
More Than Meets the Eye Regarding Cancer Metabolism


Abstract
:
1. Introduction
2. Warburg Effect
3. Crabtree Effect
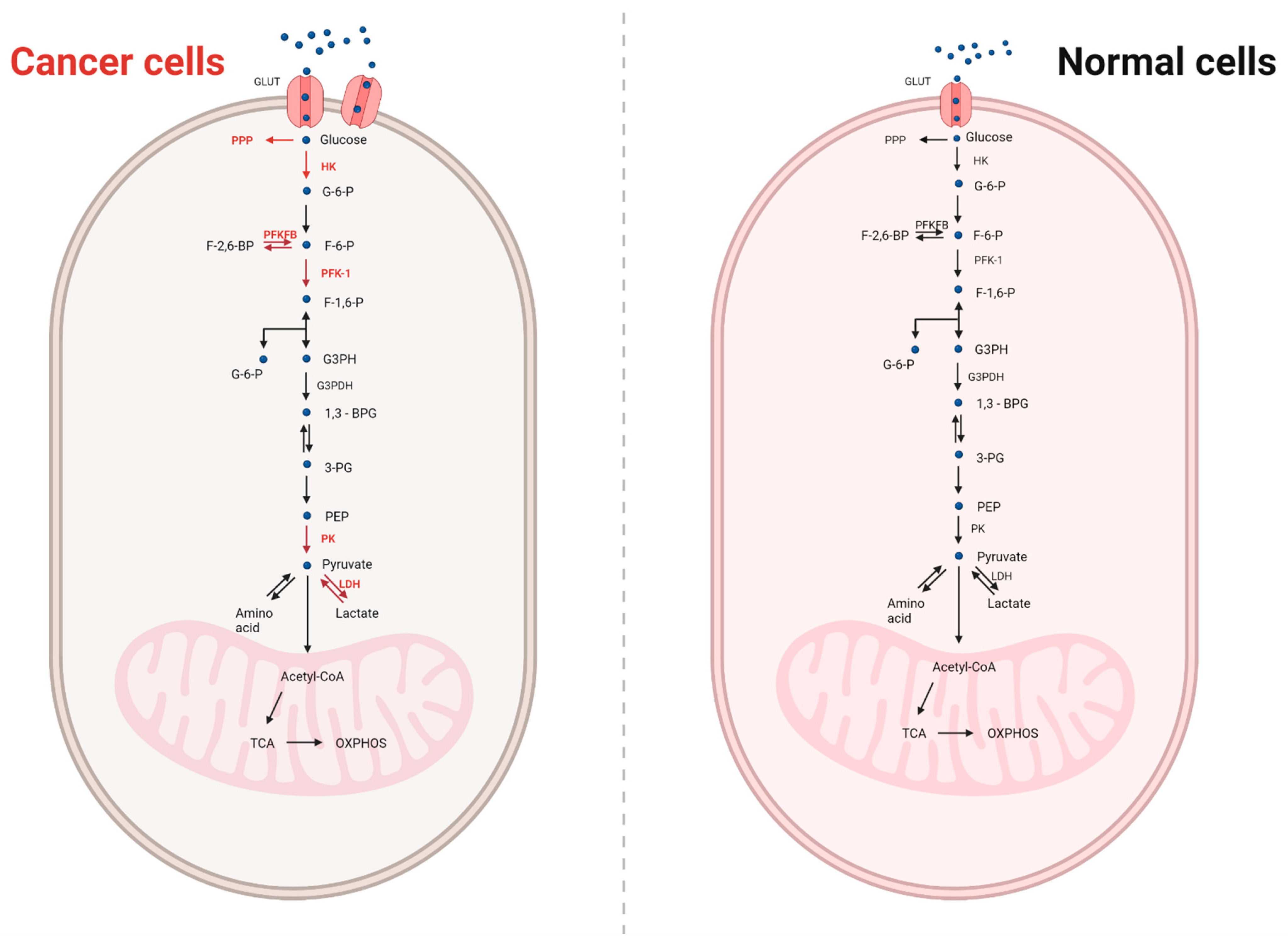
4. Metabolism in Normal and Cancer Cells
5. The Metabolic Heterogeneity of Tumours
6. The Metabolic Heterogeneity of Cancer Is Due to the Difficult Conditions
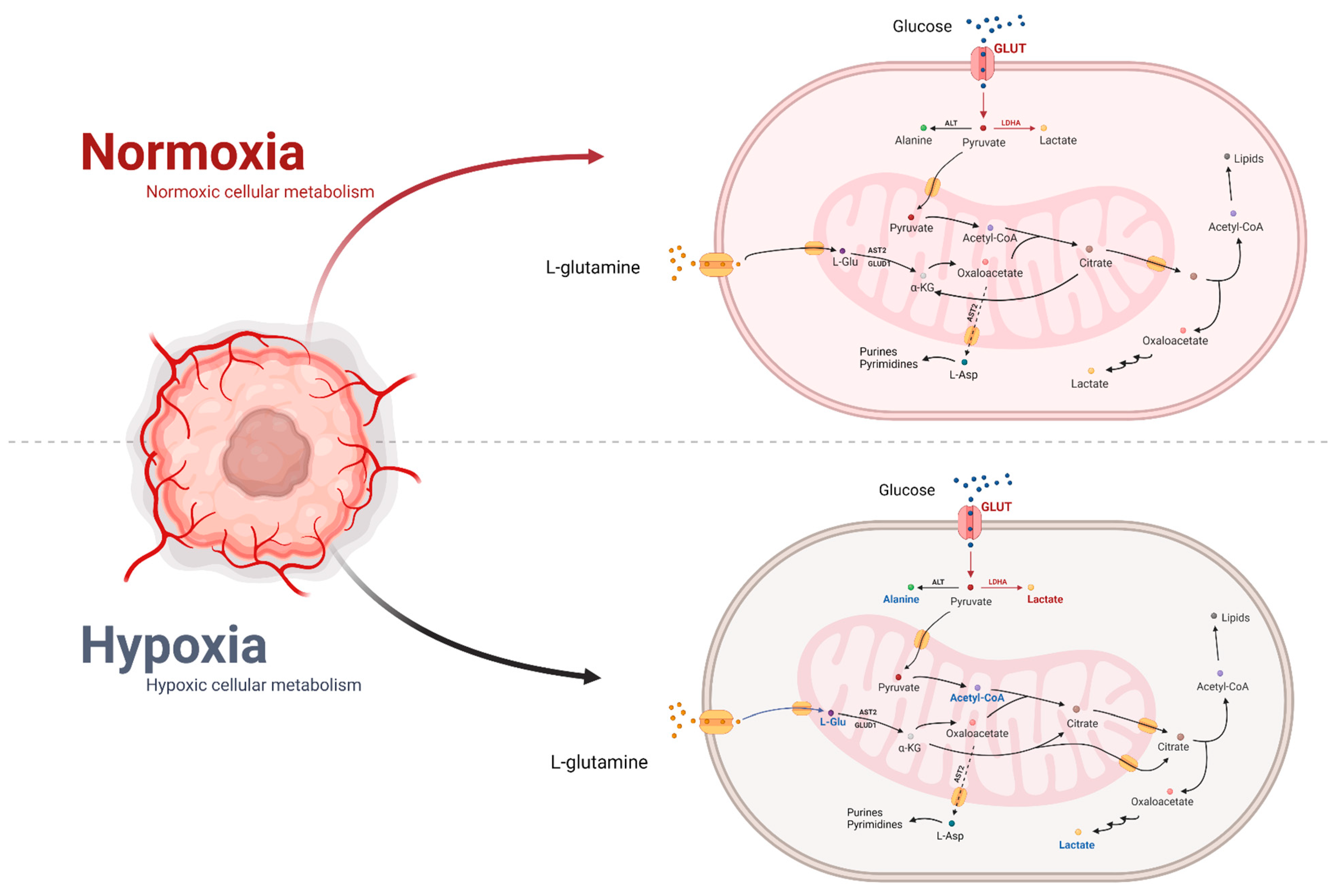
6.1. Effect of Hypoxia on Metabolism
6.2. Normoxia’s Impact on Metabolism
7. Effects of Oncogenes on Metabolism
- Mutations in PI3K component genes;
- Mutations in tumour suppressor genes;
- Signalling by receptor tyrosine kinase [93].
8. The Main Energy Source in a Cancer Cell
8.1. Glucose and Its Metabolism in Cancer
8.2. Glutamine in Cancer Metabolism
8.3. Glycolysis “Waste” Product
8.4. Ketones and Fatty Acids in Cancer
9. Crucial Transporters in Metabolism
9.1. Glucose Transporters
9.2. Monocarboxylate Transporters
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Abbreviations | Meaning |
| ACO | aconitase |
| ACSS2 | acyl-coenzyme A synthetase short-chain family member 2 |
| ADP | adenosine diphosphate |
| ANT | adenine nucleotide translocase |
| AST | aspartate aminotransferase |
| ATP | adenosine triphosphate |
| BPTES | bis-2-(5-phenylacetamide-1,2,4-thiazole-2-yl)ethyl sulfide |
| CAF | cancer-related fibroblasts |
| CD31 | cluster of differentiation 31 |
| c-Myc | c-Myc transcription factor |
| CO2 | carbon dioxide |
| COX | cytochrome c oxidase |
| CSCs | cancer stem cells |
| DNA | deoxyribonucleic acid |
| ERK1/2 | extracellular kinase 1/2 |
| F-1,6-P | fructose 1,6-bisphosphate |
| F-2,6-BP | fructose-2,6-bisphosphate |
| F-6-P | fructose 6-phosphate |
| FAD | flavin adenine dinucleotide |
| FOXO3a | forkhead box protein O3 |
| G3PH | glyceraldehyde 3-phosphate |
| G-6-P | glucose 6-phosphate |
| GLS | glutaminase |
| GLUD | glutamate dehydrogenase |
| GLUT | glucose transporters |
| GOT1/2 | glutamate oxaloacetate transaminase 1/2 |
| GPNA | L-γ-Glutamyl-p-nitroanilide |
| GSH | glutathione |
| HIF | hypoxia-inducible transcription factor |
| HO-1 | heme oxygenase 1 |
| IDH | isocytrine dehydrogenase |
| IL-8 | interleukin 8 |
| iNOS | induced nitric oxide synthase isoform |
| KD | ketogenic diet |
| KRAS | Kirsten rat sarcoma viral oncogene homolog |
| LDH | lactate dehydrogenase |
| LDHA | lactate dehydrogenase A |
| LDHB | lactate dehydrogenase B |
| L-Gln | L-glutamine |
| L-Glu | L-glutamic acid |
| MBP-1, | c-Myc binding protein 1 |
| MCT | monocarboxylate transporter |
| mTOR | mammalian target of rapamycin |
| NAD | oxidised form of dinucleotide |
| NADPH | reduced form of NADP+ |
| NEAA | non-essential amino acid |
| NFκB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| NK | natural killer cells |
| OXPHOS | oxidative phosphorylation |
| OXPHOS | oxidative phosphorylation |
| p53 | Tumour protein 53 |
| PDH | pyruvate dehydrogenase |
| PDK1 | pyruvate dehydrogenase kinase |
| PEP | phosphoenolpyruvate |
| PFK-2 | phosphofructokinase 2 |
| PFKFB | 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 |
| VEGF | vascular endothelial growth factor |
References
- Warburg, O.; Wind, F.; Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [Green Version]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [Green Version]
- Warburg, O. On the origin of cancer cells. Science (80-) 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Saman, H.; Raza, S.S.; Uddin, S.; Rasul, K. Inducing angiogenesis, a key step in cancer vascularization, and treatment approaches. Cancers 2020, 12, 1172. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; DeBerardinis, R.J. Mechanisms and Implications of Metabolic Heterogeneity in Cancer. Cell Metab. 2019, 30, 434–446. [Google Scholar] [CrossRef] [PubMed]
- Weinhouse, S. Studies on the Fate of Isotopically Labeled Metabolites in the Oxidative Metabolism of Tumors. Cancer Res. 1951, 11, 585–591. [Google Scholar]
- Epstein, T.; Gatenby, R.A.; Brown, J.S. The Warburg effect as an adaptation of cancer cells to rapid fluctuations in energy demand. PLoS ONE 2017, 12, e0185085. [Google Scholar] [CrossRef] [Green Version]
- Lunt, S.Y.; Vander Heiden, M.G. Aerobic Glycolysis: Meeting the Metabolic Requirements of Cell Proliferation. Annu. Rev. Cell Dev. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gwangwa, M.V.; Joubert, A.M.; Visagie, M.H. Crosstalk between the Warburg effect, redox regulation and autophagy induction in tumourigenesis. Cell. Mol. Biol. Lett. 2018, 23, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nogueira, V.; Hay, N. Molecular pathways: Reactive oxygen species homeostasis in cancer cells and implications for cancer therapy. Clin. Cancer Res. 2013, 19, 4309–4314. [Google Scholar] [CrossRef] [Green Version]
- Schafer, Z.T.; Grassian, A.R.; Song, L.; Jiang, Z.; Gerhart-Hines, Z.; Irie, H.Y.; Gao, S.; Puigserver, P.; Brugge, J.S. Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature 2009, 461, 109–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhup, S.; Kumar Dadhich, R.; Ettore Porporato, P.; Sonveaux, P. Multiple Biological Activities of Lactic Acid in Cancer: Influences on Tumor Growth, Angiogenesis and Metastasis. Curr. Pharm. Des. 2012, 18, 1319–1330. [Google Scholar] [CrossRef] [Green Version]
- Jurisic, V.; Radenkovic, S.; Konjevic, G. The Actual Role of LDH as Tumor Marker, Biochemical and Clinical Aspects. In Advances in Cancer Biomarkers: From Biochemistry to Clinic for a Critical Revision; Scatena, R., Ed.; Springer: Dordrecht, The Netherlands, 2015; pp. 115–124. ISBN 978-94-017-7215-0. [Google Scholar]
- Diaz-Ruiz, R.; Rigoulet, M.; Devin, A. The Warburg and Crabtree effects: On the origin of cancer cell energy metabolism and of yeast glucose repression. Biochim. Biophys. Acta Bioenerg. 2011, 1807, 568–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veech, R.L.; Lawson, J.W.; Cornell, N.W.; Krebs, H.A. Cytosolic phosphorylation potential. J. Biol. Chem. 1979, 254, 6538–6547. [Google Scholar] [CrossRef]
- Sussman, I.; Erecińska, M.; Wilson, D.F. Regulation of cellular energy metabolism. The Crabtree effect. Biochim. Biophys. Acta Bioenerg. 1980, 591, 209–223. [Google Scholar] [CrossRef]
- Díaz-Ruiz, R.; Avéret, N.; Araiza, D.; Pinson, B.; Uribe-Carvajal, S.; Devin, A.; Rigoulet, M. Mitochondrial Oxidative Phosphorylation Is Regulated by Fructose 1,6-Bisphosphate: A POSSIBLE ROLE IN CRABTREE EFFECT INDUCTION? J. Biol. Chem. 2008, 283, 26948–26955. [Google Scholar] [CrossRef] [Green Version]
- Fadaka, A.; Ajiboye, B.; Ojo, O.; Adewale, O.; Olayide, I.; Emuowhochere, R. Biology of glucose metabolization in cancer cells. J. Oncol. Sci. 2017, 3, 45–51. [Google Scholar] [CrossRef]
- Dang, C.V. Links between metabolism and cancer. Genes Dev. 2012, 26, 877–890. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.D.; Wu, H.; Huang, S.; Zhang, H.L.; Qin, C.J.; Zhao, L.H.; Fu, G.B.; Zhou, X.; Wang, X.M.; Tang, L.; et al. HBx regulates fatty acid oxidation to promote hepatocellular carcinoma survival during metabolic stress. Oncotarget 2016, 7, 6711–6726. [Google Scholar] [CrossRef] [PubMed]
- Boroughs, L.K.; Deberardinis, R.J. Metabolic pathways promoting cancer cell survival and growth. Nat. Cell Biol. 2015, 17, 351–359. [Google Scholar] [CrossRef] [Green Version]
- Antonio, M.J.; Zhang, C.; Le, A. Different Tumor Microenvironments Lead to Different Metabolic Phenotypes. In Advances in Experimental Medicine and Biology; Springer: Cham, Switzerland, 2021; Volume 1311. [Google Scholar]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [Green Version]
- Nabi, K.; Le, A. The Intratumoral Heterogeneity of Cancer Metabolism. In Advances in Experimental Medicine and Biology; Springer: Cham, Switzerland, 2021; Volume 1311. [Google Scholar]
- Nowell, P.C. The clonal evolution of tumor cell populations. Science (80-) 1976, 194, 23–28. [Google Scholar] [CrossRef]
- Greaves, M.; Maley, C.C. Clonal evolution in cancer. Nature 2012, 481, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Pavlović, M.; Radotić, K. Metabolic Reprogramming in Cancer and Metabolic Theory of CSC. In Animal and Plant Stem Cells; Springer: Cham, Switzerland, 2017. [Google Scholar]
- Prasetyanti, P.R.; Medema, J.P. Intra-tumor heterogeneity from a cancer stem cell perspective. Mol. Cancer 2017, 16, 41. [Google Scholar] [CrossRef] [Green Version]
- De Francesco, E.M.; Sotgia, F.; Lisanti, M.P. Cancer stem cells (CSCs): Metabolic strategies for their identification and eradication. Biochem. J. 2018, 475, 1611–1634. [Google Scholar] [CrossRef] [Green Version]
- González-García, I.; Solé, R.V.; Costa, J. Metapopulation dynamics and spatial heterogeneity in cancer. Proc. Natl. Acad. Sci. USA 2002, 99, 13085–13089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhani, N.; Fyles, A.; Hedley, D.; Milosevic, M. The clinical significance of hypoxia in human cancers. Semin. Nucl. Med. 2015, 45, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Vaupel, P.; Mayer, A. Hypoxia in cancer: Significance and impact on clinical outcome. Cancer Metastasis Rev. 2007, 26, 225–239. [Google Scholar] [CrossRef]
- Hashimoto, T.; Shibasaki, F. Hypoxia-Inducible Factor as an Angiogenic Master Switch. Front. Pediatr. 2015, 3, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arampatzis, A.; Mademli, L.; Reilly, T.; Lambert, M.I.; Bosquet, L.; Richalet, J.-P.; Busso, T.; Mettauer, B.; Gréciano, S.; Geny, B.; et al. Tumor Microenvironment; Springer: Cham, Switzerland, 2012; ISBN 978-3-030-34025-4. [Google Scholar]
- Majmundar, A.J.; Wong, W.J.; Simon, M.C. Hypoxia-Inducible Factors and the Response to Hypoxic Stress. Mol. Cell 2010, 40, 294–309. [Google Scholar] [CrossRef] [Green Version]
- Mcintosh, D. Book Review: Basic Clinical Radiobiology. Scott. Med. J. 2010, 55, 33. [Google Scholar] [CrossRef]
- Semenza, G.L. HIF-1: Upstream and downstream of cancer metabolism. Curr. Opin. Genet. Dev. 2010, 20, 51–56. [Google Scholar] [CrossRef] [Green Version]
- Semba, H.; Takeda, N.; Isagawa, T.; Sugiura, Y.; Honda, K.; Wake, M.; Miyazawa, H.; Yamaguchi, Y.; Miura, M.; Jenkins, D.M.R.; et al. HIF-1α-PDK1 axis-induced active glycolysis plays an essential role in macrophage migratory capacity. Nat. Commun. 2016, 7, 11635. [Google Scholar] [CrossRef] [Green Version]
- Dupuy, F.; Tabariès, S.; Andrzejewski, S.; Dong, Z.; Blagih, J.; Annis, M.G.; Omeroglu, A.; Gao, D.; Leung, S.; Amir, E.; et al. PDK1-dependent metabolic reprogramming dictates metastatic potential in breast cancer. Cell Metab. 2015, 22, 577–589. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wise, D.R.; Ward, P.S.; Shay, J.E.S.; Cross, J.R.; Gruber, J.J.; Sachdeva, U.M.; Platt, J.M.; DeMatteo, R.G.; Simon, M.C.; Thompson, C.B. Hypoxia promotes isocitrate dehydrogenasedependent carboxylation of α-ketoglutarate to citrate to support cell growth and viability. Proc. Natl. Acad. Sci. USA 2011, 108, 19611–19616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metallo, C.M.; Gameiro, P.A.; Bell, E.L.; Mattaini, K.R.; Yang, J.; Hiller, K.; Jewell, C.M.; Johnson, Z.R.; Irvine, D.J.; Guarente, L.; et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 2012, 481, 380–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samanta, D.; Semenza, G.L. Metabolic adaptation of cancer and immune cells mediated by hypoxia-inducible factors. Biochim. Biophys. Acta Rev. Cancer 2018, 1870, 15–22. [Google Scholar] [CrossRef]
- Chen, R.; Xu, M.; Nagati, J.; Garcia, J.A. Coordinate regulation of stress signaling and epigenetic events by Acss2 and HIF-2 in cancer cells. PLoS ONE 2017, 12, e0190241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, D.; Li, T.; Li, X.; Zhang, L.; Sun, L.; He, X.; Zhong, X.; Jia, D.; Song, L.; Semenza, G.L.; et al. HIF-1-mediated suppression of acyl-CoA dehydrogenases and fatty acid oxidation is critical for cancer progression. Cell Rep. 2014, 8, 1930–1942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabharwal, S.S.; Schumacker, P.T. Mitochondrial ROS in cancer: Initiators, amplifiers or an Achilles’ heel? Nat. Rev. Cancer 2014, 14, 709–721. [Google Scholar] [CrossRef] [Green Version]
- Fukuda, R.; Zhang, H.; Kim, J.W.; Shimoda, L.; Dang, C.V.; Semenza, G.L.L. HIF-1 Regulates Cytochrome Oxidase Subunits to Optimize Efficiency of Respiration in Hypoxic Cells. Cell 2007, 129, 111–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordan, J.D.; Thompson, C.B.; Simon, M.C. HIF and c-Myc: Sibling Rivals for Control of Cancer Cell Metabolism and Proliferation. Cancer Cell 2007, 12, 108–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrish, F.; Hockenbery, D. MYC and Mitochondrial Biogenesis. Cold Spring Harb. Perspect. Med. 2014, 4, a014225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tharmalingham, H.; Hoskin, P. Clinical trials targeting hypoxia. Br. J. Radiol. 2019, 92, 20170966. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, D.E.; Hadjiargyrou, M. Activation of the transcription factor HIF-1 and its target genes, VEGF, HO-1, iNOS, during fracture repair. Bone 2004, 34, 680–688. [Google Scholar] [CrossRef]
- Li, J.; Zhou, W.; Chen, W.; Wang, H.; Zhang, Y.; Yu, T. Mechanism of the hypoxia inducible factor 1/hypoxic response element pathway in rat myocardial ischemia/diazoxide post–conditioning. Mol. Med. Rep. 2020, 21, 1527–1536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graboń, W.; Otto-Ślusarczyk, D.; Chrzanowska, A.; Mielczarek-Puta, M.; Joniec-Maciejak, I.; Słabik, K.; Barańczyk-Kuźma, A. Lactate Formation in Primary and Metastatic Colon Cancer Cells at Hypoxia and Normoxia. Cell Biochem. Funct. 2016, 34, 483–490. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350. [Google Scholar] [CrossRef] [Green Version]
- Fan, J.; Kamphorst, J.J.; Mathew, R.; Chung, M.K.; White, E.; Shlomi, T.; Rabinowitz, J.D. Glutamine-driven oxidative phosphorylation is a major ATP source in transformed mammalian cells in both normoxia and hypoxia. Mol. Syst. Biol. 2013, 9, 712. [Google Scholar] [CrossRef]
- Sun, R.C.; Denko, N.C. Hypoxic regulation of glutamine metabolism through HIF1 and SIAH2 supports lipid synthesis that is necessary for tumor growth. Cell Metab. 2014, 19, 285–292. [Google Scholar] [CrossRef] [Green Version]
- DeClerck, K.; Elble, R.C. The role of hypoxia and acidosis in promoting metastasis and resistance to chemotherapy. Front. Biosci. 2010, 15, 213–225. [Google Scholar] [CrossRef] [Green Version]
- Filipp, F.V.; Scott, D.A.; Ronai, Z.A.; Osterman, A.L.; Smith, J.W. Reverse TCA cycle flux through isocitrate dehydrogenases 1 and 2 is required for lipogenesis in hypoxic melanoma cells. Pigment Cell Melanoma Res. 2012, 25, 375–383. [Google Scholar] [CrossRef] [Green Version]
- Zimna, A.; Kurpisz, M. Hypoxia-Inducible factor-1 in physiological and pathophysiological angiogenesis: Applications and therapies. BioMed Res. Int. 2015, 2015, 549412. [Google Scholar] [CrossRef] [Green Version]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef]
- Nagao, A.; Kobayashi, M.; Koyasu, S.; Chow, C.C.T.; Harada, H. HIF-1-dependent reprogramming of glucose metabolic pathway of cancer cells and its therapeutic significance. Int. J. Mol. Sci. 2019, 20, 238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaub, F.X.; Dhankani, V.; Berger, A.C.; Trivedi, M.; Richardson, A.B.; Shaw, R.; Zhao, W.; Zhang, X.; Ventura, A.; Liu, Y.; et al. Pan-cancer Alterations of the MYC Oncogene and Its Proximal Network across the Cancer Genome Atlas. Cell Syst. 2018, 6, 282–300.e2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettersson, A.; Gerke, T.; Penney, K.L.; Lis, R.T.; Stack, E.C.; Pertega-Gomes, N.; Zadra, G.; Tyekucheva, S.; Giovannucci, E.L.; Mucci, L.A.; et al. MYC overexpression at the protein and mRNA level and cancer outcomes among men treated with radical prostatectomy for prostate cancer. Cancer Epidemiol. Biomark. Prev. 2018, 27, 201–207. [Google Scholar] [CrossRef] [Green Version]
- He, W.-L.; Weng, X.-T.; Wang, J.-L.; Lin, Y.-K.; Liu, T.-W.; Zhou, Q.-Y.; Hu, Y.; Pan, Y.; Chen, X.-L. Association Between c-Myc and Colorectal Cancer Prognosis: A Meta-Analysis. Front. Physiol. 2018, 9, 1549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sesen, J.; Dahan, P.; Scotland, S.J.; Saland, E.; Dang, V.T.; Lemarié, A.; Tyler, B.M.; Brem, H.; Toulas, C.; Moyal, E.C.J.; et al. Metformin inhibits growth of human glioblastoma cells and enhances therapeutic response. PLoS ONE 2015, 10, e0123721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stine, Z.E.; Walton, Z.E.; Altman, B.J.; Hsieh, A.L.; Dang, C.V. MYC, metabolism, and cancer. Cancer Discov. 2015, 5, 1024–1039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanian, A.; Miller, D.M. Structural analysis of α-enolase: Mapping the functional domains involved in down-regulation of the c-myc protooncogene. J. Biol. Chem. 2000, 275, 5958–5965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sedoris, K.C.; Thomas, S.D.; Miller, D.M. Hypoxia induces differential translation of enolase/MBP-1. BMC Cancer 2010, 10, 157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dang, C.V.; Kim, J.W.; Gao, P.; Yustein, J. The interplay between MYC and HIF in cancer. Nat. Rev. Cancer 2008, 8, 51–56. [Google Scholar] [CrossRef]
- Miller, D.M.; Thomas, S.D.; Islam, A.; Muench, D.; Sedoris, K. c-Myc and cancer metabolism. Clin. Cancer Res. 2012, 18, 5546–5553. [Google Scholar] [CrossRef] [Green Version]
- Anastasiou, D.; Cantley, L.C. Breathless cancer cells get fat on glutamine. Cell Res. 2012, 22, 443–446. [Google Scholar] [CrossRef] [Green Version]
- Katt, W.P.; Cerione, R.A. Glutaminase regulation in cancer cells: A druggable chain of events. Drug Discov. Today 2014, 19, 450–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, A.J.L.; Shurubor, Y.I.; Dorai, T.; Pinto, J.T.; Isakova, E.P.; Deryabina, Y.I.; Denton, T.T.; Krasnikov, B.F. ω-Amidase: An underappreciated, but important enzyme in l-glutamine and l-asparagine metabolism; Relevance to sulfur and nitrogen metabolism, tumor biology and hyperammonemic diseases. Amino Acids 2016, 48, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Heiden, M.G.V.; Cantley, L.C.; Thompson, C.B. Understanding the warburg effect: The metabolic requirements of cell proliferation. Science (80-) 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Eu, J.Q.; Kong, L.R.; Wang, L.; Lim, Y.C.; Goh, B.C.; Wong, A.L.A. Targeting metabolism in cancer cells and the tumour microenvironment for cancer therapy. Molecules 2020, 25, 4831. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Goswami, D.; Adiseshaiah, P.P.; Burgan, W.; Yi, M.; Guerin, T.M.; Kozlov, S.V.; Nissley, D.V.; McCormick, F. Undermining glutaminolysis bolsters chemotherapy while NRF2 promotes chemoresistance in KRAS-driven pancreatic cancers. Cancer Res. 2020, 80, 1630–1643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardee, T.S.; Anderson, R.G.; Pladna, K.M.; Isom, S.; Ghiraldeli, L.P.; Miller, L.D.; Chou, J.W.; Jin, G.; Zhang, W.; Ellis, L.R.; et al. A phase i study of cpi-613 in combination with high-dose cytarabine and mitoxantrone for relapsed or refractory acute myeloid leukemia. Clin. Cancer Res. 2018, 24, 2060–2073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardee, T.S.; Lee, K.; Luddy, J.; Maturo, C.; Rodriguez, R.; Isom, S.; Miller, L.D.; Stadelman, K.M.; Levitan, D.; Hurd, D.; et al. A phase I study of the first-in-class antimitochondrial metabolism agent, CPI-613, in patients with advanced hematologic malignancies. Clin. Cancer Res. 2014, 20, 5255–5264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watts, J.M.; Baer, M.R.; Yang, J.; Prebet, T.; Lee, S.; Schiller, G.J.; Dinner, S.; Pigneux, A.; Montesinos, P.; Wang, E.S.; et al. Olutasidenib (FT-2102), an IDH1m Inhibitor As a Single Agent or in Combination with Azacitidine, Induces Deep Clinical Responses with Mutation Clearance in Patients with Acute Myeloid Leukemia Treated in a Phase 1 Dose Escalation and Expansion Study. Blood 2019, 134 (Suppl. 1), 231. [Google Scholar] [CrossRef]
- De Botton, S.; Yee, K.W.L.; Recher, C.; Wei, A.; Montesinos, P.; Taussig, D.; Pigneux, A.; Braun, T.; Curti, A.; Esteve, J.; et al. Effect of olutasidenib (FT-2102) on complete remissions in patients with relapsed/refractory (R/R) m IDH1 acute myeloid leukemia (AML): Results from a planned interim analysis of a phase 2 clinical trial. J. Clin. Oncol. 2021, 39, 7006. [Google Scholar] [CrossRef]
- Falchook, G.; Infante, J.; Arkenau, H.T.; Patel, M.R.; Dean, E.; Borazanci, E.; Brenner, A.; Cook, N.; Lopez, J.; Pant, S.; et al. First-in-human study of the safety, pharmacokinetics, and pharmacodynamics of first-in-class fatty acid synthase inhibitor TVB-2640 alone and with a taxane in advanced tumors. EClinicalMedicine 2021, 34, 100797. [Google Scholar] [CrossRef]
- Diaz Duque, A.E.; De Feyter, H.; Kemble, G.; McCulloch, W.; Brenner, A.J. A phase 2 study to determine the efficacy and safety of TVB-2640 in combination with bevacizumab in patients with first relapse of high grade astrocytoma. J. Clin. Oncol. 2017, 35, TPS2080. [Google Scholar] [CrossRef]
- Dinardo, C.D.; Stein, A.S.; Stein, E.M.; Fathi, A.T.; Schuh, A.C.; Montesinos Fernández, P.; Odenike, O.; Kantarjian, H.M.; Stone, R.M.; Collins, R.; et al. Mutant IDH (mIDH) inhibitors, ivosidenib or enasidenib, with azacitidine (AZA) in patients with acute myeloid leukemia (AML). J. Clin. Oncol. 2018, 36, 7042. [Google Scholar] [CrossRef]
- Yakisich, J.S.; Azad, N.; Kaushik, V.; Iyer, A.K.V. The Biguanides Metformin and Buformin in Combination with 2-Deoxy-glucose or WZB-117 Inhibit the Viability of Highly Resistant Human Lung Cancer Cells. Stem Cells Int. 2019, 2019, 6254269. [Google Scholar] [CrossRef]
- Li, Y.-L.; Weng, H.-C.; Hsu, L.-C. Abstract 4922: The combination of MK-2206 and WZB117 exerts a synergistic cytotoxic effect against breast cancer cells. Front. Pharmacol. 2019, 10, 1311. [Google Scholar] [CrossRef] [Green Version]
- Mukhopadhyay, S.; Saqcena, M.; Foster, D.A. Synthetic lethality in KRas-driven cancer cells created by glutamine deprivation. Oncoscience 2015, 2, 807. [Google Scholar] [CrossRef] [PubMed]
- Ying, H.; Kimmelman, A.C.; Lyssiotis, C.A.; Hua, S.; Chu, G.C.; Fletcher-Sananikone, E.; Locasale, J.W.; Son, J.; Zhang, H.; Coloff, J.L.; et al. Oncogenic kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 2012, 149, 656–670. [Google Scholar] [CrossRef] [Green Version]
- Kole, H.K.; Resnick, R.J.; Van Doren, M.; Racker, E. Regulation of 6-phosphofructo-1-kinase activity in ras-transformed rat-1 fibroblasts. Arch. Biochem. Biophys. 1991, 286, 586–590. [Google Scholar] [CrossRef]
- Racker, E.; Resnick, R.J.; Feldman, R. Glycolysis and methylaminoisobutyrate uptake in rat-1 cells transfected with ras or myc oncogenes. Proc. Natl. Acad. Sci. USA 1985, 82, 3535–3538. [Google Scholar] [CrossRef] [Green Version]
- Son, J.; Lyssiotis, C.A.; Ying, H.; Wang, X.; Hua, S.; Ligorio, M.; Perera, R.M.; Ferrone, C.R.; Mullarky, E.; Shyh-Chang, N.; et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 2013, 496, 101–105. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Vander Heiden, M.G.; McCormick, F. The metabolic landscape of RAS-driven cancers from biology to therapy. Nat. Cancer 2021, 2, 271–283. [Google Scholar] [CrossRef] [PubMed]
- Ricoult, S.J.H.; Yecies, J.L.; Ben-Sahra, I.; Manning, B.D. Oncogenic PI3K and K-Ras stimulate de novo lipid synthesis through mTORC1 and SREBP. Oncogene 2016, 35, 1250–1260. [Google Scholar] [CrossRef] [Green Version]
- Park, J.K.; Coffey, N.J.; Limoges, A.; Le, A. The Heterogeneity of Lipid Metabolism in Cancer. In Advances in Experimental Medicine and Biology; Springer: Cham, Switzerland, 2021; Volume 1311. [Google Scholar]
- Simons, A.L.; Orcutt, K.P.; Madsen, J.M.; Scarbrough, P.M.; Spitz, D.R. The role of akt pathway signaling in glucose metabolism and metabolic oxidative stress. In Oxidative Stress in Cancer Biology and Therapy; Springer: Cham, Switzerland, 2012. [Google Scholar]
- Upadhyay, M.; Samal, J.; Kandpal, M.; Singh, O.V.; Vivekanandan, P. The Warburg effect: Insights from the past decade. Pharmacol. Ther. 2013, 137, 318–330. [Google Scholar] [CrossRef] [PubMed]
- Zhivotovsky, B.; Orrenius, S. The Warburg Effect returns to the cancer stage. Semin. Cancer Biol. 2009, 19, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Abraham, A.G.; O’Neill, E. PI3K/Akt-mediated regulation of p53 in cancer. Biochem. Soc. Trans. 2014, 42, 798–803. [Google Scholar] [CrossRef] [PubMed]
- Gnanapradeepan, K.; Basu, S.; Barnoud, T.; Budina-Kolomets, A.; Kung, C.P.; Murphy, M.E. The p53 tumor suppressor in the control of metabolism and ferroptosis. Front. Endocrinol. (Lausanne) 2018, 9, 124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simabuco, F.M.; Morale, M.G.; Pavan, I.C.B.; Morelli, A.P.; Silva, F.R.; Tamura, R.E. p53 and metabolism: From mechanism to therapeutics. Oncotarget 2018, 9, 23780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lacroix, M.; Riscal, R.; Arena, G.; Linares, L.K.; Le Cam, L. Metabolic functions of the tumor suppressor p53: Implications in normal physiology, metabolic disorders, and cancer. Mol. Metab. 2020, 33, 2–22. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Xiong, H.; Wu, F.; Zhang, Y.; Wang, J.; Zhao, L.; Guo, X.; Chang, L.J.; Zhang, Y.; You, M.J.; et al. Hexokinase 2-Mediated Warburg Effect Is Required for PTEN- and p53-Deficiency-Driven Prostate Cancer Growth. Cell Rep. 2014, 8, 1461–1474. [Google Scholar] [CrossRef] [Green Version]
- Hitosugi, T.; Zhou, L.; Elf, S.; Fan, J.; Kang, H.B.; Seo, J.H.; Shan, C.; Dai, Q.; Zhang, L.; Xie, J.; et al. Phosphoglycerate Mutase 1 Coordinates Glycolysis and Biosynthesis to Promote Tumor Growth. Cancer Cell 2012, 22, 585–600. [Google Scholar] [CrossRef] [Green Version]
- Schwartzenberg-Bar-Yoseph, F.; Armoni, M.; Karnieli, E. The Tumor Suppressor p53 Down-Regulates Glucose Transporters GLUT1 and GLUT4 Gene Expression. Cancer Res. 2004, 64, 2627–2633. [Google Scholar] [CrossRef] [Green Version]
- Janku, F.; Yap, T.A.; Meric-Bernstam, F. Targeting the PI3K pathway in cancer: Are we making headway? Nat. Rev. Clin. Oncol. 2018, 15, 273–291. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Anjomani-Virmouni, S.; Koundouros, N.; Dimitriadi, M.; Choo-Wing, R.; Valle, A.; Zheng, Y.; Chiu, Y.H.; Agnihotri, S.; Zadeh, G.; et al. PARK2 Depletion Connects Energy and Oxidative Stress to PI3K/Akt Activation via PTEN S-Nitrosylation. Mol. Cell 2017, 65, 999–1013. [Google Scholar] [CrossRef] [Green Version]
- Shaw, R.J. Glucose metabolism and cancer. Curr. Opin. Cell Biol. 2006, 18, 598–608. [Google Scholar] [CrossRef] [PubMed]
- Hamanaka, R.B.; Chandel, N.S. Targeting glucose metabolism for cancer therapy. J. Exp. Med. 2012, 209, 211–215. [Google Scholar] [CrossRef] [Green Version]
- Fitzgerald, G.; Soro-Arnaiz, I.; De Bock, K. The Warburg effect in endothelial cells and its potential as an anti-angiogenic target in cancer. Front. Cell Dev. Biol. 2018, 6, 100. [Google Scholar] [CrossRef] [Green Version]
- Adekola, K.; Rosen, S.T.; Shanmugam, M. Glucose transporters in cancer metabolism. Curr. Opin. Oncol. 2012, 24, 650. [Google Scholar] [CrossRef]
- Yang, L.; Venneti, S.; Nagrath, D. Glutaminolysis: A Hallmark of Cancer Metabolism. Annu. Rev. Biomed. Eng. 2017, 19, 163–194. [Google Scholar] [CrossRef]
- Alberghina, L.; Gaglio, D. Redox control of glutamine utilization in cancer. Cell Death Dis. 2014, 5, e1561. [Google Scholar] [CrossRef] [Green Version]
- Teuwen, L.A.; Geldhof, V.; Carmeliet, P. How glucose, glutamine and fatty acid metabolism shape blood and lymph vessel development. Dev. Biol. 2019, 447, 90–102. [Google Scholar] [CrossRef]
- Hensley, C.T.; Wasti, A.T.; DeBerardinis, R.J. Glutamine and cancer: Cell biology, physiology, and clinical opportunities. J. Clin. Investig. 2013, 123, 3678–3684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Traverso, N.; Ricciarelli, R.; Nitti, M.; Marengo, B.; Furfaro, A.L.; Pronzato, M.A.; Marinari, U.M.; Domenicotti, C. Role of glutathione in cancer progression and chemoresistance. Oxid. Med. Cell. Longev. 2013, 2013, 972913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef]
- Hassanein, M.; Qian, J.; Hoeksema, M.D.; Wang, J.; Jacobovitz, M.; Ji, X.; Harris, F.T.; Harris, B.K.; Boyd, K.L.; Chen, H.; et al. Targeting SLC1a5-mediated glutamine dependence in non-small cell lung cancer. Int. J. Cancer 2015, 137, 1587–1597. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Cui, H. Targeting glutamine induces apoptosis: A cancer therapy approach. Int. J. Mol. Sci. 2015, 16, 22830–22855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gross, M.I.; Demo, S.D.; Dennison, J.B.; Chen, L.; Chernov-Rogan, T.; Goyal, B.; Janes, J.R.; Laidig, G.J.; Lewis, E.R.; Li, J.; et al. Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer. Mol. Cancer Ther. 2014, 13, 890–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, L.; Sheng, X.; Clark, L.H.; Zhang, L.; Guo, H.; Jones, H.M.; Willson, A.K.; Gehrig, P.A.; Zhou, C.; Bae-Jump, V.L. Glutaminase inhibitor compound 968 inhibits cell proliferation and sensitizes paclitaxel in ovarian cancer. Am. J. Transl. Res. 2016, 8, 4265. [Google Scholar]
- Dos Reis, L.M.; Adamoski, D.; Souza, R.O.O.; Ascenção, C.F.R.; De Oliveira, K.R.S.; Corrêa-Da-Silva, F.; De Sá Patroni, F.M.; Dias, M.M.; Consonni, S.R.; De Moraes-Vieira, P.M.M.; et al. Dual inhibition of glutaminase and carnitine palmitoyltransferase decreases growth and migration of glutaminase inhibition-resistant triple-negative breast cancer cells. J. Biol. Chem. 2019, 294, 9342–9357. [Google Scholar] [CrossRef] [PubMed]
- Katt, W.P.; Antonyak, M.A.; Cerione, R.A. Simultaneously targeting tissue transglutaminase and kidney type glutaminase sensitizes cancer cells to acid toxicity and offers new opportunities for therapeutic intervention. Mol. Pharm. 2015, 12, 46–55. [Google Scholar] [CrossRef] [Green Version]
- Saqcena, M.; Mukhopadhyay, S.; Hosny, C.; Alhamed, A.; Chatterjee, A.; Foster, D.A. Blocking anaplerotic entry of glutamine into the TCA cycle sensitizes K-Ras mutant cancer cells to cytotoxic drugs. Oncogene 2015, 34, 2672–2680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- San-Millán, I.; Brooks, G.A. Reexamining cancer metabolism: Lactate production for carcinogenesis could be the purpose and explanation of the Warburg Effect. Carcinogenesis 2017, 38, 119–133. [Google Scholar] [CrossRef] [PubMed]
- Sonveaux, P.; Végran, F.; Schroeder, T.; Wergin, M.C.; Verrax, J.; Rabbani, Z.N.; De Saedeleer, C.J.; Kennedy, K.M.; Diepart, C.; Jordan, B.F.; et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J. Clin. Investig. 2008, 118, 3930–3942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rawat, D.; Chhonker, S.K.; Naik, R.A.; Mehrotra, A.; Trigun, S.K.; Koiri, R.K. Lactate as a signaling molecule: Journey from dead end product of glycolysis to tumor survival. Front. Biosci. Landmark 2019, 24, 366–381. [Google Scholar] [CrossRef]
- Romero-Garcia, S.; Moreno-Altamirano, M.M.B.; Prado-Garcia, H.; Sánchez-García, F.J. Lactate contribution to the tumor microenvironment: Mechanisms, effects on immune cells and therapeutic relevance. Front. Immunol. 2016, 7, 52. [Google Scholar] [CrossRef] [Green Version]
- de la Cruz-López, K.G.; Castro-Muñoz, L.J.; Reyes-Hernández, D.O.; García-Carrancá, A.; Manzo-Merino, J. Lactate in the Regulation of Tumor Microenvironment and Therapeutic Approaches. Front. Oncol. 2019, 9, 1143. [Google Scholar] [CrossRef] [Green Version]
- Doherty, J.R.; Cleveland, J.L. Targeting lactate metabolism for cancer therapeutics. J. Clin. Investig. 2013, 123, 3685–3692. [Google Scholar] [CrossRef]
- Morandi, A.; Giannoni, E.; Chiarugi, P. Nutrient Exploitation within the Tumor–Stroma Metabolic Crosstalk. Trends Cancer 2016, 2, 736–746. [Google Scholar] [CrossRef]
- Hirschhaeuser, F.; Sattler, U.G.A.; Mueller-Klieser, W. Lactate: A metabolic key player in cancer. Cancer Res. 2011, 71, 6921–6925. [Google Scholar] [CrossRef] [Green Version]
- Arneth, B. Tumor Microenvironment. Medicina (Kaunas) 2019, 56, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gascard, P.; Tlsty, T.D. Carcinoma-associated fibroblasts: Orchestrating the composition of malignancy. Genes Dev. 2016, 30, 1002–1019. [Google Scholar] [CrossRef] [PubMed]
- Schurr, A. Lactate, Not Pyruvate, Is the End Product of Glucose Metabolism via Glycolysis. In Carbohydrate; InTech: Rijeka, Croatia, 2017. [Google Scholar]
- Andersen, A.P.; Flinck, M.; Oernbo, E.K.; Pedersen, N.B.; Viuff, B.M.; Pedersen, S.F. Roles of acid-extruding ion transporters in regulation of breast cancer cell growth in a 3-dimensional microenvironment. Mol. Cancer 2016, 15, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miranda-Gonçalves, V.; Granja, S.; Martinho, O.; Honavar, M.; Pojo, M.; Costa, B.M.; Pires, M.M.; Pinheiro, C.; Cordeiro, M.; Bebiano, G.; et al. Hypoxia-mediated upregulation of MCT1 expression supports the glycolytic phenotype of glioblastomas. Oncotarget 2016, 7, 46335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, B. Aerobic glycolysis and high level of lactate in cancer metabolism and microenvironment. Genes Dis. 2017, 4, 25–27. [Google Scholar] [CrossRef]
- Végran, F.; Boidot, R.; Michiels, C.; Sonveaux, P.; Feron, O. Lactate influx through the endothelial cell monocarboxylate transporter MCT1 supports an NF-kB/IL-8 pathway that drives tumor angiogenesis. Cancer Res. 2011, 71, 2550–2560. [Google Scholar] [CrossRef] [Green Version]
- Renner, K.; Bruss, C.; Schnell, A.; Koehl, G.; Becker, H.M.; Fante, M.; Menevse, A.N.; Kauer, N.; Blazquez, R.; Hacker, L.; et al. Restricting Glycolysis Preserves T Cell Effector Functions and Augments Checkpoint Therapy. Cell Rep. 2019, 29, 135–150. [Google Scholar] [CrossRef] [Green Version]
- Goetze, K.; Walenta, S.; Ksiazkiewicz, M.; Kunz-Schughart, L.A.; Mueller-Klieser, W. Lactate enhances motility of tumor cells and inhibits monocyte migration and cytokine release. Int. J. Oncol. 2011, 39, 453–463. [Google Scholar] [CrossRef]
- Chang, C.H.; Curtis, J.D.; Maggi, L.B.; Faubert, B.; Villarino, A.V.; O’Sullivan, D.; Huang, S.C.C.; Van Der Windt, G.J.W.; Blagih, J.; Qiu, J.; et al. XPosttranscriptional control of T cell effector function by aerobic glycolysis. Cell 2013, 153, 1239–1251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calcinotto, A.; Filipazzi, P.; Grioni, M.; Iero, M.; De Milito, A.; Ricupito, A.; Cova, A.; Canese, R.; Jachetti, E.; Rossetti, M.; et al. Modulation of microenvironment acidity reverses anergy in human and murine tumor-infiltrating T lymphocytes. Cancer Res. 2012, 72, 2746–2756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, S.; Wang, H.; Liu, J.; Jiye, A.A.; Zhou, F.; Wang, G. Multi-dimensional roles of ketone bodies in cancer biology: Opportunities for cancer therapy. Pharmacol. Res. 2019, 150, 104500. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Outschoorn, U.E.; Peiris-Pagés, M.; Pestell, R.G.; Sotgia, F.; Lisanti, M.P. Cancer metabolism: A therapeutic perspective. Nat. Rev. Clin. Oncol. 2017, 14, 11–31. [Google Scholar] [CrossRef]
- Newman, J.C.; Verdin, E. Ketone bodies as signaling metabolites. Trends Endocrinol. Metab. 2014, 25, 42–52. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Outschoorn, U.E.; Lisanti, M.P.; Sotgia, F. Catabolic cancer-associated fibroblasts transfer energy and biomass to anabolic cancer cells, fueling tumor growth. Semin. Cancer Biol. 2014, 25, 47–60. [Google Scholar] [CrossRef]
- Aminzadeh-Gohari, S.; Feichtinger, R.G.; Vidali, S.; Locker, F.; Rutherford, T.; O’Donnel, M.; Stöger-Kleiber, A.; Mayr, J.A.; Sperl, W.; Kofler, B. A ketogenic diet supplemented with medium-chain triglycerides enhances the anti-tumor and anti-angiogenic efficacy of chemotherapy on neuroblastoma xenografts in a CD1-nu mouse model. Oncotarget 2017, 8, 64728. [Google Scholar] [CrossRef] [Green Version]
- Allen, B.G.; Bhatia, S.K.; Buatti, J.M.; Brandt, K.E.; Lindholm, K.E.; Button, A.M.; Szweda, L.I.; Smith, B.J.; Spitz, D.R.; Fath, M.A. Ketogenic diets enhance oxidative stress and radio-chemo-therapy responses in lung cancer xenografts. Clin. Cancer Res. 2013, 19, 3905–3913. [Google Scholar] [CrossRef] [Green Version]
- Tan-Shalaby, J. Ketogenic Diets and Cancer: Emerging Evidence. Fed. Pract. 2017, 34 (Suppl. 1), 37S–42S. [Google Scholar]
- Weber, D.D.; Aminzadeh-Gohari, S.; Tulipan, J.; Catalano, L.; Feichtinger, R.G.; Kofler, B. Ketogenic diet in the treatment of cancer—Where do we stand? Mol. Metab. 2020, 33, 102–121. [Google Scholar] [CrossRef]
- Masino, S.A. Ketogenic Diet And Metabolic Therapies; Oxford University Press: New York, NY, USA, 2017. [Google Scholar]
- Currie, E.; Schulze, A.; Zechner, R.; Walther, T.C.; Farese, R.V. Cellular fatty acid metabolism and cancer. Cell Metab. 2013, 18, 153–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A.; et al. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2016, 44, D279–D285. [Google Scholar] [CrossRef]
- Mueckler, M.; Thorens, B. The SLC2 (GLUT) family of membrane transporters. Mol. Asp. Med. 2013, 34, 121–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joost, H.-G.; Thorens, B. The extended GLUT-family of sugar/polyol transport facilitators: Nomenclature, sequence characteristics, and potential function of its novel members. Mol. Membr. Biol. 2001, 18, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Augustin, R. The protein family of glucose transport facilitators: It’s not only about glucose after all. IUBMB Life 2010, 62, 315–333. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.-Q.; Keating, A.F. Functional properties and genomics of glucose transporters. Curr. Genomics 2007, 8, 113–128. [Google Scholar] [CrossRef]
- Samih, N.; Hovsepian, S.; Notel, F.; Prorok, M.; Zattara-Cannoni, H.; Mathieu, S.; Lombardo, D.; Fayet, G.; El-Battari, A. The impact of N- and O-glycosylation on the functions of Glut-1 transporter in human thyroid anaplastic cells. Biochim. Biophys. Acta Gen. Subj. 2003, 1621, 92–101. [Google Scholar] [CrossRef]
- Deng, D.; Xu, C.; Sun, P.; Wu, J.; Yan, C.; Hu, M.; Yan, N. Crystal structure of the human glucose transporter GLUT1. Nature 2014, 510, 121–125. [Google Scholar] [CrossRef]
- Wieman, H.L.; Horn, S.H.; Jacobs, S.R.; Altman, B.J.; Kornbluth, S.; Rathmell, J.C. An essential role for the Glut1 PDZ-binding motif in growth factor regulation of Glut1 degradation and trafficking. Biochem. J. 2009, 418, 345–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carvalho, K.C.; Cunha, I.W.; Rocha, R.M.; Ayala, F.R.; Cajaíba, M.M.; Begnami, M.D.; Vilela, R.S.; Paiva, G.R.; Andrade, R.G.; Soares, F.A. GLUT1 expression in malignant tumors and its use as an immunodiagnostic marker. Clinics 2011, 66, 965–972. [Google Scholar] [CrossRef] [Green Version]
- Szablewski, L. Expression of glucose transporters in cancers. Biochim. Biophys. Acta Rev. Cancer 2013, 1835, 164–169. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Ye, C.; Chen, C.; Xiong, H.; Xie, B.; Zhou, J.; Chen, Y.; Zheng, S.; Wang, L. Glucose transporter GLUT1 expression and clinical outcome in solid tumors: A systematic review and meta-analysis. Oncotarget 2017, 8, 16875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halestrap, A.P.; Wilson, M.C. The monocarboxylate transporter family-Role and regulation. IUBMB Life 2012, 64, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.S.; Morris, M.E. Monocarboxylate Transporters: Therapeutic Targets and Prognostic Factors in Disease. Clin. Pharmacol. Ther. 2016, 100, 454–463. [Google Scholar] [CrossRef]
- Baltazar, F.; Pinheiro, C.; Morais-Santos, F.; Azevedo-Silva, J.; Queirós, O.; Preto, A.; Casal, M. Monocarboxylate transporters as targets and mediators in cancer therapy response. Histol. Histopathol. 2014, 29, 1511–1524. [Google Scholar]
- Payen, V.L.; Mina, E.; Van Hée, V.F.; Porporato, P.E.; Sonveaux, P. Monocarboxylate transporters in cancer. Mol. Metab. 2020, 33, 48–66. [Google Scholar] [CrossRef]
- Zhang, G.; Zhang, Y.; Dong, D.; Wang, F.; Ma, X.; Guan, F.; Sun, L. MCT1 regulates aggressive and metabolic phenotypes in bladder cancer. J. Cancer 2018, 9, 2492. [Google Scholar] [CrossRef]
- Granja, S.; Tavares-Valente, D.; Queirós, O.; Baltazar, F. Value of pH regulators in the diagnosis, prognosis and treatment of cancer. Semin. Cancer Biol. 2017, 43, 17–34. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Main Activity | Type of Cancer | References |
|---|---|---|---|
| CPI-613 | inhibits pyruvate dehydrogenase (PDH) and a-ketogluterate dehydrogenase (KGDH) | Hematologic Malignancies | [77,78] |
| Olutasidenib (FT-2102) | Mutant isocitrate dehydrogenase 1 inhibitor | Acute Myeloid Leukemia | [79,80] |
| TVB-2640 | Fatty Acid Synthase inhibitor | Non-small Cell Lung Cancer, Breast Cancer, Ovarian Cancer, Astrocytoma | [81,82] |
| Enasidenib (AG-221) | Mutant Isocitrate Dehydrogenase 2 Inhibitor | Acute Myeloid Leukemia | [83] |
| WZB117 | GLUT1 inhibitor | Lung cancer, Breast cancer | [84,85] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kubicka, A.; Matczak, K.; Łabieniec-Watała, M. More Than Meets the Eye Regarding Cancer Metabolism. Int. J. Mol. Sci. 2021, 22, 9507. https://doi.org/10.3390/ijms22179507
Kubicka A, Matczak K, Łabieniec-Watała M. More Than Meets the Eye Regarding Cancer Metabolism. International Journal of Molecular Sciences. 2021; 22(17):9507. https://doi.org/10.3390/ijms22179507
Chicago/Turabian StyleKubicka, Anna, Karolina Matczak, and Magdalena Łabieniec-Watała. 2021. "More Than Meets the Eye Regarding Cancer Metabolism" International Journal of Molecular Sciences 22, no. 17: 9507. https://doi.org/10.3390/ijms22179507