
Is Multiple System Atrophy a Prion-like Disorder?


{kind=link}
Abstract
:1. Introduction
2. Self-Propagation of Prionoids
3. In Vivo and In Vitro Data
4. Prion-Like Properties of αSyn
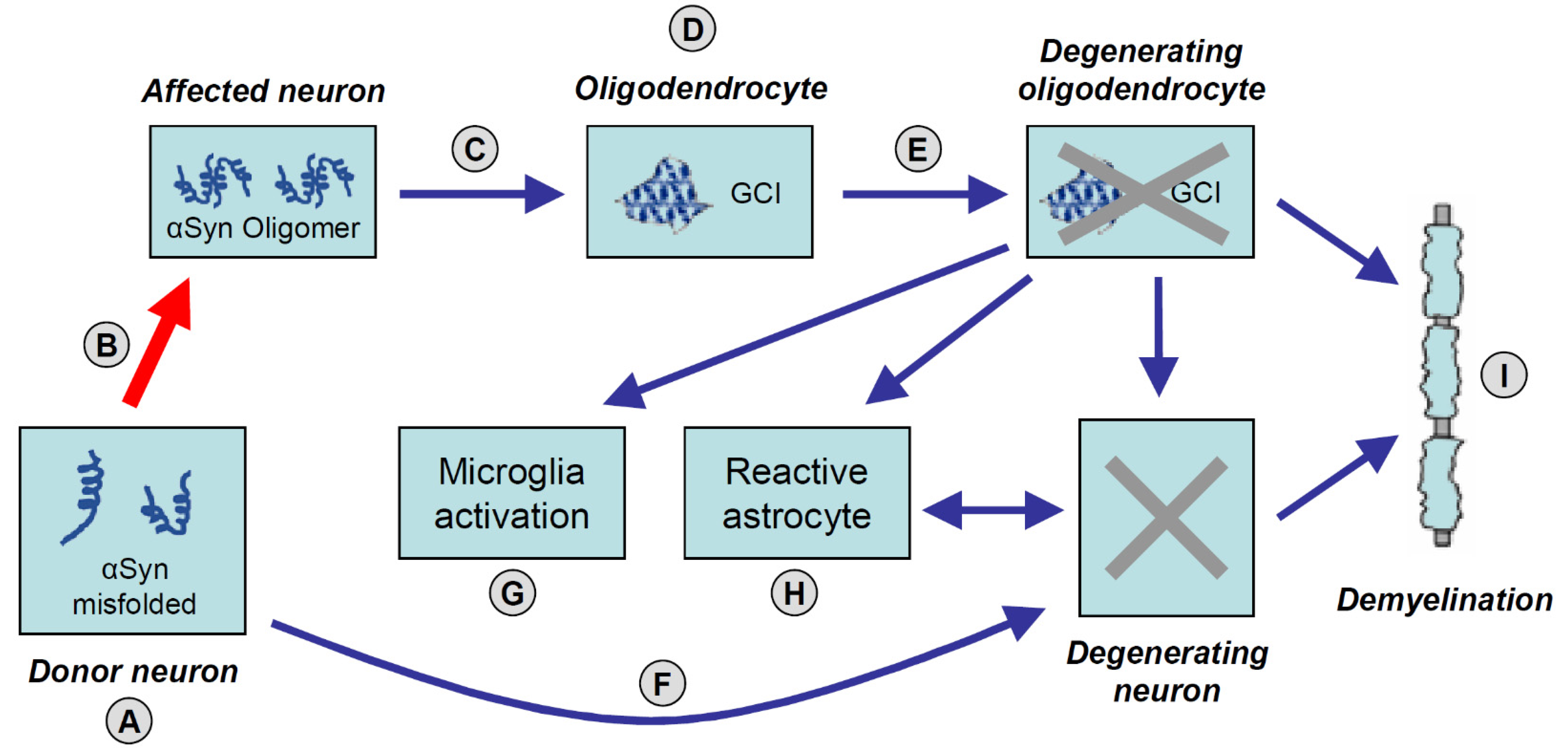
5. Multiple System Atrophy: A “Prion” Disease?
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Aβ | β-amyloid |
| αSyn | α-synuclein |
| CNS | central nervous system |
| DLB | Lewy body dementia |
| GCIs | glial cytoplasmic inclusions |
| LB | Lewy body |
| LB | Lewy body |
| MSA | Multiple system atrophy |
| PD | Parkinson’s disease |
| PFFs | pre-formed fibrils |
| PI | post-injection |
| PrPC | cellular prion protein |
| PrPSc | pathologic PrP isoform |
| tg | transgenic |
| WT | wild-type |
References
- Jellinger, K.A. Neuropathological findings in multiple system atrophy with cognitive impairment. J. Neural Transm. 2020, 127, 1031–1039. [Google Scholar] [CrossRef] [PubMed]
- Krismer, F.; Wenning, G.K. Multiple system atrophy: Insights into a rare and debilitating movement disorder. Nat. Rev. Neurol. 2017, 13, 232–243. [Google Scholar] [CrossRef]
- Gilman, S.; Wenning, G.K.; Low, P.A.; Brooks, D.J.; Mathias, C.J.; Trojanowski, J.Q.; Wood, N.W.; Colosimo, C.; Durr, A.; Fowler, C.J.; et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology 2008, 71, 670–676. [Google Scholar] [CrossRef] [PubMed]
- Campese, N.; Fanciulli, A.; Stefanova, N.; Haybaeck, J.; Kiechl, S.; Wenning, G.K. Neuropathology of multiple system atrophy: Kurt Jellinger’s legacy. J. Neural Transm. 2021. [Google Scholar] [CrossRef]
- Trojanowski, J.Q.; Revesz, T. Proposed neuropathological criteria for the post mortem diagnosis of multiple system atrophy. Neuropathol. Appl. Neurobiol. 2007, 33, 615–620. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Goedert, M. Synucleinopathies: Past, present and future. Neuropathol. Appl. Neurobiol. 2016, 42, 3–5. [Google Scholar] [CrossRef] [Green Version]
- Koga, S.; Dickson, D.W. Recent advances in neuropathology, biomarkers and therapeutic approach of multiple system atrophy. J. Neurol. Neurosurg. Psychiatry 2018, 89, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Kaji, S.; Maki, T.; Kinoshita, H.; Uemura, N.; Ayaki, T.; Kawamoto, Y.; Furuta, T.; Urushitani, M.; Hasegawa, M.; Kinoshita, Y.; et al. Pathological endogenous alpha-synuclein accumulation in oligodendrocyte precursor cells potentially induces inclusions in multiple system atrophy. Stem Cell Rep. 2018, 10, 356–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cykowski, M.D.; Coon, E.A.; Powell, S.Z.; Jenkins, S.M.; Benarroch, E.E.; Low, P.A.; Schmeichel, A.M.; Parisi, J.E. Expanding the spectrum of neuronal pathology in multiple system atrophy. Brain 2015, 138, 2293–2309. [Google Scholar] [CrossRef] [PubMed]
- Monzio Compagnoni, G.; Di Fonzo, A. Understanding the pathogenesis of multiple system atrophy: State of the art and future perspectives. Acta Neuropathol. Commun. 2019, 7, 113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jellinger, K.A. Multiple system atrophy: An oligodendroglioneural synucleinopathy. J. Alzheimers Dis. 2018, 62, 1141–1179. [Google Scholar] [CrossRef] [Green Version]
- Herrera-Vaquero, M.; Heras-Garvin, A.; Krismer, F.; Deleanu, R.; Boesch, S.; Wenning, G.K.; Stefanova, N. Signs of early cellular dysfunction in multiple system atrophy. Neuropathol. Appl. Neurobiol. 2021, 47, 268–282. [Google Scholar] [CrossRef] [PubMed]
- Bettencourt, C.; Miki, Y.; Piras, I.S.; de Silva, R.; Foti, S.C.; Talboom, J.S.; Revesz, T.; Lashley, T.; Balazs, R.; Viré, E.; et al. MOBP and HIP1 in multiple system atrophy: New alpha-synuclein partners in glial cytoplasmic inclusions implicated in the disease pathogenesis. Neuropathol. Appl. Neurobiol. 2021, 47, 640–652. [Google Scholar] [CrossRef] [PubMed]
- Peelaerts, W.; Bousset, L.; Baekelandt, V.; Melki, R. Alpha-synuclein strains and seeding in Parkinson’s disease, incidental Lewy body disease, dementia with Lewy bodies and multiple system atrophy: Similarities and differences. Cell Tissue Res. 2018, 373, 195–212. [Google Scholar] [CrossRef]
- Vargas, J.Y.; Grudina, C.; Zurzolo, C. The prion-like spreading of alpha-synuclein: From in vitro to in vivo models of Parkinson’s disease. Ageing Res. Rev. 2019, 50, 89–101. [Google Scholar] [CrossRef] [PubMed]
- Vasili, E.; Dominguez-Meijide, A.; Outeiro, T.F. Spreading of alpha-synuclein and tau: A systematic comparison of the mechanisms involved. Front. Mol. Neurosci. 2019, 12, 107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarutani, A.; Arai, T.; Murayama, S.; Hisanaga, S.I.; Hasegawa, M. Potent prion-like behaviors of pathogenic alpha-synuclein and evaluation of inactivation methods. Acta Neuropathol. Commun. 2018, 6, 29. [Google Scholar] [CrossRef]
- Visanji, N.P.; Brooks, P.L.; Hazrati, L.N.; Lang, A.E. The prion hypothesis in Parkinson’s disease: Braak to the future. Acta Neuropathol. Commun. 2013, 1, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhillon, J.S.; Trejo-Lopez, J.A.; Riffe, C.; McFarland, N.R.; Hiser, W.M.; Giasson, B.I.; Yachnis, A.T. Dissecting alpha-synuclein inclusion pathology diversity in multiple system atrophy: Implications for the prion-like transmission hypothesis. Lab. Investig. 2019, 99, 982–992. [Google Scholar] [CrossRef]
- Steiner, J.A.; Quansah, E.; Brundin, P. The concept of alpha-synuclein as a prion-like protein: Ten years after. Cell Tissue Res. 2018, 373, 161–173. [Google Scholar] [CrossRef]
- Karpowicz, R.J., Jr.; Trojanowski, J.Q.; Lee, V.M. Transmission of alpha-synuclein seeds in neurodegenerative disease: Recent developments. Lab. Investig. 2019, 99, 971–981. [Google Scholar] [CrossRef] [PubMed]
- Brás, I.C.; Outeiro, T.F. Alpha-synuclein: Mechanisms of release and pathology progression in synucleinopathies. Cells 2021, 10, 375. [Google Scholar] [CrossRef]
- Jaunmuktane, Z.; Brandner, S. The role of prion-like mechanisms in neurodegenerative diseases. Neuropathol. Appl. Neurobiol. 2020, 46, 522–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veys, L.; Van Houcke, J.; Aerts, J.; Van Pottelberge, S.; Mahieu, M.; Coens, A.; Melki, R.; Moechars, D.; De Muynck, L.; De Groef, L. Absence of uptake and prion-like spreading of alpha-synuclein and tau after intravitreal injection of preformed fibrils. Front. Aging Neurosci. 2021, 12, 614587. [Google Scholar] [CrossRef] [PubMed]
- Woerman, A.L.; Patel, S.; Kazmi, S.A.; Oehler, A.; Lee, J.; Mordes, D.A.; Olson, S.H.; Prusiner, S.B. Kinetics of alpha-synuclein prions preceding neuropathological inclusions in multiple system atrophy. PLoS Pathog. 2020, 16, e1008222. [Google Scholar] [CrossRef] [Green Version]
- Prusiner, S.B.; Woerman, A.L.; Mordes, D.A.; Watts, J.C.; Rampersaud, R.; Berry, D.B.; Patel, S.; Oehler, A.; Lowe, J.K.; Kravitz, S.N.; et al. Evidence for alpha-synuclein prions causing multiple system atrophy in humans with parkinsonism. Proc. Natl. Acad. Sci. USA 2015, 112, E5308–E5317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woerman, A.L.; Watts, J.C.; Aoyagi, A.; Giles, K.; Middleton, L.T.; Prusiner, S.B. Alpha-synuclein: Multiple system atrophy prions. Cold Spring Harb. Perspect. Med. 2018, 8, a024588. [Google Scholar] [CrossRef]
- Woerman, A.L.; Oehler, A.; Kazmi, S.A.; Lee, J.; Halliday, G.M.; Middleton, L.T.; Gentleman, S.M.; Mordes, D.A.; Spina, S.; Grinberg, L.T.; et al. Multiple system atrophy prions retain strain specificity after serial propagation in two different Tg(SNCA*A53T) mouse lines. Acta Neuropathol. 2019, 137, 437–454. [Google Scholar] [CrossRef]
- Watts, J.C.; Giles, K.; Oehler, A.; Middleton, L.; Dexter, D.T.; Gentleman, S.M.; DeArmond, S.J.; Prusiner, S.B. Transmission of multiple system atrophy prions to transgenic mice. Proc. Natl. Acad. Sci. USA 2013, 110, 19555–19560. [Google Scholar] [CrossRef] [Green Version]
- Chu, Y.; Kordower, J.H. The prion hypothesis of Parkinson’s disease. Curr. Neurol. Neurosci. Rep. 2015, 15, 28. [Google Scholar] [CrossRef]
- Ma, J.; Gao, J.; Wang, J.; Xie, A. Prion-like mechanisms in Parkinson’s disease. Front. Neurosci. 2019, 13, 552. [Google Scholar] [CrossRef] [PubMed]
- Melki, R. Alpha-synuclein and the prion hypothesis in Parkinson’s disease. Rev. Neurol. 2018, 174, 644–652. [Google Scholar] [CrossRef]
- Olanow, C.W. Do prions cause Parkinson disease? The evidence accumulates. Ann. Neurol. 2014, 75, 331–333. [Google Scholar] [CrossRef]
- Rey, N.L.; George, S.; Brundin, P. Review: Spreading the word: Precise animal models and validated methods are vital when evaluating prion-like behaviour of alpha-synuclein. Neuropathol. Appl. Neurobiol. 2016, 42, 51–76. [Google Scholar] [CrossRef] [PubMed]
- Tamguney, G.; Korczyn, A.D. A critical review of the prion hypothesis of human synucleinopathies. Cell Tissue Res. 2018, 373, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Leak, R.K.; Frosch, M.P.; Beach, T.G.; Halliday, G.M. Alpha-synuclein: Prion or prion-like? Acta Neuropathol. 2019, 138, 509–514. [Google Scholar] [CrossRef] [PubMed]
- Wenning, G.; Trojanowski, J.Q.; Kaufmann, H.; Wisniewski, T.; Rocca, W.A.; Low, P.A. Is multiple system atrophy an infectious disease? Ann. Neurol. 2018, 83, 10–12. [Google Scholar] [CrossRef] [Green Version]
- Meissner, W.G.; Fernagut, P.O.; Dehay, B.; Peran, P.; Traon, A.P.; Foubert-Samier, A.; Lopez Cuina, M.; Bezard, E.; Tison, F.; Rascol, O. Multiple system atrophy: Recent developments and future perspectives. Mov. Disord. 2019, 34, 1629–1642. [Google Scholar] [CrossRef]
- Scialò, C.; De Cecco, E.; Manganotti, P.; Legname, G. Prion and prion-like protein strains: Deciphering the molecular basis of heterogeneity in neurodegeneration. Viruses 2019, 11, 261. [Google Scholar] [CrossRef] [Green Version]
- Hass, E.W.; Sorrentino, Z.A.; Lloyd, G.M.; McFarland, N.R.; Prokop, S.; Giasson, B.I. Robust a-synuclein pathology in select brainstem neuronal populations is a potential instigator of multiple system atrophy. Acta Neuropathol. Commun. 2021, 9, 80. [Google Scholar] [CrossRef]
- Hass, E.W.; Sorrentino, Z.A.; Xia, Y.; Lloyd, G.M.; Trojanowski, J.Q.; Prokop, S.; Giasson, B.I. Disease-, region- and cell type specific diversity of alpha-synuclein carboxy terminal truncations in synucleinopathies. Acta Neuropathol. Commun. 2021, 9, 146. [Google Scholar] [CrossRef]
- Uchihara, T.; Giasson, B.I. Propagation of alpha-synuclein pathology: Hypotheses, discoveries, and yet unresolved questions from experimental and human brain studies. Acta Neuropathol. 2016, 131, 49–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makin, S. Pathology: The prion principle. Nature 2016, 538, S13–S16. [Google Scholar] [CrossRef]
- Brundin, P.; Melki, R. Prying into the prion hypothesis for Parkinson’s disease. J. Neurosci. 2017, 37, 9808–9818. [Google Scholar] [CrossRef] [PubMed]
- Surmeier, D.J.; Obeso, J.A.; Halliday, G.M. Parkinson’s disease is not simply a prion disorder. J. Neurosci. 2017, 37, 9799–9807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siddique, Y.H. Does human alpha synuclein behave like prions? CNS Neurol. Disord. Drug Targets 2021. [Google Scholar] [CrossRef]
- Zheng, H.; Shi, C.; Luo, H.; Fan, L.; Yang, Z.; Hu, X.; Zhang, Z.; Zhang, S.; Hu, Z.; Fan, Y.; et al. Alpha-synuclein in Parkinson’s disease: Does a prion-like mechanism of propagation from periphery to the brain play a role? Neuroscientist 2021, 27, 367–387. [Google Scholar] [CrossRef]
- Woerman, A.L.; Kazmi, S.A.; Patel, S.; Aoyagi, A.; Oehler, A.; Widjaja, K.; Mordes, D.A.; Olson, S.H.; Prusiner, S.B. Familial Parkinson’s point mutation abolishes multiple system atrophy prion replication. Proc. Natl. Acad. Sci. USA 2018, 115, 409–414. [Google Scholar] [CrossRef] [Green Version]
- Watts, J.C. Calling alpha-synuclein a prion is scientifically justifiable. Acta Neuropathol. 2019, 138, 505–508. [Google Scholar] [CrossRef]
- Prusiner, S.B. Novel proteinaceous infectious particles cause scrapie. Science 1982, 216, 136–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prusiner, S.B. Biology and genetics of prions causing neurodegeneration. Annu. Rev. Genet. 2013, 47, 601–623. [Google Scholar] [CrossRef] [Green Version]
- Colin, M.; Dujardin, S.; Schraen-Maschke, S.; Meno-Tetang, G.; Duyckaerts, C.; Courade, J.P.; Buee, L. From the prion-like propagation hypothesis to therapeutic strategies of anti-tau immunotherapy. Acta Neuropathol. 2020, 139, 3–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghaemmaghami, S. Biology and genetics of PrP prion strains. In Prion Diseases; Prusiner, S.B., Ed.; Cold Spring Harbor Laboratory Press: Long Island, NY, USA, 2017; pp. 45–56. [Google Scholar]
- Tanaka, M.; Chien, P.; Naber, N.; Cooke, R.; Weissman, J.S. Conformational variations in an infectious protein determine prion strain differences. Nature 2004, 428, 323–328. [Google Scholar] [CrossRef]
- Telling, G.C.; Parchi, P.; DeArmond, S.J.; Cortelli, P.; Montagna, P.; Gabizon, R.; Mastrianni, J.; Lugaresi, E.; Gambetti, P.; Prusiner, S.B. Evidence for the conformation of the pathologic isoform of the prion protein enciphering and propagating prion diversity. Science 1996, 274, 2079–2082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lau, A.; So, R.W.; Lau, H.H.; Sang, J.C.; Ruiz-Riquelme, A.; Fleck, S.C.; Stuart, E.; Menon, S.; Visanji, N.P.; Meisl, G.; et al. Alpha-synuclein strains target distinct brain regions and cell types. Nat. Neurosci. 2020, 23, 21–31. [Google Scholar] [CrossRef]
- Prusiner, S.B. An introduction to prion diseases. In Prion Diseases; Prusiner, S.B., Ed.; Cold Spring Harbor Laboratory Press: Long Island, NY, USA, 2017; pp. 1–29. [Google Scholar]
- Woerman, A.L. Strain diversity in neurodegenerative disease: An argument for a personalized medicine approach to diagnosis and treatment. Acta Neuropathol. 2021, 142, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Uemura, N.; Uemura, M.T.; Luk, K.C.; Lee, V.M.; Trojanowski, J.Q. Cell-to-cell transmission of tau and alpha-synuclein. Trends Mol. Med. 2020, 26, 936–952. [Google Scholar] [CrossRef]
- Holec, S.A.; Woerman, A.L. Evidence of distinct alpha-synuclein strains underlying disease heterogeneity. Acta Neuropathol. 2020, 142, 73–86. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, Z.A.; Giasson, B.I. The emerging role of alpha-synuclein truncation in aggregation and disease. J. Biol. Chem. 2020, 295, 10224–10244. [Google Scholar] [CrossRef] [PubMed]
- Schweighauser, M.; Shi, Y.; Tarutani, A.; Kametani, F.; Murzin, A.G.; Ghetti, B.; Matsubara, T.; Tomita, T.; Ando, T.; Hasegawa, K.; et al. Structures of alpha-synuclein filaments from multiple system atrophy. Nature 2020, 585, 464–469. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Guo, J.J.; Su, J.H.; Svanbergsson, A.; Yuan, L.; Haikal, C.; Li, W.; Gouras, G.; Li, J.Y. Differential seeding and propagating efficiency of a-synuclein strains generated in different conditions. Transl. Neurodegener. 2021, 10, 20. [Google Scholar] [CrossRef] [PubMed]
- Van der Perren, A.; Gelders, G.; Fenyi, A.; Bousset, L.; Brito, F.; Peelaerts, W.; Van den Haute, C.; Gentleman, S.; Melki, R.; Baekelandt, V. The structural differences between patient-derived alpha-synuclein strains dictate characteristics of Parkinson’s disease, multiple system atrophy and dementia with Lewy bodies. Acta Neuropathol. 2020, 139, 977–1000. [Google Scholar] [CrossRef]
- Guo, J.L.; Lee, V.M. Cell-to-cell transmission of pathogenic proteins in neurodegenerative diseases. Nat. Med. 2014, 20, 130–138. [Google Scholar] [CrossRef] [Green Version]
- Ugalde, C.L.; Finkelstein, D.I.; Lawson, V.A.; Hill, A.F. Pathogenic mechanisms of prion protein, amyloid-beta and alpha-synuclein misfolding: The prion concept and neurotoxicity of protein oligomers. J. Neurochem. 2016, 139, 162–180. [Google Scholar] [CrossRef]
- Chu, Y.; Muller, S.; Tavares, A.; Barret, O.; Alagille, D.; Seibyl, J.; Tamagnan, G.; Marek, K.; Luk, K.C.; Trojanowski, J.Q.; et al. Intrastriatal alpha-synuclein fibrils in monkeys: Spreading, imaging and neuropathological changes. Brain 2019, 142, 3565–3579. [Google Scholar] [CrossRef]
- Luk, K.C.; Kehm, V.; Carroll, J.; Zhang, B.; O’Brien, P.; Trojanowski, J.Q.; Lee, V.M. Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 2012, 338, 949–953. [Google Scholar] [CrossRef] [Green Version]
- Bassil, F.; Brown, H.J.; Pattabhiraman, S.; Iwasyk, J.E.; Maghames, C.M.; Meymand, E.S.; Cox, T.O.; Riddle, D.M.; Zhang, B.; Trojanowski, J.Q.; et al. Amyloid-beta (Abeta) plaques promote seeding and spreading of alpha-synuclein and tau in a mouse model of Lewy body disorders with Abeta pathology. Neuron 2020, 105, 260–275.e6. [Google Scholar] [CrossRef]
- Badiola, N.; de Oliveira, R.M.; Herrera, F.; Guardia-Laguarta, C.; Gonçalves, S.A.; Pera, M.; Suárez-Calvet, M.; Clarimon, J.; Outeiro, T.F.; Lleó, A. Tau enhances alpha-synuclein aggregation and toxicity in cellular models of synucleinopathy. PLoS ONE 2011, 6, e26609. [Google Scholar] [CrossRef]
- Masuda-Suzukake, M.; Hasegawa, M. Prion-like propagation of pathological alpha-synuclein in vivo. Yakugaku Zasshi 2019, 139, 1007–1013. [Google Scholar] [CrossRef] [PubMed]
- Bernis, M.E.; Babila, J.T.; Breid, S.; Wusten, K.A.; Wullner, U.; Tamguney, G. Prion-like propagation of human brain-derived alpha-synuclein in transgenic mice expressing human wild-type alpha-synuclein. Acta Neuropathol. Commun. 2015, 3, 75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Candelise, N.; Schmitz, M.; Llorens, F.; Villar-Pique, A.; Cramm, M.; Thom, T.; da Silva Correia, S.M.; da Cunha, J.E.; Mobius, W.; Outeiro, T.F.; et al. Seeding variability of different alpha synuclein strains in synucleinopathies. Ann. Neurol. 2019, 85, 691–703. [Google Scholar] [CrossRef] [PubMed]
- Shearer, L.J.; Petersen, N.O.; Woodside, M.T. Internalization of alpha-synuclein oligomers into SH-SY5Y cells. Biophys. J. 2021, 120, 877–885. [Google Scholar] [CrossRef]
- Oueslati, A.; Ximerakis, M.; Vekrellis, K. Protein transmission, seeding and degradation: Key steps for a-synuclein prion-like propagation. Exp. Neurobiol. 2014, 23, 324–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trinkaus, V.A.; Riera-Tur, I.; Martínez-Sánchez, A.; Bäuerlein, F.J.; Guo, Q.; Arzberger, T.; Baumeister, W.; Dudanova, I.; Hipp, M.S.; Hartl, F.U.; et al. In situ architecture of neuronal alpha-synuclein inclusions. Nat. Commun. 2021, 12, 2110. [Google Scholar] [CrossRef]
- Jan, A.; Gonçalves, N.P.; Vaegter, C.B.; Jensen, P.H.; Ferreira, N. The prion-like spreading of alpha-synuclein in Parkinson’s disease: Update on models and hypotheses. Int. J. Mol. Sci. 2021, 22, 8338. [Google Scholar] [CrossRef]
- Thom, T.; Schmitz, M.; Fischer, A.L.; Correia, A.; Correia, S.; Llorens, F.; Pique, A.V.; Möbius, W.; Domingues, R.; Zafar, S.; et al. Cellular prion protein mediates alpha-synuclein uptake, localization, and toxicity in vitro and in vivo. Mov. Disord. 2021. [Google Scholar] [CrossRef]
- Hijaz, B.A.; Volpicelli-Daley, L.A. Initiation and propagation of alpha-synuclein aggregation in the nervous system. Mol. Neurodegener. 2020, 15, 19. [Google Scholar] [CrossRef]
- Ayers, J.I.; Paras, N.A.; Prusiner, S.B. Expanding spectrum of prion diseases. Emerg. Top. Life Sci. 2020, 4, 155–167. [Google Scholar]
- Scheckel, C.; Aguzzi, A. Prions, prionoids and protein misfolding disorders. Nat. Rev. Genet. 2018, 19, 405–418. [Google Scholar] [CrossRef] [Green Version]
- Kraus, A.; Groveman, B.R.; Caughey, B. Prions and the potential transmissibility of protein misfolding diseases. Annu. Rev. Microbiol. 2013, 67, 543–564. [Google Scholar] [CrossRef] [Green Version]
- Walker, L.C.; Jucker, M. Neurodegenerative diseases: Expanding the prion concept. Annu. Rev. Neurosci. 2015, 38, 87–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collinge, J. Mammalian prions and their wider relevance in neurodegenerative diseases. Nature 2016, 539, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Irwin, D.J.; Abrams, J.Y.; Schonberger, L.B.; Leschek, E.W.; Mills, J.L.; Lee, V.M.; Trojanowski, J.Q. Evaluation of potential infectivity of Alzheimer and Parkinson disease proteins in recipients of cadaver-derived human growth hormone. JAMA Neurol. 2013, 70, 462–468. [Google Scholar] [CrossRef] [PubMed]
- Caughey, B.; Kraus, A. Transmissibility versus Pathogenicity of Self-Propagating Protein Aggregates. Viruses 2019, 11, 1044. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, D.L.; Barria, M.A. Prion diseases: A unique transmissible agent or a model for neurodegenerative diseases? Biomolecules 2021, 11, 207. [Google Scholar] [CrossRef]
- Schwarzman, A.L.; Senkevich, K.A.; Emelyanov, A.K.; Pchelina, S.N. Prion properties of alpha-synuclein. Mol. Biol. 2019, 53, 335–341. [Google Scholar] [CrossRef]
- Watts, J.C.; Prusiner, S.B. Beta-Amyloid prions and the pathobiology of Alzheimer’s disease. Cold Spring Harb. Perspect. Med. 2018, 8, a023507. [Google Scholar] [CrossRef] [Green Version]
- Aguzzi, A. Cell biology: Beyond the prion principle. Nature 2009, 459, 924–925. [Google Scholar] [CrossRef]
- Verma, A. Prions, prion-like prionoids, and neurodegenerative disorders. Ann. Indian Acad Neurol. 2016, 19, 169–174. [Google Scholar] [CrossRef]
- Wells, C.; Brennan, S.E.; Keon, M.; Saksena, N.K. Prionoid proteins in the pathogenesis of neurodegenerative diseases. Front. Mol. Neurosci. 2019, 12, 271. [Google Scholar] [CrossRef]
- Mavroeidi, P.; Xilouri, M. Neurons and glia interplay in alpha-synucleinopathies. Int. J. Mol. Sci. 2021, 22, 4994. [Google Scholar] [CrossRef]
- Jucker, M.; Walker, L.C. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 2013, 501, 45–51. [Google Scholar] [CrossRef] [Green Version]
- Prusiner, S.B. Cell biology. A unifying role for prions in neurodegenerative diseases. Science 2012, 336, 1511–1513. [Google Scholar] [CrossRef] [Green Version]
- Duyckaerts, C.; Clavaguera, F.; Potier, M.C. The prion-like propagation hypothesis in Alzheimer’s and Parkinson’s disease. Curr. Opin. Neurol. 2019, 32, 266–271. [Google Scholar] [CrossRef] [PubMed]
- Heras-Garvin, A.; Stefanova, N. From synaptic protein to prion: The long and controversial journey of alpha-synuclein. Front. Synaptic Neurosci. 2020, 12, 584536. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Masuda-Suzukake, M.; Falcon, B. Like prions: The propagation of aggregated tau and alpha-synuclein in neurodegeneration. Brain 2017, 140, 266–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kordower, J.H.; Chu, Y.; Hauser, R.A.; Freeman, T.B.; Olanow, C.W. Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nat. Med. 2008, 14, 504–506. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Y.; Englund, E.; Holton, J.L.; Soulet, D.; Hagell, P.; Lees, A.J.; Lashley, T.; Quinn, N.P.; Rehncrona, S.; Bjorklund, A.; et al. Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat. Med. 2008, 14, 501–503. [Google Scholar] [CrossRef]
- El-Agnaf, O.M.; Salem, S.A.; Paleologou, K.E.; Cooper, L.J.; Fullwood, N.J.; Gibson, M.J.; Curran, M.D.; Court, J.A.; Mann, D.M.; Ikeda, S.; et al. Alpha-synuclein implicated in Parkinson’s disease is present in extracellular biological fluids, including human plasma. FASEB J. 2003, 17, 1945–1947. [Google Scholar] [CrossRef]
- Hasegawa, M.; Nonaka, T.; Masuda-Suzukake, M. Alpha-synuclein: Experimental pathology. Cold Spring Harb. Perspect. Med. 2016, 6, a024273. [Google Scholar] [CrossRef] [Green Version]
- Henderson, M.X.; Cornblath, E.J.; Darwich, A.; Zhang, B.; Brown, H.; Gathagan, R.J.; Sandler, R.M.; Bassett, D.S.; Trojanowski, J.Q.; Lee, V.M. Spread of alpha-synuclein pathology through the brain connectome is modulated by selective vulnerability and predicted by network analysis. Nat. Neurosci. 2019, 22, 1248–1257. [Google Scholar] [CrossRef]
- Dehay, B.; Vila, M.; Bezard, E.; Brundin, P.; Kordower, J.H. Alpha-synuclein propagation: New insights from animal models. Mov. Disord. 2016, 31, 161–168. [Google Scholar] [CrossRef]
- Hasegawa, M.; Nonaka, T.; Masuda-Suzukake, M. Prion-like mechanisms and potential therapeutic targets in neurodegenerative disorders. Pharmacol. Ther. 2017, 172, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Stopschinski, B.E.; Diamond, M.I. The prion model for progression and diversity of neurodegenerative diseases. Lancet Neurol. 2017, 16, 323–332. [Google Scholar] [CrossRef]
- Valdinocci, D.; Radford, R.A.; Siow, S.M.; Chung, R.S.; Pountney, D.L. Potential modes of intercellular alpha-synuclein transmission. Int. J. Mol. Sci. 2017, 18, 469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desplats, P.; Lee, H.J.; Bae, E.J.; Patrick, C.; Rockenstein, E.; Crews, L.; Spencer, B.; Masliah, E.; Lee, S.J. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc. Natl. Acad. Sci. USA 2009, 106, 13010–13015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woerman, A.L.; Stohr, J.; Aoyagi, A.; Rampersaud, R.; Krejciova, Z.; Watts, J.C.; Ohyama, T.; Patel, S.; Widjaja, K.; Oehler, A.; et al. Propagation of prions causing synucleinopathies in cultured cells. Proc. Natl. Acad. Sci. USA 2015, 112, E4949–E4958. [Google Scholar] [CrossRef] [Green Version]
- Dujardin, S.; Lécolle, K.; Caillierez, R.; Bégard, S.; Zommer, N.; Lachaud, C.; Carrier, S.; Dufour, N.; Aurégan, G.; Winderickx, J.; et al. Neuron-to-neuron wild-type tau protein transfer through a trans-synaptic mechanism: Relevance to sporadic tauopathies. Acta Neuropathol. Commun. 2014, 2, 14. [Google Scholar] [CrossRef]
- Clavaguera, F.; Akatsu, H.; Fraser, G.; Crowther, R.A.; Frank, S.; Hench, J.; Probst, A.; Winkler, D.T.; Reichwald, J.; Staufenbiel, M.; et al. Brain homogenates from human tauopathies induce tau inclusions in mouse brain. Proc. Natl. Acad. Sci. USA 2013, 110, 9535–9540. [Google Scholar] [CrossRef] [Green Version]
- Clavaguera, F.; Lavenir, I.; Falcon, B.; Frank, S.; Goedert, M.; Tolnay, M. “Prion-like” templated misfolding in tauopathies. Brain Pathol. 2013, 23, 342–349. [Google Scholar] [CrossRef]
- Olsson, T.T.; Klementieva, O.; Gouras, G.K. Prion-like seeding and nucleation of intracellular amyloid-beta. Neurobiol. Dis. 2018, 113, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Gomes, L.A.; Hipp, S.A.; Rijal Upadhaya, A.; Balakrishnan, K.; Ospitalieri, S.; Koper, M.J.; Largo-Barrientos, P.; Uytterhoeven, V.; Reichwald, J.; Rabe, S.; et al. Abeta-induced acceleration of Alzheimer-related tau-pathology spreading and its association with prion protein. Acta Neuropathol. 2019, 138, 913–941. [Google Scholar] [CrossRef]
- Fornari, S.; Schäfer, A.; Kuhl, E.; Goriely, A. Spatially-extended nucleation-aggregation-fragmentation models for the dynamics of prion-like neurodegenerative protein-spreading in the brain and its connectome. J. Theor. Biol. 2020, 486, 110102. [Google Scholar] [CrossRef] [PubMed]
- Masuda-Suzukake, M.; Nonaka, T.; Hosokawa, M.; Oikawa, T.; Arai, T.; Akiyama, H.; Mann, D.M.; Hasegawa, M. Prion-like spreading of pathological alpha-synuclein in brain. Brain 2013, 136, 1128–1138. [Google Scholar] [CrossRef]
- Masuda-Suzukake, M.; Nonaka, T.; Hosokawa, M.; Kubo, M.; Shimozawa, A.; Akiyama, H.; Hasegawa, M. Pathological alpha-synuclein propagates through neural networks. Acta Neuropathol. Commun. 2014, 2, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, H.K.; Ho, H.A.; Pérez-Acuña, D.; Lee, S.J. Modeling a-synuclein propagation with preformed fibril injections. J. Mov. Disord. 2019, 12, 139–151. [Google Scholar] [CrossRef]
- Reyes, J.F.; Sackmann, C.; Hoffmann, A.; Svenningsson, P.; Winkler, J.; Ingelsson, M.; Hallbeck, M. Binding of a-synuclein oligomers to Cx32 facilitates protein uptake and transfer in neurons and oligodendrocytes. Acta Neuropathol. 2019, 138, 23–47. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, N.; Gram, H.; Sorrentino, Z.A.; Gregersen, E.; Schmidt, S.I.; Reimer, L.; Betzer, C.; Perez-Gozalbo, C.; Beltoja, M.; Nagaraj, M.; et al. Multiple system atrophy-associated oligodendroglial protein p25α stimulates formation of novel α-synuclein strain with enhanced neurodegenerative potential. Acta Neuropathol. 2021, 142, 87–115. [Google Scholar] [CrossRef]
- Lohmann, S.; Bernis, M.E.; Tachu, B.J.; Ziemski, A.; Grigoletto, J.; Tamguney, G. Oral and intravenous transmission of alpha-synuclein fibrils to mice. Acta Neuropathol. 2019, 138, 515–533. [Google Scholar] [CrossRef] [Green Version]
- Sacino, A.N.; Brooks, M.; Thomas, M.A.; McKinney, A.B.; Lee, S.; Regenhardt, R.W.; McGarvey, N.H.; Ayers, J.I.; Notterpek, L.; Borchelt, D.R.; et al. Intramuscular injection of alpha-synuclein induces CNS alpha-synuclein pathology and a rapid-onset motor phenotype in transgenic mice. Proc. Natl. Acad. Sci. USA 2014, 111, 10732–10737. [Google Scholar] [CrossRef] [Green Version]
- Breid, S.; Bernis, M.E.; Babila, J.T.; Garza, M.C.; Wille, H.; Tamguney, G. Neuroinvasion of alpha-synuclein prionoids after intraperitoneal and intraglossal inoculation. J. Virol. 2016, 90, 9182–9193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayers, J.I.; Brooks, M.M.; Rutherford, N.J.; Howard, J.K.; Sorrentino, Z.A.; Riffe, C.J.; Giasson, B.I. Robust central nervous system pathology in transgenic mice following peripheral injection of alpha-synuclein fibrils. J. Virol. 2017, 91, e02095-16. [Google Scholar] [CrossRef] [Green Version]
- Van Den Berge, N.; Ferreira, N.; Gram, H.; Mikkelsen, T.W.; Alstrup, A.K.; Casadei, N.; Tsung-Pin, P.; Riess, O.; Nyengaard, J.R.; Tamguney, G.; et al. Evidence for bidirectional and trans-synaptic parasympathetic and sympathetic propagation of alpha-synuclein in rats. Acta Neuropathol. 2019, 138, 535–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Kwon, S.H.; Kam, T.I.; Panicker, N.; Karuppagounder, S.S.; Lee, S.; Lee, J.H.; Kim, W.R.; Kook, M.; Foss, C.A.; et al. Transneuronal propagation of pathologic alpha-synuclein from the gut to the brain models Parkinson’s disease. Neuron 2019, 103, 627–641.e7. [Google Scholar] [CrossRef] [PubMed]
- Breen, D.P.; Halliday, G.M.; Lang, A.E. Gut-brain axis and the spread of alpha-synuclein pathology: Vagal highway or dead end? Mov. Disord. 2019, 34, 307–316. [Google Scholar] [CrossRef]
- Recasens, A.; Dehay, B.; Bove, J.; Carballo-Carbajal, I.; Dovero, S.; Perez-Villalba, A.; Fernagut, P.O.; Blesa, J.; Parent, A.; Perier, C.; et al. Lewy body extracts from Parkinson disease brains trigger alpha-synuclein pathology and neurodegeneration in mice and monkeys. Ann. Neurol. 2014, 75, 351–362. [Google Scholar] [CrossRef]
- Kawakami, I.; Motoda, A.; Hashimoto, M.; Shimozawa, A.; Masuda-Suzukake, M.; Ohtani, R.; Takase, M.; Kumashiro, M.; Samejima, K.; Hasegawa, M. Progression of phosphorylated a-synuclein in Macaca fuscata. Brain Pathol. 2021, 31, e12952. [Google Scholar] [CrossRef] [PubMed]
- Shimozawa, A.; Ono, M.; Takahara, D.; Tarutani, A.; Imura, S.; Masuda-Suzukake, M.; Higuchi, M.; Yanai, K.; Hisanaga, S.I.; Hasegawa, M. Propagation of pathological alpha-synuclein in marmoset brain. Acta Neuropathol. Commun. 2017, 5, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogata, J.; Takemoto, D.; Shimonaka, S.; Imai, Y.; Hattori, N. Alpha-synuclein seeding assay using cultured cells. Methods Mol. Biol. 2021, 2322, 27–39. [Google Scholar]
- Aguzzi, A.; Lakkaraju, A.K. Cell biology of prions and prionoids: A status report. Trends Cell Biol. 2016, 26, 40–51. [Google Scholar] [CrossRef]
- Peng, C.; Gathagan, R.J.; Covell, D.J.; Medellin, C.; Stieber, A.; Robinson, J.L.; Zhang, B.; Pitkin, R.M.; Olufemi, M.F.; Luk, K.C.; et al. Cellular milieu imparts distinct pathological alpha-synuclein strains in alpha-synucleinopathies. Nature 2018, 557, 558–563. [Google Scholar] [CrossRef]
- Peng, C.; Gathagan, R.J.; Lee, V.M. Distinct alpha-synuclein strains and implications for heterogeneity among alpha-synucleinopathies. Neurobiol. Dis. 2018, 109, 209–218. [Google Scholar] [CrossRef]
- Uemura, N.; Uemura, M.T.; Lo, A.; Bassil, F.; Zhang, B.; Luk, K.C.; Lee, V.M.; Takahashi, R.; Trojanowski, J.Q. Slow progressive accumulation of oligodendroglial alpha-synuclein (alpha-syn) pathology in synthetic alpha-syn fibril-induced mouse models of synucleinopathy. J. Neuropathol. Exp. Neurol. 2019, 78, 877–890. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.W.; Johnson, J.M.; Solano, S.M.; Hollingsworth, Z.R.; Standaert, D.G.; Young, A.B. Absence of alpha-synuclein mRNA expression in normal and multiple system atrophy oligodendroglia. J. Neural Transm. 2005, 112, 1613–1624. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, T.R.; Holmes, B.B.; Furman, J.L.; Dhavale, D.D.; Su, B.W.; Song, E.S.; Cairns, N.J.; Kotzbauer, P.T.; Diamond, M.I. Parkinson’s disease and multiple system atrophy have distinct alpha-synuclein seed characteristics. J. Biol. Chem. 2019, 294, 1045–1058. [Google Scholar] [CrossRef] [Green Version]
- Lövestam, S.; Schweighauser, M.; Matsubara, T.; Murayama, S.; Tomita, T.; Ando, T.; Hasegawa, K.; Yoshida, M.; Tarutani, A.; Hasegawa, M.; et al. Seeded assembly in vitro does not replicate the structures of a-synuclein filaments from multiple system atrophy. FEBS Openbio 2021, 11, 999–1013. [Google Scholar] [CrossRef]
- Giasson, B.I.; Duda, J.E.; Quinn, S.M.; Zhang, B.; Trojanowski, J.Q.; Lee, V.M. Neuronal alpha-synucleinopathy with severe movement disorder in mice expressing A53T human alpha-synuclein. Neuron 2002, 34, 521–533. [Google Scholar] [CrossRef] [Green Version]
- Peelaerts, W.; Bousset, L.; Van der Perren, A.; Moskalyuk, A.; Pulizzi, R.; Giugliano, M.; Van den Haute, C.; Melki, R.; Baekelandt, V. Alpha-synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature 2015, 522, 340–344. [Google Scholar] [CrossRef]
- Bousset, L.; Melki, R. Infectious properties of protein aggregates involved in neurodegenerative diseases. Biol. Aujourdhui 2013, 207, 55–59. [Google Scholar] [CrossRef]
- Peelaerts, W.; Baekelandt, V. Alpha-synuclein strains and the variable pathologies of synucleinopathies. J. Neurochem. 2016, 139 (Suppl. S1), 256–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarutani, A.; Hasegawa, M. Prion-like propagation of alpha-synuclein in neurodegenerative diseases. Prog. Mol. Biol. Transl. Sci. 2019, 168, 323–348. [Google Scholar]
- Fearon, C.; Farrell, M.A. Disease-specific strains of alpha-synuclein in multiple system atrophy and Parkinson’s disease: But why? Mov. Disord. 2020, 35, 756–757. [Google Scholar] [CrossRef] [PubMed]
- Shahnawaz, M.; Mukherjee, A.; Pritzkow, S.; Mendez, N.; Rabadia, P.; Liu, X.; Hu, B.; Schmeichel, A.; Singer, W.; Wu, G.; et al. Discriminating alpha-synuclein strains in Parkinson’s disease and multiple system atrophy. Nature 2020, 578, 273–277. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, J.K.; Trejo-Lopez, J.A.; Riffe, C.; Levites, Y.; Sacino, A.N.; Borchelt, D.R.; Yachnis, A.Y.; Giasson, B.I. Comparative analyses of the in vivo induction and transmission of alpha-synuclein pathology in transgenic mice by MSA brain lysate and recombinant alpha-synuclein fibrils. Acta Neuropathol. Commun. 2019, 7, 80. [Google Scholar] [CrossRef]
- Sargent, D.; Verchere, J.; Lazizzera, C.; Gaillard, D.; Lakhdar, L.; Streichenberger, N.; Morignat, E.; Betemps, D.; Baron, T. ‘Prion-like’ propagation of the synucleinopathy of M83 transgenic mice depends on the mouse genotype and type of inoculum. J. Neurochem. 2017, 143, 126–135. [Google Scholar] [CrossRef] [Green Version]
- Krejciova, Z.; Carlson, G.A.; Giles, K.; Prusiner, S.B. Replication of multiple system atrophy prions in primary astrocyte cultures from transgenic mice expressing human alpha-synuclein. Acta Neuropathol. Commun. 2019, 7, 81. [Google Scholar] [CrossRef]
- Sacino, A.N.; Ayers, J.I.; Brooks, M.M.; Chakrabarty, P.; Hudson, V.J., 3rd; Howard, J.K.; Golde, T.E.; Giasson, B.I.; Borchelt, D.R. Non-prion-type transmission in A53T alpha-synuclein transgenic mice: A normal component of spinal homogenates from naive non-transgenic mice induces robust alpha-synuclein pathology. Acta Neuropathol. 2016, 131, 151–154. [Google Scholar] [CrossRef] [Green Version]
- Sanders, D.W.; Kaufman, S.K.; DeVos, S.L.; Sharma, A.M.; Mirbaha, H.; Li, A.; Barker, S.J.; Foley, A.C.; Thorpe, J.R.; Serpell, L.C.; et al. Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron 2014, 82, 1271–1288. [Google Scholar] [CrossRef] [Green Version]
- Aulic, S.; Masperone, L.; Narkiewicz, J.; Isopi, E.; Bistaffa, E.; Ambrosetti, E.; Pastore, B.; De Cecco, E.; Scaini, D.; Zago, P.; et al. Alpha-Synuclein amyloids hijack prion protein to gain cell entry, facilitate cell-to-cell spreading and block prion replication. Sci. Rep. 2017, 7, 10050. [Google Scholar] [CrossRef]
- De Cecco, E.; Legname, G. The role of the prion protein in the internalization of alpha-synuclein amyloids. Prion 2018, 12, 23–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haik, S.; Privat, N.; Adjou, K.T.; Sazdovitch, V.; Dormont, D.; Duyckaerts, C.; Hauw, J.J. Alpha-synuclein-immunoreactive deposits in human and animal prion diseases. Acta Neuropathol. 2002, 103, 516–520. [Google Scholar]
- La Vitola, P.; Beeg, M.; Balducci, C.; Santamaria, G.; Restelli, E.; Colombo, L.; Caldinelli, L.; Pollegioni, L.; Gobbi, M.; Chiesa, R.; et al. Cellular prion protein neither binds to alpha-synuclein oligomers nor mediates their detrimental effects. Brain 2019, 142, 249–254. [Google Scholar] [CrossRef]
- Gelpi, E.; Colom-Cadena, M. Oligomers: A hot topic for neurodegeneration and a note of caution for experimental models. Brain 2019, 142, 228–230. [Google Scholar] [CrossRef]
- Visanji, N.P.; Lang, A.E.; Kovacs, G.G. Beyond the synucleinopathies: Alpha synuclein as a driving force in neurodegenerative comorbidities. Transl. Neurodegener. 2019, 8, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bistaffa, E.; Rossi, M.; De Luca, C.M.G.; Cazzaniga, F.; Carletta, O.; Campagnani, I.; Tagliavini, F.; Legname, G.; Giaccone, G.; Moda, F. Prion efficiently replicates in alpha-synuclein knockout mice. Mol. Neurobiol. 2019, 56, 7448–7457. [Google Scholar] [CrossRef] [PubMed]
- Bartz, J.C. Environmental and host factors that contribute to prion strain evolution. Acta Neuropathol. 2021, 142, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Chelban, V.; Manole, A.; Pihlstrom, L.; Schottlaender, L.; Efthymiou, S.; O’Conner, E.; Meissner, W.G.; Holton, J.L.; Houlden, H. Analysis of the prion protein gene in multiple system atrophy. Neurobiol. Aging 2017, 49, 216.e15–216.e18. [Google Scholar] [CrossRef] [Green Version]
- De Pablo-Fernandez, E.; Cerdan Santacruz, D.; Warner, T.T.; Holton, J.L. No evidence of iatrogenic human transmission in autopsy confirmed multiple system atrophy. Mov. Disord. 2018, 33, 1183–1184. [Google Scholar] [CrossRef] [PubMed]
- Rajput, A.H.; Ferguson, L.W.; Robinson, C.A.; Guella, I.; Farrer, M.J.; Rajput, A. Conjugal parkinsonism—Clinical, pathology and genetic study. No evidence of person-to-person transmission. Parkinsonism Relat. Disord. 2016, 31, 87–90. [Google Scholar] [CrossRef]
- Coon, E.A.; Rocca, W.; Melson, C.S.; Ahlskog, J.E.; Matsumoto, J.Y.; Low, P.A.; Singer, W. Conjugal multiple system atrophy: Chance, shared risk factors, or evidence of transmissibility? Parkinsonism Relat. Disord. 2019, 67, 10–13. [Google Scholar] [CrossRef]
- Asher, D.M.; Belay, E.; Bigio, E.; Brandner, S.; Brubaker, S.A.; Caughey, B.; Clark, B.; Damon, I.; Diamond, M.; Freund, M.; et al. Risk of transmissibility from neurodegenerative disease-associated proteins: Experimental knowns and unknowns. J. Neuropathol. Exp. Neurol. 2020, 79, 1141–1146. [Google Scholar] [CrossRef]
- Rey, N.L.; George, S.; Steiner, J.A.; Madaj, Z.; Luk, K.C.; Trojanowski, J.Q.; Lee, V.M.; Brundin, P. Spread of aggregates after olfactory bulb injection of alpha-synuclein fibrils is associated with early neuronal loss and is reduced long term. Acta Neuropathol. 2018, 135, 65–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jellinger, K.A.; Wenning, G.K.; Stefanova, N. Is Multiple System Atrophy a Prion-like Disorder? Int. J. Mol. Sci. 2021, 22, 10093. https://doi.org/10.3390/ijms221810093
Jellinger KA, Wenning GK, Stefanova N. Is Multiple System Atrophy a Prion-like Disorder? International Journal of Molecular Sciences. 2021; 22(18):10093. https://doi.org/10.3390/ijms221810093
Chicago/Turabian StyleJellinger, Kurt A., Gregor K. Wenning, and Nadia Stefanova. 2021. "Is Multiple System Atrophy a Prion-like Disorder?" International Journal of Molecular Sciences 22, no. 18: 10093. https://doi.org/10.3390/ijms221810093