, 8, 11247. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.Y.; Su, T.H.; Lau, H.H. Estrogen for the prevention of recurrent urinary tract infections in postmenopausal women: A meta-analysis of randomized controlled trials. Int. Urogynecol. J. 2021, 32, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Stanton, A.; Mowbray, C.; Lanz, M.; Brown, K.; Hilton, P.; Tyson-Capper, A.; Pickard, R.S.; Ali, A.S.M.; Hall, J. Topical Estrogen Treatment Augments the Vaginal Response to Escherichia coli Flagellin. Sci. Rep. 2020, 10, 8473. [Google Scholar] [CrossRef]
- Abramenko, N.; Vellieux, F.; Tesarova, P.; Kejik, Z.; Kaplanek, R.; Lacina, L.; Dvorankova, B.; Rosel, D.; Brabek, J.; Tesar, A.; et al. Estrogen Receptor Modulators in Viral Infections Such as SARS-CoV-2: Therapeutic Consequences. Int. J. Mol. Sci. 2021, 22, 6551. [Google Scholar] [CrossRef]
- Yang, X.; Guo, Y.; He, J.; Zhang, F.; Sun, X.; Yang, S.; Dong, H. Estrogen and estrogen receptors in the modulation of gastrointestinal epithelial secretion. Oncotarget 2017, 8, 97683–97692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Condliffe, S.B.; Doolan, C.M.; Harvey, B.J. 17beta-oestradiol acutely regulates Cl- secretion in rat distal colonic epithelium. J. Physiol 2001, 530, 47–54. [Google Scholar] [CrossRef]
- Alzamora, R.; O’Mahony, F.; Harvey, B.J. Estrogen inhibits chloride secretion caused by cholera and Escherichia coli enterotoxins in female rat distal colon. Steroids 2011, 76, 867–876. [Google Scholar] [CrossRef] [PubMed]
- Sampson, T.R.; Mazmanian, S.K. Control of brain development, function, and behavior by the microbiome. Cell Host Microbe 2015, 17, 565–576. [Google Scholar] [CrossRef] [Green Version]
- Callan, N.G.L.; Mitchell, E.S.; Heitkemper, M.M.; Woods, N.F. Abdominal pain during the menopause transition and early postmenopause: Observations from the Seattle Midlife Women’s Health Study. Womens Midlife Health 2019, 5, 2. [Google Scholar] [CrossRef]
- Flores, R.; Shi, J.; Fuhrman, B.; Xu, X.; Veenstra, T.D.; Gail, M.H.; Gajer, P.; Ravel, J.; Goedert, J.J. Fecal microbial determinants of fecal and systemic estrogens and estrogen metabolites: A cross-sectional study. J. Transl. Med. 2012, 10, 253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakatsu, C.H.; Armstrong, A.; Clavijo, A.P.; Martin, B.R.; Barnes, S.; Weaver, C.M. Fecal bacterial community changes associated with isoflavone metabolites in postmenopausal women after soy bar consumption. PLoS ONE 2014, 9, e108924. [Google Scholar] [CrossRef]
- Sau, L.; Olmstead, C.M.; Cui, L.J.; Chen, A.; Shah, R.S.; Kelley, S.T.; Thackray, V.G. Alterations in Gut Microbiota Do Not Play a Causal Role in Diet-independent Weight Gain Caused by Ovariectomy. J. Endocr. Soc. 2021, 5, bvaa173. [Google Scholar] [CrossRef] [PubMed]
- Lei, Z.; Wu, H.; Yang, Y.; Hu, Q.; Lei, Y.; Liu, W.; Nie, Y.; Yang, L.; Zhang, X.; Yang, C.; et al. Ovariectomy Impaired Hepatic Glucose and Lipid Homeostasis and Altered the Gut Microbiota in Mice With Different Diets. Front. Endocrinol. 2021, 12, 708838. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Wang, B.; Wang, S.; Qian, X.; Li, X.; Zhao, J.; Zhang, H.; Chen, W.; Wang, G. Modulation of the Gut Microbiota Structure with Probiotics and Isoflavone Alleviates Metabolic Disorder in Ovariectomized Mice. Nutrients 2021, 13, 1793. [Google Scholar] [CrossRef]
- Chen, K.L.A.; Liu, X.; Zhao, Y.C.; Hieronymi, K.; Rossi, G.; Auvil, L.S.; Welge, M.; Bushell, C.; Smith, R.L.; Carlson, K.E.; et al. Long-Term Administration of Conjugated Estrogen and Bazedoxifene Decreased Murine Fecal beta-Glucuronidase Activity Without Impacting Overall Microbiome Community. Sci. Rep. 2018, 8, 8166. [Google Scholar] [CrossRef] [Green Version]
- Urban, A.S.; Pavlov, K.V.; Kamynina, A.V.; Okhrimenko, I.S.; Arseniev, A.S.; Bocharov, E.V. Structural Studies Providing Insights into Production and Conformational Behavior of Amyloid-beta Peptide Associated with Alzheimer’s Disease Development. Molecules 2021, 26, 2897. [Google Scholar] [CrossRef]
- Guyon, A.; Rousseau, J.; Lamothe, G.; Tremblay, J.P. The protective mutation A673T in amyloid precursor protein gene decreases Abeta peptides production for 14 forms of Familial Alzheimer’s Disease in SH-SY5Y cells. PLoS ONE 2020, 15, e0237122. [Google Scholar] [CrossRef]
- Tackenberg, C.; Nitsch, R.M. The secreted APP ectodomain sAPPalpha, but not sAPPbeta, protects neurons against Abeta oligomer-induced dendritic spine loss and increased tau phosphorylation. Mol. Brain 2019, 12, 27. [Google Scholar] [CrossRef] [Green Version]
- Manthey, D.; Heck, S.; Engert, S.; Behl, C. Estrogen induces a rapid secretion of amyloid beta precursor protein via the mitogen-activated protein kinase pathway. Eur. J. Biochem. 2001, 268, 4285–4291. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Huang, Y.; Zhu, Y.C.; Yao, T. Estrogen stimulates release of secreted amyloid precursor protein from primary rat cortical neurons via protein kinase C pathway. Acta Pharmacol. Sin. 2005, 26, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Sochocka, M.; Zwolinska, K.; Leszek, J. The Infectious Etiology of Alzheimer’s Disease. Curr. Neuropharmacol. 2017, 15, 996–1009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, A.F.; Morris, J.A.; Wray, M. Pathogenesis of Alzheimer’s disease: Multiple interacting causes against which amyloid precursor protein protects. Med. Hypotheses 2020, 143, 110035. [Google Scholar] [CrossRef] [PubMed]
- Felberbaum, R.; Kupker, W. COVID-19 from the perspective of a gynecological endocrinologist. Gynakol. Endokrinol. 2021, 1–4. [Google Scholar] [CrossRef]
- Tschiffely, A.E.; Schuh, R.A.; Prokai-Tatrai, K.; Ottinger, M.A.; Prokai, L. An exploratory investigation of brain-selective estrogen treatment in males using a mouse model of Alzheimer’s disease. Horm. Behav. 2018, 98, 16–21. [Google Scholar] [CrossRef]
- Tschiffely, A.E.; Schuh, R.A.; Prokai-Tatrai, K.; Prokai, L.; Ottinger, M.A. A comparative evaluation of treatments with 17beta-estradiol and its brain-selective prodrug in a double-transgenic mouse model of Alzheimer’s disease. Horm. Behav. 2016, 83, 39–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenfield, J.P.; Leung, L.W.; Cai, D.; Kaasik, K.; Gross, R.S.; Rodriguez-Boulan, E.; Greengard, P.; Xu, H. Estrogen lowers Alzheimer beta-amyloid generation by stimulating trans-Golgi network vesicle biogenesis. J. Biol. Chem 2002, 277, 12128–12136. [Google Scholar] [CrossRef] [Green Version]
- Jayaraman, A.; Carroll, J.C.; Morgan, T.E.; Lin, S.; Zhao, L.; Arimoto, J.M.; Murphy, M.P.; Beckett, T.L.; Finch, C.E.; Brinton, R.D.; et al. 17beta-estradiol and progesterone regulate expression of beta-amyloid clearance factors in primary neuron cultures and female rat brain. Endocrinology 2012, 153, 5467–5479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, L.; Yao, J.; Mao, Z.; Chen, S.; Wang, Y.; Brinton, R.D. 17beta-Estradiol regulates insulin-degrading enzyme expression via an ERbeta/PI3-K pathway in hippocampus: Relevance to Alzheimer’s prevention. Neurobiol. Aging 2011, 32, 1949–1963. [Google Scholar] [CrossRef] [Green Version]
- Merlo, S.; Sortino, M.A. Estrogen activates matrix metalloproteinases-2 and -9 to increase beta amyloid degradation. Mol. Cell Neurosci. 2012, 49, 423–429. [Google Scholar] [CrossRef]
- Liang, K.; Yang, L.; Yin, C.; Xiao, Z.; Zhang, J.; Liu, Y.; Huang, J. Estrogen stimulates degradation of beta-amyloid peptide by up-regulating neprilysin. J. Biol. Chem. 2010, 285, 935–942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mputhia, Z.; Hone, E.; Tripathi, T.; Sargeant, T.; Martins, R.; Bharadwaj, P. Autophagy Modulation as a Treatment of Amyloid Diseases. Molecules 2019, 24, 3372. [Google Scholar] [CrossRef] [Green Version]
- Pike, C.J. Estrogen modulates neuronal Bcl-xL expression and beta-amyloid-induced apoptosis: Relevance to Alzheimer’s disease. J. Neurochem. 1999, 72, 1552–1563. [Google Scholar] [CrossRef]
- Zhang, Y.; Champagne, N.; Beitel, L.K.; Goodyer, C.G.; Trifiro, M.; LeBlanc, A. Estrogen and androgen protection of human neurons against intracellular amyloid beta1-42 toxicity through heat shock protein 70. J. Neurosci. 2004, 24, 5315–5321. [Google Scholar] [CrossRef] [PubMed]
- Nilsen, J.; Chen, S.; Irwin, R.W.; Iwamoto, S.; Brinton, R.D. Estrogen protects neuronal cells from amyloid beta-induced apoptosis via regulation of mitochondrial proteins and function. BMC Neurosci. 2006, 7, 74. [Google Scholar] [CrossRef] [Green Version]
- Marin, R.; Guerra, B.; Hernandez-Jimenez, J.G.; Kang, X.L.; Fraser, J.D.; Lopez, F.J.; Alonso, R. Estradiol prevents amyloid-beta peptide-induced cell death in a cholinergic cell line via modulation of a classical estrogen receptor. Neuroscience 2003, 121, 917–926. [Google Scholar] [CrossRef]
- Kwakowsky, A.; Potapov, K.; Kim, S.; Peppercorn, K.; Tate, W.P.; Abraham, I.M. Treatment of beta amyloid 1-42 (Abeta(1-42))-induced basal forebrain cholinergic damage by a non-classical estrogen signaling activator in vivo. Sci. Rep. 2016, 6, 21101. [Google Scholar] [CrossRef] [Green Version]
- Lai, Y.J.; Zhu, B.L.; Sun, F.; Luo, D.; Ma, Y.L.; Luo, B.; Tang, J.; Xiong, M.J.; Liu, L.; Long, Y.; et al. Estrogen receptor alpha promotes Cav1.2 ubiquitination and degradation in neuronal cells and in APP/PS1 mice. Aging Cell 2019, 18, e12961. [Google Scholar] [CrossRef]
- Ramirez-Barrantes, R.; Carvajal-Zamorano, K.; Rodriguez, B.; Cordova, C.; Lozano, C.; Simon, F.; Diaz, P.; Munoz, P.; Marchant, I.; Latorre, R.; et al. TRPV1-Estradiol Stereospecific Relationship Underlies Cell Survival in Oxidative Cell Death. Front. Physiol. 2020, 11, 444. [Google Scholar] [CrossRef]
- Pan, Q.; Guo, K.; Xue, M.; Tu, Q. Estrogen protects neuroblastoma cell from amyloid-beta 42 (Abeta42)-induced apoptosis via TXNIP/TRX axis and AMPK signaling. Neurochem. Int. 2020, 135, 104685. [Google Scholar] [CrossRef] [PubMed]
- Tsubaki, H.; Tooyama, I.; Walker, D.G. Thioredoxin-Interacting Protein (TXNIP) with Focus on Brain and Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 9357. [Google Scholar] [CrossRef] [PubMed]
- Manoharan, S.; Guillemin, G.J.; Abiramasundari, R.S.; Essa, M.M.; Akbar, M.; Akbar, M.D. The Role of Reactive Oxygen Species in the Pathogenesis of Alzheimer’s Disease, Parkinson’s Disease, and Huntington’s Disease: A Mini Review. Oxid. Med. Cell Longev. 2016, 2016, 8590578. [Google Scholar] [CrossRef] [PubMed]
- Lejri, I.; Agapouda, A.; Grimm, A.; Eckert, A. Mitochondria- and Oxidative Stress-Targeting Substances in Cognitive Decline-Related Disorders: From Molecular Mechanisms to Clinical Evidence. Oxid. Med. Cell Longev. 2019, 2019, 9695412. [Google Scholar] [CrossRef] [Green Version]
- Massaad, C.A. Neuronal and vascular oxidative stress in Alzheimer’s disease. Curr. Neuropharmacol. 2011, 9, 662–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cervellati, C.; Wood, P.L.; Romani, A.; Valacchi, G.; Squerzanti, M.; Sanz, J.M.; Ortolani, B.; Zuliani, G. Oxidative challenge in Alzheimer’s disease: State of knowledge and future needs. J. Investig. Med. 2016, 64, 21–32. [Google Scholar] [CrossRef]
- Abramov, A.Y.; Canevari, L.; Duchen, M.R. Beta-amyloid peptides induce mitochondrial dysfunction and oxidative stress in astrocytes and death of neurons through activation of NADPH oxidase. J. Neurosci. 2004, 24, 565–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wojsiat, J.; Zoltowska, K.M.; Laskowska-Kaszub, K.; Wojda, U. Oxidant/Antioxidant Imbalance in Alzheimer’s Disease: Therapeutic and Diagnostic Prospects. Oxid. Med. Cell Longev. 2018, 2018, 6435861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.Y.; Andoh, T.; Murphy, D.L.; Chiueh, C.C. 17beta-estradiol activates ICI 182,780-sensitive estrogen receptors and cyclic GMP-dependent thioredoxin expression for neuroprotection. FASEB J. 2003, 17, 947–948. [Google Scholar] [CrossRef]
- Ayres, S.; Abplanalp, W.; Liu, J.H.; Subbiah, M.T. Mechanisms involved in the protective effect of estradiol-17beta on lipid peroxidation and DNA damage. Am. J. Physiol. 1998, 274, E1002–E1008. [Google Scholar]
- Behl, C.; Skutella, T.; Lezoualc’h, F.; Post, A.; Widmann, M.; Newton, C.J.; Holsboer, F. Neuroprotection against oxidative stress by estrogens: Structure-activity relationship. Mol. Pharmacol. 1997, 51, 535–541. [Google Scholar] [CrossRef]
- Gridley, K.E.; Green, P.S.; Simpkins, J.W. Low concentrations of estradiol reduce beta-amyloid (25-35)-induced toxicity, lipid peroxidation and glucose utilization in human SK-N-SH neuroblastoma cells. Brain Res. 1997, 778, 158–165. [Google Scholar] [CrossRef]
- Celsi, F.; Ferri, A.; Casciati, A.; D’Ambrosi, N.; Rotilio, G.; Costa, A.; Volonte, C.; Carri, M.T. Overexpression of superoxide dismutase 1 protects against beta-amyloid peptide toxicity: Effect of estrogen and copper chelators. Neurochem. Int. 2004, 44, 25–33. [Google Scholar] [CrossRef]
- Pena-Bautista, C.; Baquero, M.; Vento, M.; Chafer-Pericas, C. Free radicals in Alzheimer’s disease: Lipid peroxidation biomarkers. Clin. Chim. Acta 2019, 491, 85–90. [Google Scholar] [CrossRef]
- Chang, Y.T.; Chang, W.N.; Tsai, N.W.; Huang, C.C.; Kung, C.T.; Su, Y.J.; Lin, W.C.; Cheng, B.C.; Su, C.M.; Chiang, Y.F.; et al. The roles of biomarkers of oxidative stress and antioxidant in Alzheimer’s disease: A systematic review. Biomed. Res. Int. 2014, 2014, 182303. [Google Scholar] [CrossRef]
- Schonfeld, P.; Reiser, G. How the brain fights fatty acids’ toxicity. Neurochem. Int. 2021, 148, 105050. [Google Scholar] [CrossRef] [PubMed]
- Dani, M.; Wood, M.; Mizoguchi, R.; Fan, Z.; Walker, Z.; Morgan, R.; Hinz, R.; Biju, M.; Kuruvilla, T.; Brooks, D.J.; et al. Microglial activation correlates in vivo with both tau and amyloid in Alzheimer’s disease. Brain 2018, 141, 2740–2754. [Google Scholar] [CrossRef]
- Fan, Z.; Brooks, D.J.; Okello, A.; Edison, P. An early and late peak in microglial activation in Alzheimer’s disease trajectory. Brain 2017, 140, 792–803. [Google Scholar] [CrossRef] [PubMed]
- Varnum, M.M.; Ikezu, T. The classification of microglial activation phenotypes on neurodegeneration and regeneration in Alzheimer’s disease brain. Arch. Immunol. Ther. Exp. 2012, 60, 251–266. [Google Scholar] [CrossRef]
- Sierra, A.; Gottfried-Blackmore, A.; Milner, T.A.; McEwen, B.S.; Bulloch, K. Steroid hormone receptor expression and function in microglia. Glia 2008, 56, 659–674. [Google Scholar] [CrossRef]
- Ghisletti, S.; Meda, C.; Maggi, A.; Vegeto, E. 17beta-estradiol inhibits inflammatory gene expression by controlling NF-kappaB intracellular localization. Mol. Cell Biol. 2005, 25, 2957–2968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruce-Keller, A.J.; Keeling, J.L.; Keller, J.N.; Huang, F.F.; Camondola, S.; Mattson, M.P. Antiinflammatory effects of estrogen on microglial activation. Endocrinology 2000, 141, 3646–3656. [Google Scholar] [CrossRef]
- Zhao, T.Z.; Ding, Q.; Hu, J.; He, S.M.; Shi, F.; Ma, L.T. GPER expressed on microglia mediates the anti-inflammatory effect of estradiol in ischemic stroke. Brain Behav. 2016, 6, e00449. [Google Scholar] [CrossRef] [Green Version]
- Shindo, S.; Chen, S.H.; Gotoh, S.; Yokobori, K.; Hu, H.; Ray, M.; Moore, R.; Nagata, K.; Martinez, J.; Hong, J.S.; et al. Estrogen receptor alpha phosphorylated at Ser216 confers inflammatory function to mouse microglia. Cell Commun. Signal. 2020, 18, 117. [Google Scholar] [CrossRef]
- Li, K.X.; Sun, Q.; Wei, L.L.; Du, G.H.; Huang, X.; Wang, J.K. ERalpha Gene Promoter Methylation in Cognitive Function and Quality of Life of Patients With Alzheimer Disease. J. Geriatr. Psychiatry Neurol. 2019, 32, 221–228. [Google Scholar] [CrossRef]
- Gamache, J.; Yun, Y.; Chiba-Falek, O. Sex-dependent effect of APOE on Alzheimer’s disease and other age-related neurodegenerative disorders. Dis. Model. Mech. 2020, 13, dmm045211. [Google Scholar] [CrossRef]
- Ratnakumar, A.; Zimmerman, S.E.; Jordan, B.A.; Mar, J.C. Estrogen activates Alzheimer’s disease genes. Alzheimers Dement. 2019, 5, 906–917. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.M.; Irwin, R.W.; Brinton, R.D. Activation of estrogen receptor alpha increases and estrogen receptor beta decreases apolipoprotein E expression in hippocampus in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2006, 103, 16983–16988. [Google Scholar] [CrossRef] [Green Version]
- Reiman, E.M.; Arboleda-Velasquez, J.F.; Quiroz, Y.T.; Huentelman, M.J.; Beach, T.G.; Caselli, R.J.; Chen, Y.; Su, Y.; Myers, A.J.; Hardy, J.; et al. Exceptionally low likelihood of Alzheimer’s dementia in APOE2 homozygotes from a 5000-person neuropathological study. Nat. Commun. 2020, 11, 667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nathan, B.P.; Barsukova, A.G.; Shen, F.; McAsey, M.; Struble, R.G. Estrogen facilitates neurite extension via apolipoprotein E in cultured adult mouse cortical neurons. Endocrinology 2004, 145, 3065–3073. [Google Scholar] [CrossRef] [Green Version]
- Zaretsky, D.V.; Zaretskaia, M.V. Mini-review: Amyloid degradation toxicity hypothesis of Alzheimer’s disease. Neurosci. Lett. 2021, 756, 135959. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; Nahon-Crystal, E.; Shteinfer-Kuzmine, A.; Gupta, R. VDAC1, mitochondrial dysfunction, and Alzheimer’s disease. Pharmacol. Res. 2018, 131, 87–101. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, C.M.; Gonzalez, M.; Diaz, M.; Alonso, R.; Ferrer, I.; Santpere, G.; Puig, B.; Meyer, G.; Marin, R. VDAC and ERalpha interaction in caveolae from human cortex is altered in Alzheimer’s disease. Mol. Cell Neurosci. 2009, 42, 172–183. [Google Scholar] [CrossRef]
- Herrera, J.L.; Diaz, M.; Hernandez-Fernaud, J.R.; Salido, E.; Alonso, R.; Fernandez, C.; Morales, A.; Marin, R. Voltage-dependent anion channel as a resident protein of lipid rafts: Post-transductional regulation by estrogens and involvement in neuronal preservation against Alzheimer’s disease. J. Neurochem. 2011, 116, 820–827. [Google Scholar] [CrossRef]
- Long, J.; He, P.; Shen, Y.; Li, R. New evidence of mitochondria dysfunction in the female Alzheimer’s disease brain: Deficiency of estrogen receptor-beta. J. Alzheimers Dis. 2012, 30, 545–558. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Hou, Y.; Song, X.; Wang, L.; Zhang, F.; Zhang, H.; Yu, H.; Zhou, Y. Estrogen Deficiency Induces Mitochondrial Damage Prior to Emergence of Cognitive Deficits in a Postmenopausal Mouse Model. Front. Aging Neurosci. 2021, 13, 713819. [Google Scholar] [CrossRef]
- Mosconi, L.; Berti, V.; Quinn, C.; McHugh, P.; Petrongolo, G.; Osorio, R.S.; Connaughty, C.; Pupi, A.; Vallabhajosula, S.; Isaacson, R.S.; et al. Perimenopause and emergence of an Alzheimer’s bioenergetic phenotype in brain and periphery. PLoS ONE 2017, 12, e0185926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosconi, L.; Berti, V.; Dyke, J.; Schelbaum, E.; Jett, S.; Loughlin, L.; Jang, G.; Rahman, A.; Hristov, H.; Pahlajani, S.; et al. Menopause impacts human brain structure, connectivity, energy metabolism, and amyloid-beta deposition. Sci. Rep. 2021, 11, 10867. [Google Scholar] [CrossRef] [PubMed]
- Ragonese, P.; D’Amelio, M.; Salemi, G.; Aridon, P.; Gammino, M.; Epifanio, A.; Morgante, L.; Savettieri, G. Risk of Parkinson disease in women: Effect of reproductive characteristics. Neurology 2004, 62, 2010–2014. [Google Scholar] [CrossRef]
- Walf, A.A.; Koonce, C.J.; Frye, C.A. Progestogens’ effects and mechanisms for object recognition memory across the lifespan. Behav. Brain Res. 2015, 294, 50–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almey, A.; Milner, T.A.; Brake, W.G. Estrogen receptors in the central nervous system and their implication for dopamine-dependent cognition in females. Horm. Behav. 2015, 74, 125–138. [Google Scholar] [CrossRef] [Green Version]
- Almey, A.; Filardo, E.J.; Milner, T.A.; Brake, W.G. Estrogen receptors are found in glia and at extranuclear neuronal sites in the dorsal striatum of female rats: Evidence for cholinergic but not dopaminergic colocalization. Endocrinology 2012, 153, 5373–5383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitterling, K.L.; Spencer, J.L.; Dziedzic, N.; Shenoy, S.; McCarthy, K.; Waters, E.M.; McEwen, B.S.; Milner, T.A. Cellular and subcellular localization of estrogen and progestin receptor immunoreactivities in the mouse hippocampus. J. Comp. Neurol. 2010, 518, 2729–2743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Funakoshi, T.; Yanai, A.; Shinoda, K.; Kawano, M.M.; Mizukami, Y. G protein-coupled receptor 30 is an estrogen receptor in the plasma membrane. Biochem. Biophys. Res. Commun. 2006, 346, 904–910. [Google Scholar] [CrossRef] [PubMed]
- Campos, F.L.; Cristovao, A.C.; Rocha, S.M.; Fonseca, C.P.; Baltazar, G. GDNF contributes to oestrogen-mediated protection of midbrain dopaminergic neurones. J. Neuroendocrinol. 2012, 24, 1386–1397. [Google Scholar] [CrossRef] [PubMed]
- Castilla-Cortazar, I.; Aguirre, G.A.; Femat-Roldan, G.; Martin-Estal, I.; Espinosa, L. Is insulin-like growth factor-1 involved in Parkinson’s disease development? J. Transl. Med. 2020, 18, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.; Li, J.; Qiu, S.; Wen, H.; Du, J. Hormone replacement therapy and Parkinson’s disease risk in women: A meta-analysis of 14 observational studies. Neuropsychiatr. Dis. Treat. 2015, 11, 59–66. [Google Scholar] [PubMed] [Green Version]
- Barron, A.M.; Pike, C.J. Sex hormones, aging, and Alzheimer’s disease. Front. Biosci. 2012, 4, 976–997. [Google Scholar]
- Carroll, J.C.; Rosario, E.R.; Chang, L.; Stanczyk, F.Z.; Oddo, S.; LaFerla, F.M.; Pike, C.J. Progesterone and estrogen regulate Alzheimer-like neuropathology in female 3xTg-AD mice. J. Neurosci. 2007, 27, 13357–13365. [Google Scholar] [CrossRef]
- Savolainen-Peltonen, H.; Rahkola-Soisalo, P.; Hoti, F.; Vattulainen, P.; Gissler, M.; Ylikorkala, O.; Mikkola, T.S. Use of postmenopausal hormone therapy and risk of Alzheimer’s disease in Finland: Nationwide case-control study. BMJ 2019, 364, l665. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Wang, J.M.; Irwin, R.W.; Yao, J.; Liu, L.; Brinton, R.D. Allopregnanolone promotes regeneration and reduces beta-amyloid burden in a preclinical model of Alzheimer’s disease. PLoS ONE 2011, 6, e24293. [Google Scholar]
- Irwin, R.W.; Brinton, R.D. Allopregnanolone as regenerative therapeutic for Alzheimer’s disease: Translational development and clinical promise. Prog. Neurobiol. 2014, 113, 40–55. [Google Scholar] [CrossRef]
- Wang, T.; Yao, J.; Chen, S.; Mao, Z.; Brinton, R.D. Allopregnanolone Reverses Bioenergetic Deficits in Female Triple Transgenic Alzheimer’s Mouse Model. Neurotherapeutics 2020, 17, 178–188. [Google Scholar] [CrossRef]
- Adeosun, S.O.; Hou, X.; Jiao, Y.; Zheng, B.; Henry, S.; Hill, R.; He, Z.; Pani, A.; Kyle, P.; Ou, X.; et al. Allopregnanolone reinstates tyrosine hydroxylase immunoreactive neurons and motor performance in an MPTP-lesioned mouse model of Parkinson’s disease. PLoS ONE 2012, 7, e50040. [Google Scholar] [CrossRef]
- Chen, Z.C.; Wang, T.T.; Bian, W.; Ye, X.; Li, M.Y.; Du, J.J.; Zhou, P.; Cui, H.R.; Ding, Y.Q.; Ren, Y.H.; et al. Allopregnanolone restores the tyrosine hydroxylase-positive neurons and motor performance in a 6-OHDA-injected mouse model. CNS Neurosci. Ther. 2020, 26, 1069–1082. [Google Scholar] [CrossRef]
- Okereke, O.; Kang, J.H.; Ma, J.; Hankinson, S.E.; Pollak, M.N.; Grodstein, F. Plasma IGF-I levels and cognitive performance in older women. Neurobiol. Aging 2007, 28, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Salzmann, A.; James, S.N.; Williams, D.M.; Richards, M.; Cadar, D.; Schott, J.M.; Coath, W.; Sudre, C.H.; Chaturvedi, N.; Garfield, V. Investigating the Relationship Between IGF-I, IGF-II, and IGFBP-3 Concentrations and Later-Life Cognition and Brain Volume. J. Clin. Endocrinol. Metab. 2021, 106, 1617–1629. [Google Scholar] [CrossRef]
- Doi, T.; Shimada, H.; Makizako, H.; Tsutsumimoto, K.; Hotta, R.; Nakakubo, S.; Suzuki, T. Association of insulin-like growth factor-1 with mild cognitive impairment and slow gait speed. Neurobiol. Aging 2015, 36, 942–947. [Google Scholar] [CrossRef]
- Alvarez, A.; Cacabelos, R.; Sanpedro, C.; Garcia-Fantini, M.; Aleixandre, M. Serum TNF-alpha levels are increased and correlate negatively with free IGF-I in Alzheimer disease. Neurobiol. Aging 2007, 28, 533–536. [Google Scholar] [CrossRef]
- Mustafa, A.; Lannfelt, L.; Lilius, L.; Islam, A.; Winblad, B.; Adem, A. Decreased plasma insulin-like growth factor-I level in familial Alzheimer’s disease patients carrying the Swedish APP 670/671 mutation. Dement. Geriatr. Cogn. Disord. 1999, 10, 446–451. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Miyazaki, A.; Katagiri, T.; Yamamoto, H.; Idei, T.; Iguchi, T. Relationship between serum insulin-like growth factor-1 levels and Alzheimer’s disease and vascular dementia. J. Am. Geriatr. Soc. 2005, 53, 1748–1753. [Google Scholar] [CrossRef]
- Duron, E.; Funalot, B.; Brunel, N.; Coste, J.; Quinquis, L.; Viollet, C.; Belmin, J.; Jouanny, P.; Pasquier, F.; Treluyer, J.M.; et al. Insulin-like growth factor-I and insulin-like growth factor binding protein-3 in Alzheimer’s disease. J. Clin. Endocrinol. Metab. 2012, 97, 4673–4681. [Google Scholar] [CrossRef] [Green Version]
- Westwood, A.J.; Beiser, A.; Decarli, C.; Harris, T.B.; Chen, T.C.; He, X.M.; Roubenoff, R.; Pikula, A.; Au, R.; Braverman, L.E.; et al. Insulin-like growth factor-1 and risk of Alzheimer dementia and brain atrophy. Neurology 2014, 82, 1613–1619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vardy, E.R.; Rice, P.J.; Bowie, P.C.; Holmes, J.D.; Grant, P.J.; Hooper, N.M. Increased circulating insulin-like growth factor-1 in late-onset Alzheimer’s disease. J. Alzheimers Dis. 2007, 12, 285–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vargas, T.; Martinez-Garcia, A.; Antequera, D.; Vilella, E.; Clarimon, J.; Mateo, I.; Sanchez-Juan, P.; Rodriguez-Rodriguez, E.; Frank, A.; Rosich-Estrago, M.; et al. IGF-I gene variability is associated with an increased risk for AD. Neurobiol. Aging 2011, 32, 556.e3–556.e11. [Google Scholar] [CrossRef] [PubMed]
- Trueba-Saiz, A.; Cavada, C.; Fernandez, A.M.; Leon, T.; Gonzalez, D.A.; Fortea Ormaechea, J.; Lleo, A.; Del Ser, T.; Nunez, A.; Torres-Aleman, I. Loss of serum IGF-I input to the brain as an early biomarker of disease onset in Alzheimer mice. Transl. Psychiatry 2013, 3, e330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostrowski, P.P.; Barszczyk, A.; Forstenpointner, J.; Zheng, W.; Feng, Z.P. Meta-Analysis of Serum Insulin-Like Growth Factor 1 in Alzheimer’s Disease. PLoS ONE 2016, 11, e0155733. [Google Scholar] [CrossRef] [Green Version]
- De la Monte, S.M.; Wands, J.R. Alzheimer’s disease is type 3 diabetes-evidence reviewed. J. Diabetes Sci. Technol. 2008, 2, 1101–1113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Picillo, M.; Pivonello, R.; Santangelo, G.; Pivonello, C.; Savastano, R.; Auriemma, R.; Amboni, M.; Scannapieco, S.; Pierro, A.; Colao, A.; et al. Serum IGF-1 is associated with cognitive functions in early, drug-naive Parkinson’s disease. PLoS ONE 2017, 12, e0186508. [Google Scholar] [CrossRef] [Green Version]
- Fan, D.; Pitcher, T.; Dalrymple-Alford, J.; MacAskill, M.; Anderson, T.; Guan, J. Changes of plasma cGP/IGF-1 molar ratio with age is associated with cognitive status of Parkinson disease. Alzheimers Dement. 2020, 12, e12025. [Google Scholar] [CrossRef]
- Al-Delaimy, W.K.; von Muhlen, D.; Barrett-Connor, E. Insulinlike growth factor-1, insulinlike growth factor binding protein-1, and cognitive function in older men and women. J. Am. Geriatr. Soc. 2009, 57, 1441–1446. [Google Scholar] [CrossRef]
- Perice, L.; Barzilai, N.; Verghese, J.; Weiss, E.F.; Holtzer, R.; Cohen, P.; Milman, S. Lower circulating insulin-like growth factor-I is associated with better cognition in females with exceptional longevity without compromise to muscle mass and function. Aging 2016, 8, 2414–2424. [Google Scholar] [CrossRef] [Green Version]
- Frederiksen, H.; Johannsen, T.H.; Andersen, S.E.; Albrethsen, J.; Landersoe, S.K.; Petersen, J.H.; Andersen, A.N.; Vestergaard, E.T.; Schorring, M.E.; Linneberg, A.; et al. Sex-specific Estrogen Levels and Reference Intervals from Infancy to Late Adulthood Determined by LC-MS/MS. J. Clin. Endocrinol. Metab. 2020, 105, 754–768. [Google Scholar] [CrossRef]
- Morinaga, A.; Ono, K.; Takasaki, J.; Ikeda, T.; Hirohata, M.; Yamada, M. Effects of sex hormones on Alzheimer’s disease-associated beta-amyloid oligomer formation in vitro. Exp. Neurol. 2011, 228, 298–302. [Google Scholar] [CrossRef]
- Palmisano, B.T.; Zhu, L.; Stafford, J.M. Role of Estrogens in the Regulation of Liver Lipid Metabolism. Adv. Exp. Med. Biol. 2017, 1043, 227–256. [Google Scholar]
- Dimache, A.M.; Salaru, D.L.; Sascau, R.; Statescu, C. The Role of High Triglycerides Level in Predicting Cognitive Impairment: A Review of Current Evidence. Nutrients 2021, 13, 2118. [Google Scholar] [CrossRef] [PubMed]
- Beekman, J.M.; Allan, G.F.; Tsai, S.Y.; Tsai, M.J.; O’Malley, B.W. Transcriptional activation by the estrogen receptor requires a conformational change in the ligand binding domain. Mol. Endocrinol. 1993, 7, 1266–1274. [Google Scholar] [PubMed]
- Fliss, A.E.; Benzeno, S.; Rao, J.; Caplan, A.J. Control of estrogen receptor ligand binding by Hsp90. J. Steroid Biochem. Mol. Biol. 2000, 72, 223–230. [Google Scholar] [CrossRef]
- Bustamante-Barrientos, F.A.; Mendez-Ruette, M.; Ortloff, A.; Luz-Crawford, P.; Rivera, F.J.; Figueroa, C.D.; Molina, L.; Batiz, L.F. The Impact of Estrogen and Estrogen-Like Molecules in Neurogenesis and Neurodegeneration: Beneficial or Harmful? Front. Cell Neurosci. 2021, 15, 636176. [Google Scholar] [CrossRef]
- Klinge, C.M. Estrogen receptor interaction with estrogen response elements. Nucleic Acids Res. 2001, 29, 2905–2919. [Google Scholar] [CrossRef] [Green Version]
- Sohrabji, F.; Miranda, R.C.; Toran-Allerand, C.D. Identification of a putative estrogen response element in the gene encoding brain-derived neurotrophic factor. Proc. Natl. Acad. Sci. USA 1995, 92, 11110–11114. [Google Scholar] [CrossRef] [Green Version]
- Kight, K.E.; McCarthy, M.M. Sex differences and estrogen regulation of BDNF gene expression, but not propeptide content, in the developing hippocampus. J. Neurosci. Res. 2017, 95, 345–354. [Google Scholar] [CrossRef] [Green Version]
- Kwakowsky, A.; Milne, M.R.; Waldvogel, H.J.; Faull, R.L. Effect of Estradiol on Neurotrophin Receptors in Basal Forebrain Cholinergic Neurons: Relevance for Alzheimer’s Disease. Int. J. Mol. Sci. 2016, 17, 2122. [Google Scholar] [CrossRef] [Green Version]
- Krolick, K.N.; Zhu, Q.; Shi, H. Effects of Estrogens on Central Nervous System Neurotransmission: Implications for Sex Differences in Mental Disorders. Prog. Mol. Biol. Transl. Sci. 2018, 160, 105–171. [Google Scholar]
- Vail, G.; Roepke, T.A. Membrane-initiated estrogen signaling via Gq-coupled GPCR in the central nervous system. Steroids 2019, 142, 77–83. [Google Scholar] [CrossRef]
- Revankar, C.M.; Cimino, D.F.; Sklar, L.A.; Arterburn, J.B.; Prossnitz, E.R. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science 2005, 307, 1625–1630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, H.; Zhang, Q.; Yang, L.; Dong, Y.; Khan, M.; Yang, F.; Brann, D.W.; Wang, R. Reprint of "GPR30 mediates estrogen rapid signaling and neuroprotection". Mol. Cell Endocrinol. 2014, 389, 92–98. [Google Scholar] [CrossRef]
- Kim, J.; Szinte, J.S.; Boulware, M.I.; Frick, K.M. 17beta-Estradiol and Agonism of G-protein-Coupled Estrogen Receptor Enhance Hippocampal Memory via Different Cell-Signaling Mechanisms. J. Neurosci. 2016, 36, 3309–3321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Weiland, H.; Schofbanker, M.; Zhang, W. Estrogen Receptors Alpha and Beta Mediate Synaptic Transmission in the PFC and Hippocampus of Mice. Int. J. Mol. Sci. 2021, 22, 1485. [Google Scholar] [CrossRef]
- Frick, K.M. Molecular mechanisms underlying the memory-enhancing effects of estradiol. Horm. Behav. 2015, 74, 4–18. [Google Scholar] [CrossRef] [Green Version]
- Filardo, E.J.; Quinn, J.A.; Bland, K.I.; Frackelton, A.R., Jr. Estrogen-induced activation of Erk-1 and Erk-2 requires the G protein-coupled receptor homolog, GPR30, and occurs via trans-activation of the epidermal growth factor receptor through release of HB-EGF. Mol. Endocrinol. 2000, 14, 1649–1660. [Google Scholar] [CrossRef]
- Filardo, E.J.; Quinn, J.A.; Frackelton, A.R., Jr.; Bland, K.I. Estrogen action via the G protein-coupled receptor, GPR30: Stimulation of adenylyl cyclase and cAMP-mediated attenuation of the epidermal growth factor receptor-to-MAPK signaling axis. Mol. Endocrinol. 2002, 16, 70–84. [Google Scholar] [CrossRef] [PubMed]
- Shi, D.; Zhao, P.; Cui, L.; Li, H.; Sun, L.; Niu, J.; Chen, M. Inhibition of PI3K/AKT molecular pathway mediated by membrane estrogen receptor GPER accounts for cryptotanshinone induced antiproliferative effect on breast cancer SKBR-3 cells. BMC Pharmacol. Toxicol. 2020, 21, 32. [Google Scholar] [CrossRef] [PubMed]
- Evans, N.J.; Bayliss, A.L.; Reale, V.; Evans, P.D. Characterisation of Signalling by the Endogenous GPER1 (GPR30) Receptor in an Embryonic Mouse Hippocampal Cell Line (mHippoE-18). PLoS ONE 2016, 11, e0152138. [Google Scholar] [CrossRef] [Green Version]
- Bourque, M.; Morissette, M.; Di Paolo, T. Neuroprotection in Parkinsonian-treated mice via estrogen receptor alpha activation requires G protein-coupled estrogen receptor 1. Neuropharmacology 2015, 95, 343–352. [Google Scholar] [CrossRef]
- Cheng, Q.; Meng, J.; Wang, X.S.; Kang, W.B.; Tian, Z.; Zhang, K.; Liu, G.; Zhao, J.N. G-1 exerts neuroprotective effects through G protein-coupled estrogen receptor 1 following spinal cord injury in mice. Biosci. Rep. 2016, 36, 36. [Google Scholar] [CrossRef] [Green Version]
- Brotfain, E.; Gruenbaum, S.E.; Boyko, M.; Kutz, R.; Zlotnik, A.; Klein, M. Neuroprotection by Estrogen and Progesterone in Traumatic Brain Injury and Spinal Cord Injury. Curr. Neuropharmacol. 2016, 14, 641–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briz, V.; Baudry, M. Estrogen Regulates Protein Synthesis and Actin Polymerization in Hippocampal Neurons through Different Molecular Mechanisms. Front. Endocrinol. 2014, 5, 22. [Google Scholar] [CrossRef]
- Yuan, L.J.; Wang, X.W.; Wang, H.T.; Zhang, M.; Sun, J.W.; Chen, W.F. G protein-coupled estrogen receptor is involved in the neuroprotective effect of IGF-1 against MPTP/MPP(+)-induced dopaminergic neuronal injury. J. Steroid Biochem. Mol. Biol. 2019, 192, 105384. [Google Scholar] [CrossRef]
- Wang, X.W.; Yuan, L.J.; Yang, Y.; Zhang, M.; Chen, W.F. IGF-1 inhibits MPTP/MPP(+)-induced autophagy on dopaminergic neurons through the IGF-1R/PI3K-Akt-mTOR pathway and GPER. Am. J. Physiol. Endocrinol. Metab. 2020, 319, E734–E743. [Google Scholar] [CrossRef]
- Briski, K.P.; Ali, M.H.; Napit, P.R. Sex-specific acclimation of A2 noradrenergic neuron dopamine-beta-hydroxylase and estrogen receptor variant protein and 5’-AMP-Activated protein kinase reactivity to recurring hypoglycemia in rat. J. Chem. Neuroanat. 2020, 109, 101845. [Google Scholar] [CrossRef] [PubMed]
- Velarde, M.C. Mitochondrial and sex steroid hormone crosstalk during aging. Longev. Healthspan. 2014, 3, 2. [Google Scholar] [CrossRef] [Green Version]
- Dobolyi, A.; Leko, A.H. The insulin-like growth factor-1 system in the adult mammalian brain and its implications in central maternal adaptation. Front. Neuroendocrinol. 2019, 52, 181–194. [Google Scholar] [CrossRef] [PubMed]
- Sonntag, W.E.; Bennett, S.A.; Khan, A.S.; Thornton, P.L.; Xu, X.; Ingram, R.L.; Brunso-Bechtold, J.K. Age and insulin-like growth factor-1 modulate N-methyl-D-aspartate receptor subtype expression in rats. Brain Res. Bull. 2000, 51, 331–338. [Google Scholar] [CrossRef]
- Lichtenwalner, R.J.; Forbes, M.E.; Bennett, S.A.; Lynch, C.D.; Sonntag, W.E.; Riddle, D.R. Intracerebroventricular infusion of insulin-like growth factor-I ameliorates the age-related decline in hippocampal neurogenesis. Neuroscience 2001, 107, 603–613. [Google Scholar] [CrossRef]
- Poirier, R.; Fernandez, A.M.; Torres-Aleman, I.; Metzger, F. Early brain amyloidosis in APP/PS1 mice with serum insulin-like growth factor-I deficiency. Neurosci. Lett. 2012, 509, 101–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carro, E.; Trejo, J.L.; Gerber, A.; Loetscher, H.; Torrado, J.; Metzger, F.; Torres-Aleman, I. Therapeutic actions of insulin-like growth factor I on APP/PS2 mice with severe brain amyloidosis. Neurobiol. Aging 2006, 27, 1250–1257. [Google Scholar] [CrossRef] [Green Version]
- Carro, E.; Trejo, J.L.; Gomez-Isla, T.; LeRoith, D.; Torres-Aleman, I. Serum insulin-like growth factor I regulates brain amyloid-beta levels. Nat. Med. 2002, 8, 1390–1397. [Google Scholar] [CrossRef] [PubMed]
- George, C.; Gontier, G.; Lacube, P.; Francois, J.C.; Holzenberger, M.; Aid, S. The Alzheimer’s disease transcriptome mimics the neuroprotective signature of IGF-1 receptor-deficient neurons. Brain 2017, 140, 2012–2027. [Google Scholar] [CrossRef] [Green Version]
- Gontier, G.; George, C.; Chaker, Z.; Holzenberger, M.; Aid, S. Blocking IGF Signaling in Adult Neurons Alleviates Alzheimer’s Disease Pathology through Amyloid-beta Clearance. J. Neurosci. 2015, 35, 11500–11513. [Google Scholar] [CrossRef] [PubMed]
- Sohrabi, M.; Floden, A.M.; Manocha, G.D.; Klug, M.G.; Combs, C.K. IGF-1R Inhibitor Ameliorates Neuroinflammation in an Alzheimer’s Disease Transgenic Mouse Model. Front. Cell Neurosci. 2020, 14, 200. [Google Scholar] [CrossRef] [PubMed]
- Lanz, T.A.; Salatto, C.T.; Semproni, A.R.; Marconi, M.; Brown, T.M.; Richter, K.E.; Schmidt, K.; Nelson, F.R.; Schachter, J.B. Peripheral elevation of IGF-1 fails to alter Abeta clearance in multiple in vivo models. Biochem. Pharmacol. 2008, 75, 1093–1103. [Google Scholar] [CrossRef]
- Selles, M.C.; Fortuna, J.T.S.; Zappa-Villar, M.F.; de Faria, Y.P.R.; Souza, A.S.; Suemoto, C.K.; Leite, R.E.P.; Rodriguez, R.D.; Grinberg, L.T.; Reggiani, P.C.; et al. Adenovirus-Mediated Transduction of Insulin-Like Growth Factor 1 Protects Hippocampal Neurons from the Toxicity of Abeta Oligomers and Prevents Memory Loss in an Alzheimer Mouse Model. Mol. Neurobiol. 2020, 57, 1473–1483. [Google Scholar] [CrossRef]
- Sevigny, J.J.; Ryan, J.M.; van Dyck, C.H.; Peng, Y.; Lines, C.R.; Nessly, M.L.; Group, M.K.P.S. Growth hormone secretagogue MK-677: No clinical effect on AD progression in a randomized trial. Neurology 2008, 71, 1702–1708. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Wang, T.; Wang, Q.; Guo, L.; Du, H. MK0677, a Ghrelin Mimetic, Improves Neurogenesis but Fails to Prevent Hippocampal Lesions in a Mouse Model of Alzheimer’s Disease Pathology. J. Alzheimers Dis. 2019, 72, 467–478. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, S.L. Progesterone for the treatment of central nervous system disorders: The many signaling roads for a single molecule. Neural. Regen. Res. 2020, 15, 1846–1847. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Chen, Z.; Han, X.; Wu, H.; Yu, Y.; Wu, J.; Liu, S.; Hou, Y. Progesterone attenuates Abeta(25-35)-induced neuronal toxicity via JNK inactivation and progesterone receptor membrane component 1-dependent inhibition of mitochondrial apoptotic pathway. J. Steroid. Biochem. Mol. Biol. 2015, 154, 302–311. [Google Scholar] [CrossRef]
- Jolivel, V.; Brun, S.; Biname, F.; Benyounes, J.; Taleb, O.; Bagnard, D.; De Seze, J.; Patte-Mensah, C.; Mensah-Nyagan, A.G. Microglial Cell Morphology and Phagocytic Activity Are Critically Regulated by the Neurosteroid Allopregnanolone: A Possible Role in Neuroprotection. Cells 2021, 10, 698. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.N.; Chen, X.S.; Su, L.; Liu, Y.L.; Cai, Q.Y.; Zhan, X.L.; Xu, Y.; Zhao, S.F.; Yao, Z.X. Progesterone alleviates neural behavioral deficits and demyelination with reduced degeneration of oligodendroglial cells in cuprizone-induced mice. PLoS ONE 2013, 8, e54590. [Google Scholar] [CrossRef] [Green Version]
- El-Etr, M.; Rame, M.; Boucher, C.; Ghoumari, A.M.; Kumar, N.; Liere, P.; Pianos, A.; Schumacher, M.; Sitruk-Ware, R. Progesterone and nestorone promote myelin regeneration in chronic demyelinating lesions of corpus callosum and cerebral cortex. Glia 2015, 63, 104–117. [Google Scholar] [CrossRef] [Green Version]
- El-Etr, M.; Akwa, Y.; Rame, M.; Schumacher, M.; Sitruk-Ware, R. Nestorone((R)), a 19nor-progesterone derivative boosts remyelination in an animal model of demyelination. CNS Neurosci. Ther. 2021, 27, 464–469. [Google Scholar] [CrossRef]
- Bouhrara, M.; Reiter, D.A.; Bergeron, C.M.; Zukley, L.M.; Ferrucci, L.; Resnick, S.M.; Spencer, R.G. Evidence of demyelination in mild cognitive impairment and dementia using a direct and specific magnetic resonance imaging measure of myelin content. Alzheimers Dement. 2018, 14, 998–1004. [Google Scholar] [CrossRef]
- Yilmaz, C.; Karali, K.; Fodelianaki, G.; Gravanis, A.; Chavakis, T.; Charalampopoulos, I.; Alexaki, V.I. Neurosteroids as regulators of neuroinflammation. Front. Neuroendocrinol. 2019, 55, 100788. [Google Scholar] [CrossRef] [PubMed]
- Aryanpour, R.; Pasbakhsh, P.; Zibara, K.; Namjoo, Z.; Beigi Boroujeni, F.; Shahbeigi, S.; Kashani, I.R.; Beyer, C.; Zendehdel, A. Progesterone therapy induces an M1 to M2 switch in microglia phenotype and suppresses NLRP3 inflammasome in a cuprizone-induced demyelination mouse model. Int. Immunopharmacol. 2017, 51, 131–139. [Google Scholar] [CrossRef]
- Gutzeit, O.; Segal, L.; Korin, B.; Iluz, R.; Khatib, N.; Dabbah-Assadi, F.; Ginsberg, Y.; Fainaru, O.; Ross, M.G.; Weiner, Z.; et al. Progesterone Attenuates Brain Inflammatory Response and Inflammation-Induced Increase in Immature Myeloid Cells in a Mouse Model. Inflammation 2021, 44, 956–964. [Google Scholar] [CrossRef]
- Litim, N.; Morissette, M.; Di Paolo, T. Effects of progesterone administered after MPTP on dopaminergic neurons of male mice. Neuropharmacology 2017, 117, 209–218. [Google Scholar] [CrossRef]
- Castelnovo, L.F.; Thomas, P. Membrane progesterone receptor alpha (mPRalpha/PAQR7) promotes migration, proliferation and BDNF release in human Schwann cell-like differentiated adipose stem cells. Mol. Cell Endocrinol. 2021, 531, 111298. [Google Scholar] [CrossRef] [PubMed]
- Acosta, J.I.; Hiroi, R.; Camp, B.W.; Talboom, J.S.; Bimonte-Nelson, H.A. An update on the cognitive impact of clinically-used hormone therapies in the female rat: Models, mazes, and mechanisms. Brain Res. 2013, 1514, 18–39. [Google Scholar] [CrossRef] [Green Version]
- Singh, M.; Su, C. Progesterone-induced neuroprotection: Factors that may predict therapeutic efficacy. Brain Res. 2013, 1514, 98–106. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.; Moore, A.B.; Castro, L.; Gao, X.; Huynh, H.L.; Klippel, M.; Flagler, N.D.; Lu, Y.; Kissling, G.E.; Dixon, D. Estrogen Regulates MAPK-Related Genes through Genomic and Nongenomic Interactions between IGF-I Receptor Tyrosine Kinase and Estrogen Receptor-Alpha Signaling Pathways in Human Uterine Leiomyoma Cells. J. Signal. Transduct. 2012, 2012, 204236. [Google Scholar] [CrossRef]
- Xu, S.; Yu, S.; Dong, D.; Lee, L.T.O. G Protein-Coupled Estrogen Receptor: A Potential Therapeutic Target in Cancer. Front. Endocrinol. 2019, 10, 725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.; Nozaki, K.; Smith, J.A.; Krause, J.S.; Banik, N.L. Cross-talk between IGF-1 and estrogen receptors attenuates intracellular changes in ventral spinal cord 4.1 motoneuron cells because of interferon-gamma exposure. J. Neurochem. 2014, 128, 904–918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, J.; Berton, T.R.; Shirley, S.H.; Lambertz, I.; Gimenez-Conti, I.B.; DiGiovanni, J.; Korach, K.S.; Conti, C.J.; Fuchs-Young, R. Developmental stage determines estrogen receptor alpha expression and non-genomic mechanisms that control IGF-1 signaling and mammary proliferation in mice. J. Clin. Investig. 2012, 122, 192–204. [Google Scholar] [CrossRef] [Green Version]
- Ding, Q.; Vaynman, S.; Akhavan, M.; Ying, Z.; Gomez-Pinilla, F. Insulin-like growth factor I interfaces with brain-derived neurotrophic factor-mediated synaptic plasticity to modulate aspects of exercise-induced cognitive function. Neuroscience 2006, 140, 823–833. [Google Scholar] [CrossRef]
- Aberg, M.A.; Aberg, N.D.; Palmer, T.D.; Alborn, A.M.; Carlsson-Skwirut, C.; Bang, P.; Rosengren, L.E.; Olsson, T.; Gage, F.H.; Eriksson, P.S. IGF-I has a direct proliferative effect in adult hippocampal progenitor cells. Mol. Cell Neurosci. 2003, 24, 23–40. [Google Scholar] [CrossRef]
- Nieto-Estevez, V.; Oueslati-Morales, C.O.; Li, L.; Pickel, J.; Morales, A.V.; Vicario-Abejon, C. Brain Insulin-Like Growth Factor-I Directs the Transition from Stem Cells to Mature Neurons During Postnatal/Adult Hippocampal Neurogenesis. Stem Cells 2016, 34, 2194–2209. [Google Scholar] [CrossRef] [Green Version]
- Cohen, E.; Paulsson, J.F.; Blinder, P.; Burstyn-Cohen, T.; Du, D.; Estepa, G.; Adame, A.; Pham, H.M.; Holzenberger, M.; Kelly, J.W.; et al. Reduced IGF-1 signaling delays age-associated proteotoxicity in mice. Cell 2009, 139, 1157–1169. [Google Scholar] [CrossRef] [Green Version]
- Yuan, L.J.; Zhang, M.; Chen, S.; Chen, W.F. Anti-inflammatory effect of IGF-1 is mediated by IGF-1R cross talk with GPER in MPTP/MPP(+)-induced astrocyte activation. Mol. Cell Endocrinol. 2021, 519, 111053. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.C.; McMahon, L.L. Estrogen-induced increase in the magnitude of long-term potentiation occurs only when the ratio of NMDA transmission to AMPA transmission is increased. J. Neurosci. 2005, 25, 7780–7791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clements, L.; Harvey, J. Activation of oestrogen receptor alpha induces a novel form of LTP at hippocampal temporoammonic-CA1 synapses. Br. J. Pharmacol. 2020, 177, 642–655. [Google Scholar] [CrossRef] [PubMed]
- Lewis, M.C.; Kerr, K.M.; Orr, P.T.; Frick, K.M. Estradiol-induced enhancement of object memory consolidation involves NMDA receptors and protein kinase A in the dorsal hippocampus of female C57BL/6 mice. Behav. Neurosci. 2008, 122, 716–721. [Google Scholar] [CrossRef] [Green Version]
- Vedder, L.C.; Smith, C.C.; Flannigan, A.E.; McMahon, L.L. Estradiol-induced increase in novel object recognition requires hippocampal NR2B-containing NMDA receptors. Hippocampus 2013, 23, 108–115. [Google Scholar] [CrossRef] [Green Version]
- Snyder, M.A.; Cooke, B.M.; Woolley, C.S. Estradiol potentiation of NR2B-dependent EPSCs is not due to changes in NR2B protein expression or phosphorylation. Hippocampus 2011, 21, 398–408. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Sokabe, M. Bidirectional modulatory effect of 17beta-estradiol on NMDA receptors via ERalpha and ERbeta in the dentate gyrus of juvenile male rats. Neuropharmacology 2013, 75, 262–273. [Google Scholar] [CrossRef]
- Boulware, M.I.; Weick, J.P.; Becklund, B.R.; Kuo, S.P.; Groth, R.D.; Mermelstein, P.G. Estradiol activates group I and II metabotropic glutamate receptor signaling, leading to opposing influences on cAMP response element-binding protein. J. Neurosci. 2005, 25, 5066–5078. [Google Scholar] [CrossRef]
- Benquet, P.; Gee, C.E.; Gerber, U. Two distinct signaling pathways upregulate NMDA receptor responses via two distinct metabotropic glutamate receptor subtypes. J. Neurosci. 2002, 22, 9679–9686. [Google Scholar] [CrossRef] [Green Version]
- Sahab-Negah, S.; Hajali, V.; Moradi, H.R.; Gorji, A. The Impact of Estradiol on Neurogenesis and Cognitive Functions in Alzheimer’s Disease. Cell Mol. Neurobiol. 2020, 40, 283–299. [Google Scholar] [CrossRef] [PubMed]
- Sha, S.; Hong, J.; Qu, W.J.; Lu, Z.H.; Li, L.; Yu, W.F.; Chen, L. Sex-related neurogenesis decrease in hippocampal dentate gyrus with depressive-like behaviors in sigma-1 receptor knockout mice. Eur. Neuropsychopharmacol. 2015, 25, 1275–1286. [Google Scholar] [CrossRef]
- Liu, S.B.; Zhao, M.G. Neuroprotective effect of estrogen: Role of nonsynaptic NR2B-containing NMDA receptors. Brain Res. Bull. 2013, 93, 27–31. [Google Scholar] [CrossRef]
- Androvicova, R.; Pfaus, J.G.; Ovsepian, S.V. Estrogen pendulum in schizophrenia and Alzheimer’s disease: Review of therapeutic benefits and outstanding questions. Neurosci. Lett. 2021, 759, 136038. [Google Scholar] [CrossRef] [PubMed]
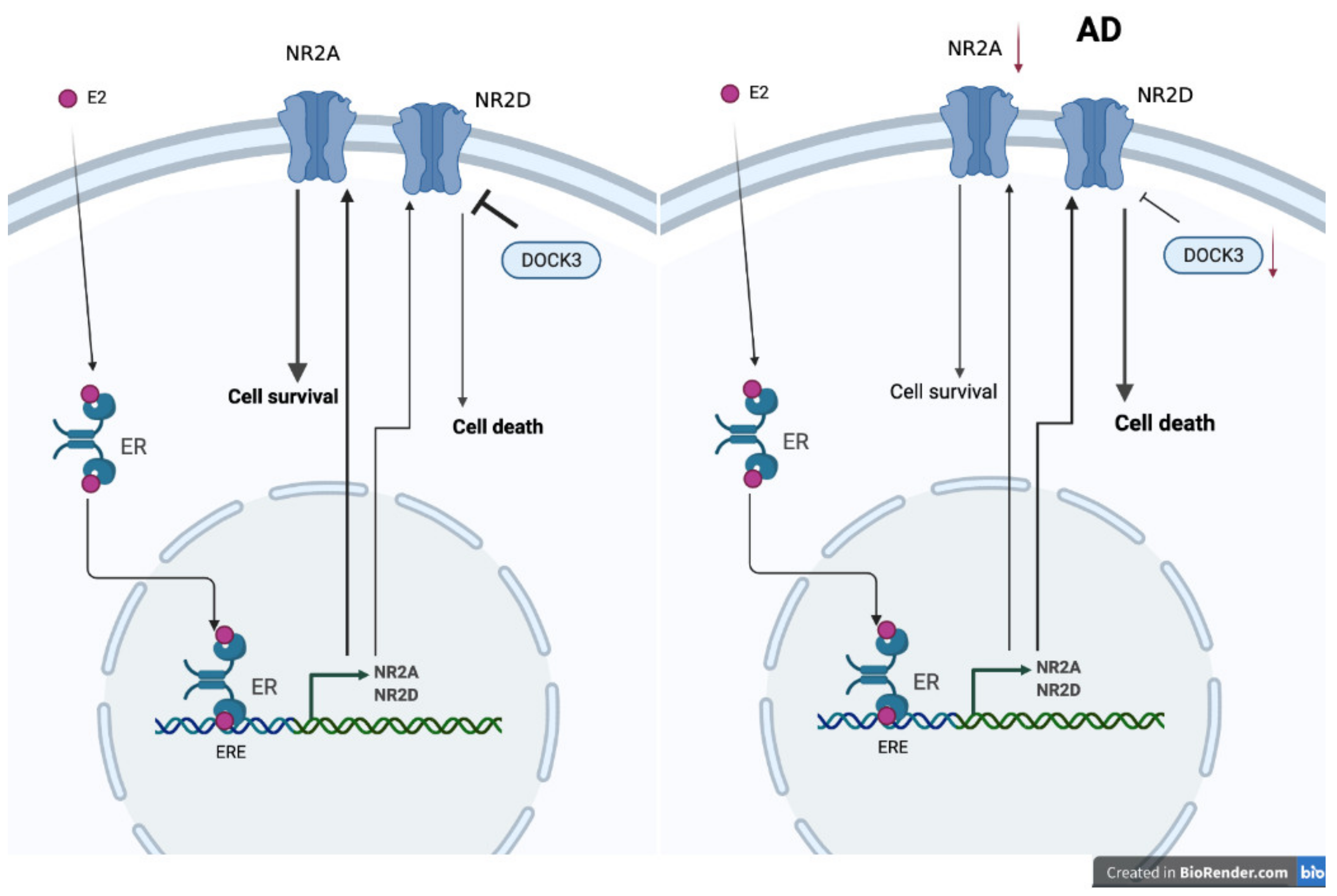
- Watanabe, T.; Inoue, S.; Hiroi, H.; Orimo, A.; Muramatsu, M. NMDA receptor type 2D gene as target for estrogen receptor in the brain. Brain Res. Mol. Brain Res. 1999, 63, 375–379. [Google Scholar] [CrossRef]
- Ikeda, K.; Fukushima, T.; Ogura, H.; Tsukui, T.; Mishina, M.; Muramatsu, M.; Inoue, S. Estrogen regulates the expression of N-methyl-D-aspartate (NMDA) receptor subunit epsilon 4 (Grin2d), that is essential for the normal sexual behavior in female mice. FEBS Lett. 2010, 584, 806–810. [Google Scholar] [CrossRef] [PubMed]
- Hynd, M.R.; Scott, H.L.; Dodd, P.R. Differential expression of N-methyl-D-aspartate receptor NR2 isoforms in Alzheimer’s disease. J. Neurochem. 2004, 90, 913–919. [Google Scholar] [CrossRef] [PubMed]
- Clayton, D.A.; Browning, M.D. Deficits in the expression of the NR2B subunit in the hippocampus of aged Fisher 344 rats. Neurobiol. Aging 2001, 22, 165–168. [Google Scholar] [CrossRef]
- Magnusson, K.R.; Kresge, D.; Supon, J. Differential effects of aging on NMDA receptors in the intermediate versus the dorsal hippocampus. Neurobiol. Aging 2006, 27, 324–333. [Google Scholar] [CrossRef]
- Jullienne, A.; Montagne, A.; Orset, C.; Lesept, F.; Jane, D.E.; Monaghan, D.T.; Maubert, E.; Vivien, D.; Ali, C. Selective inhibition of GluN2D-containing N-methyl-D-aspartate receptors prevents tissue plasminogen activator-promoted neurotoxicity both in vitro and in vivo. Mol. Neurodegener. 2011, 6, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefort, R. Reversing synapse loss in Alzheimer’s disease: Rho-guanosine triphosphatases and insights from other brain disorders. Neurotherapeutics 2015, 12, 19–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, N.; Hayashi, H.; Aida, T.; Namekata, K.; Harada, T.; Mishina, M.; Tanaka, K. Dock3 interaction with a glutamate-receptor NR2D subunit protects neurons from excitotoxicity. Mol. Brain 2013, 6, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Namekata, K.; Kimura, A.; Kawamura, K.; Guo, X.; Harada, C.; Tanaka, K.; Harada, T. Dock3 attenuates neural cell death due to NMDA neurotoxicity and oxidative stress in a mouse model of normal tension glaucoma. Cell Death Differ. 2013, 20, 1250–1256. [Google Scholar] [CrossRef]
- Liu, S.B.; Zhang, N.; Guo, Y.Y.; Zhao, R.; Shi, T.Y.; Feng, S.F.; Wang, S.Q.; Yang, Q.; Li, X.Q.; Wu, Y.M.; et al. G-protein-coupled receptor 30 mediates rapid neuroprotective effects of estrogen via depression of NR2B-containing NMDA receptors. J. Neurosci. 2012, 32, 4887–4900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, H.; Lethaby, A. Phytoestrogens for menopausal vasomotor symptoms: A Cochrane review summary. Maturitas 2014, 78, 79–81. [Google Scholar] [CrossRef]
- Zand, R.S.; Jenkins, D.J.; Diamandis, E.P. Steroid hormone activity of flavonoids and related compounds. Breast Cancer Res. Treat. 2000, 62, 35–49. [Google Scholar] [CrossRef]
- Oseni, T.; Patel, R.; Pyle, J.; Jordan, V.C. Selective estrogen receptor modulators and phytoestrogens. Planta Med. 2008, 74, 1656–1665. [Google Scholar] [CrossRef] [Green Version]
- Clement, Y.N.; Onakpoya, I.; Hung, S.K.; Ernst, E. Effects of herbal and dietary supplements on cognition in menopause: A systematic review. Maturitas 2011, 68, 256–263. [Google Scholar] [CrossRef]
- Lim, D.W.; Lee, C.; Kim, I.H.; Kim, Y.T. Anti-inflammatory effects of total isoflavones from Pueraria lobata on cerebral ischemia in rats. Molecules 2013, 18, 10404–10412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganai, A.A.; Khan, A.A.; Malik, Z.A.; Farooqi, H. Genistein modulates the expression of NF-kappaB and MAPK (p-38 and ERK1/2), thereby attenuating d-Galactosamine induced fulminant hepatic failure in Wistar rats. Toxicol. Appl. Pharmacol. 2015, 283, 139–146. [Google Scholar] [CrossRef]
- Yu, J.; Bi, X.; Yu, B.; Chen, D. Isoflavones: Anti-Inflammatory Benefit and Possible Caveats. Nutrients 2016, 8, 361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, G.A.; Park, S. Antioxidant action of soy isoflavones on oxidative stress and antioxidant enzyme activities in exercised rats. Nutr Res. Pract. 2014, 8, 618–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, J.; Yang, F.; Gong, C.; Shi, X.; Wang, G. Protective effect of daidzein against streptozotocin-induced Alzheimer’s disease via improving cognitive dysfunction and oxidative stress in rat model. J. Biochem. Mol. Toxicol. 2019, 33, e22319. [Google Scholar] [CrossRef]
- Chatterjee, G.; Roy, D.; Khemka, V.K.; Chattopadhyay, M.; Chakrabarti, S. Genistein, the Isoflavone in Soybean, Causes Amyloid Beta Peptide Accumulation in Human Neuroblastoma Cell Line: Implications in Alzheimer’s Disease. Aging Dis. 2015, 6, 456–465. [Google Scholar] [CrossRef] [Green Version]
- Bagheri, M.; Joghataei, M.T.; Mohseni, S.; Roghani, M. Genistein ameliorates learning and memory deficits in amyloid beta(1-40) rat model of Alzheimer’s disease. Neurobiol. Learn. Mem. 2011, 95, 270–276. [Google Scholar] [CrossRef] [Green Version]
- Xi, Y.D.; Li, X.Y.; Ding, J.; Yu, H.L.; Ma, W.W.; Yuan, L.H.; Wu, J.; Xiao, R. Soy isoflavone alleviates Abeta1-42-induced impairment of learning and memory ability through the regulation of RAGE/LRP-1 in neuronal and vascular tissue. Curr. Neurovasc. Res. 2013, 10, 144–156. [Google Scholar] [CrossRef] [PubMed]
- Devi, K.P.; Shanmuganathan, B.; Manayi, A.; Nabavi, S.F.; Nabavi, S.M. Molecular and Therapeutic Targets of Genistein in Alzheimer’s Disease. Mol. Neurobiol. 2017, 54, 7028–7041. [Google Scholar] [CrossRef] [PubMed]
- Son, S.H.; Do, J.M.; Yoo, J.N.; Lee, H.W.; Kim, N.K.; Yoo, H.S.; Gee, M.S.; Kim, J.H.; Seong, J.H.; Inn, K.S.; et al. Identification of ortho catechol-containing isoflavone as a privileged scaffold that directly prevents the aggregation of both amyloid beta plaques and tau-mediated neurofibrillary tangles and its in vivo evaluation. Bioorg. Chem. 2021, 113, 105022. [Google Scholar] [CrossRef]
- Lazennec, G.; Bresson, D.; Lucas, A.; Chauveau, C.; Vignon, F. ER beta inhibits proliferation and invasion of breast cancer cells. Endocrinology 2001, 142, 4120–4130. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.S.; Hasana, S.; Hossain, M.F.; Islam, M.S.; Behl, T.; Perveen, A.; Hafeez, A.; Ashraf, G.M. Molecular Genetics of Early- and Late-Onset Alzheimer’s Disease. Curr. Gene Ther. 2021, 21, 43–52. [Google Scholar] [CrossRef]
- Selkoe, D.J. Alzheimer’s disease: Genotypes, phenotypes, and treatments. Science 1997, 275, 630–631. [Google Scholar] [CrossRef] [PubMed]
- Rebeck, G.W.; Reiter, J.S.; Strickland, D.K.; Hyman, B.T. Apolipoprotein E in sporadic Alzheimer’s disease: Allelic variation and receptor interactions. Neuron 1993, 11, 575–580. [Google Scholar] [CrossRef]
- Strittmatter, W.J.; Saunders, A.M.; Schmechel, D.; Pericak-Vance, M.; Enghild, J.; Salvesen, G.S.; Roses, A.D. Apolipoprotein E: High-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 1977–1981. [Google Scholar] [CrossRef] [Green Version]
- Castellano, J.M.; Kim, J.; Stewart, F.R.; Jiang, H.; DeMattos, R.B.; Patterson, B.W.; Fagan, A.M.; Morris, J.C.; Mawuenyega, K.G.; Cruchaga, C.; et al. Human apoE isoforms differentially regulate brain amyloid-beta peptide clearance. Sci. Transl. Med. 2011, 3, 89ra57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenberg, R.N.; Lambracht-Washington, D.; Yu, G.; Xia, W. Genomics of Alzheimer Disease: A Review. JAMA Neurol. 2016, 73, 867–874. [Google Scholar] [CrossRef]
- Chen, L.H.; Fan, Y.H.; Kao, P.Y.; Ho, D.T.; Ha, J.C.; Chu, L.W.; Song, Y.Q. Genetic Polymorphisms in Estrogen Metabolic Pathway Associated with Risks of Alzheimer’s Disease: Evidence from a Southern Chinese Population. J. Am. Geriatr. Soc. 2017, 65, 332–339. [Google Scholar] [CrossRef]
- Song, Y.; Lu, Y.; Liang, Z.; Yang, Y.; Liu, X. Association between rs10046, rs1143704, rs767199, rs727479, rs1065778, rs1062033, rs1008805, and rs700519 polymorphisms in aromatase (CYP19A1) gene and Alzheimer’s disease risk: A systematic review and meta-analysis involving 11,051 subjects. Neurol. Sci. 2019, 40, 2515–2527. [Google Scholar] [CrossRef]
- Andrews, S.J.; Fulton-Howard, B.; Goate, A. Interpretation of risk loci from genome-wide association studies of Alzheimer’s disease. Lancet Neurol. 2020, 19, 326–335. [Google Scholar] [CrossRef]
- Lin, E.; Lin, C.H.; Lane, H.Y. Deep Learning with Neuroimaging and Genomics in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22. [Google Scholar] [CrossRef] [PubMed]
- Hersi, M.; Irvine, B.; Gupta, P.; Gomes, J.; Birkett, N.; Krewski, D. Risk factors associated with the onset and progression of Alzheimer’s disease: A systematic review of the evidence. Neurotoxicology 2017, 61, 143–187. [Google Scholar] [CrossRef] [PubMed]
- Kim, C. Does menopause increase diabetes risk? Strategies for diabetes prevention in midlife women. Womens Health 2012, 8, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Tini, G.; Scagliola, R.; Monacelli, F.; La Malfa, G.; Porto, I.; Brunelli, C.; Rosa, G.M. Alzheimer’s Disease and Cardiovascular Disease: A Particular Association. Cardiol. Res. Pract. 2020, 2020, 2617970. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, Y.-J.; Lin, C.-H.; Lane, H.-Y. From Menopause to Neurodegeneration—Molecular Basis and Potential Therapy. Int. J. Mol. Sci. 2021, 22, 8654. https://doi.org/10.3390/ijms22168654
Cheng Y-J, Lin C-H, Lane H-Y. From Menopause to Neurodegeneration—Molecular Basis and Potential Therapy. International Journal of Molecular Sciences. 2021; 22(16):8654. https://doi.org/10.3390/ijms22168654
Chicago/Turabian StyleCheng, Yu-Jung, Chieh-Hsin Lin, and Hsien-Yuan Lane. 2021. "From Menopause to Neurodegeneration—Molecular Basis and Potential Therapy" International Journal of Molecular Sciences 22, no. 16: 8654. https://doi.org/10.3390/ijms22168654