
Step-by-Step Immune Activation for Suicide Gene Therapy Reinforcement

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
- Act on different types of cancer, similar to classical chemotherapy or radiotherapy;
- Offer enhanced efficiency and safety due to their expression within the tumor, thus causing minimal damage outside the tumor and, at the same time, inducing overall antitumor and antimetastatic immunity;
- Remain inexpensive due to their simple production technology;
- Be used in combination with traditional chemo- and radiotherapy, as well as with newly developed immunotherapies.
2. GDEPT: Why Good Intentions Do Not Lead to Paradise in Suicide Cancer Gene Therapy
3. A Brief Description of GM–CSF, the Most Commonly Used Immunomodulator
4. The Involvement of Danger Signals and Immunogenic Cell Death Could Further Improve the Antitumor Immune Response
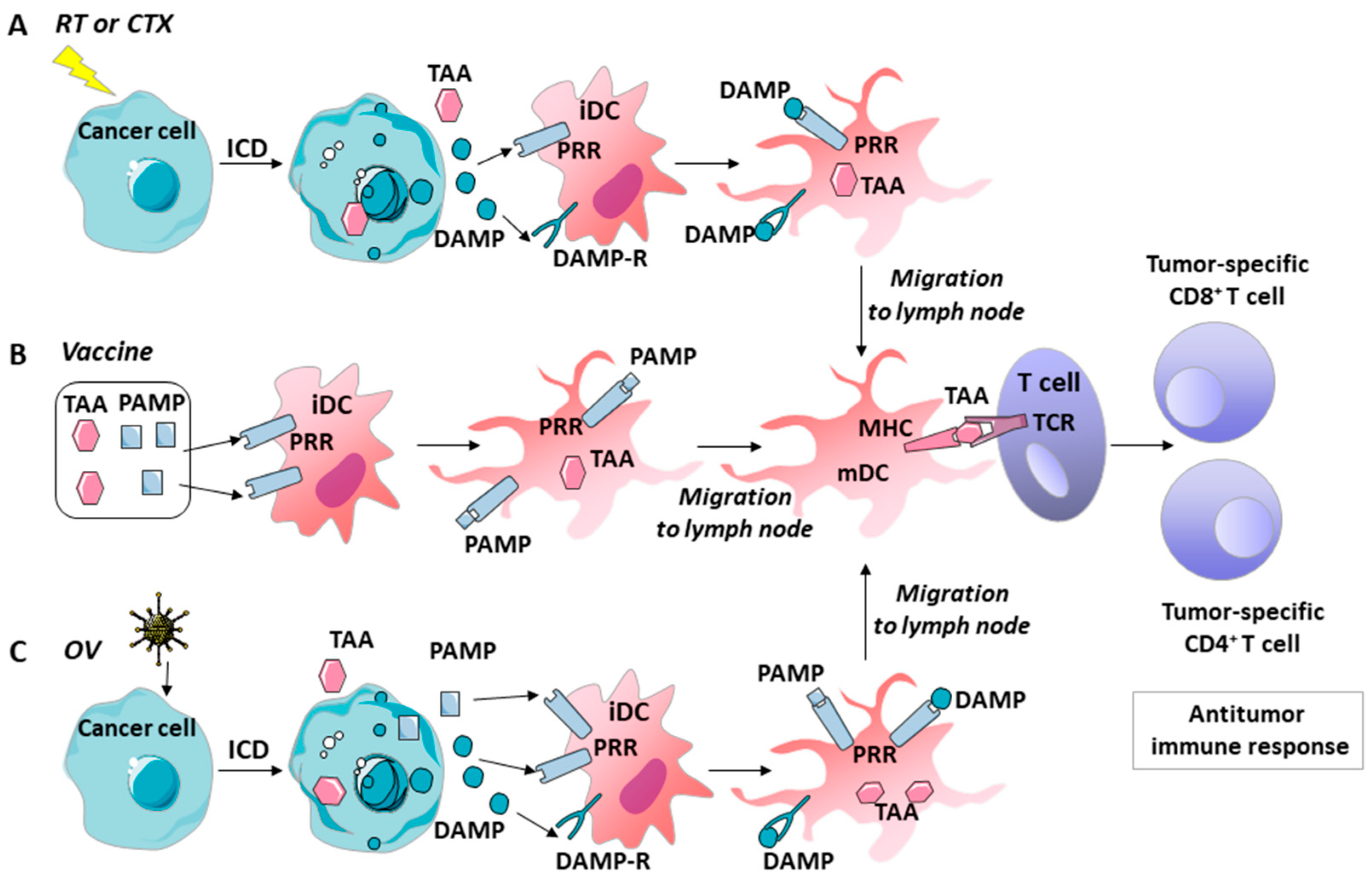
5. GM–CSF, Danger Signals, and ICD’s Role in the Immune Effects of Various Tumor Therapies
5.1. Chemotherapy
5.2. Radiotherapy
5.3. Vaccines
5.4. Oncolytic Viruses
6. Opportunities for Combinations of Chemo-, Radio-, Suicide-, and Immunotherapy
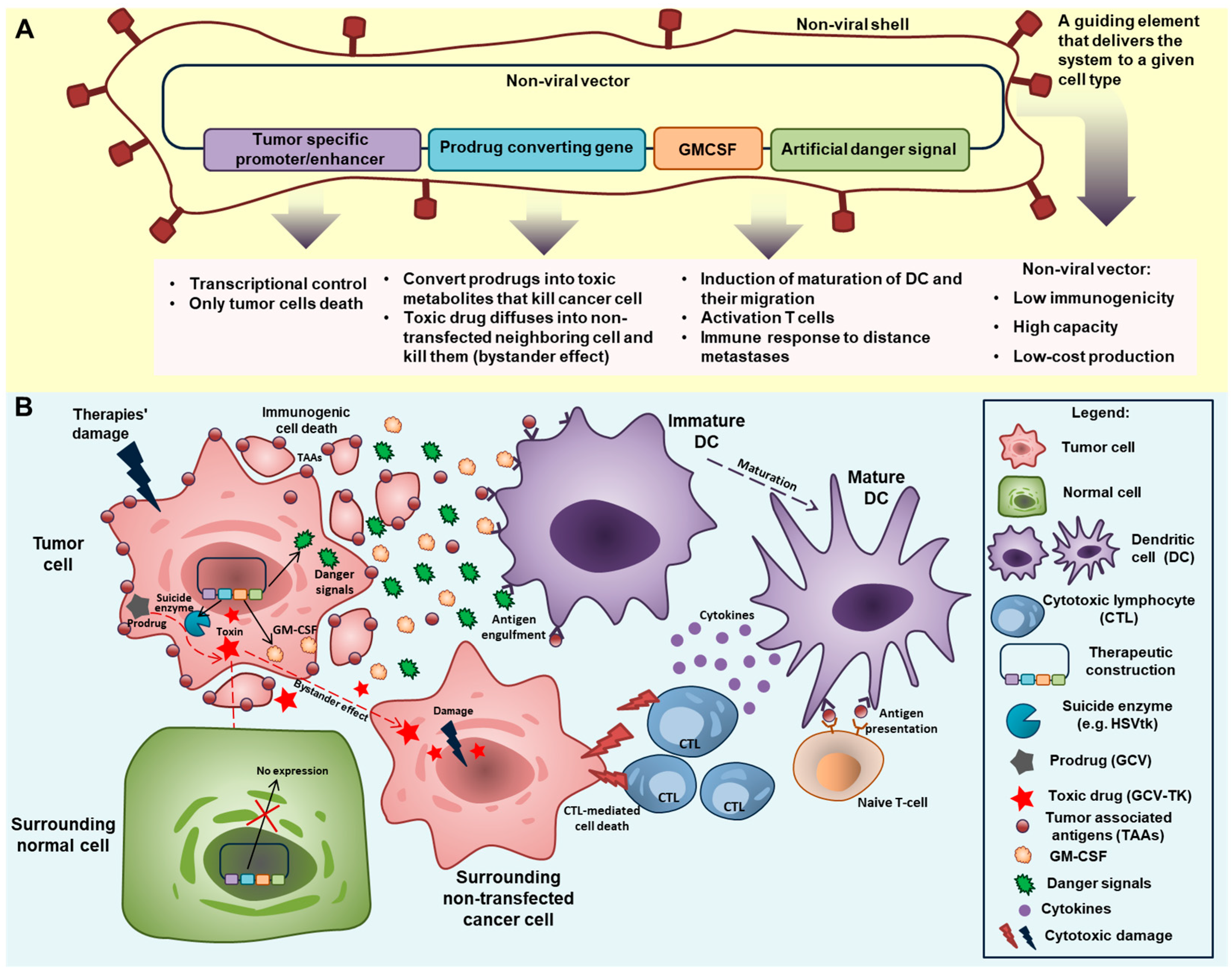
7. Regulation of Expression and Optimization of the Delivery System in Support of Anticancer Gene Therapy
7.1. Expression Regulatory Element
7.2. Non-Viral Gene-Delivery System
8. Conclusions: A Look at GDEPT’s Future Development
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bordon, Y. Immunotherapy: Checkpoint parley. Nat. Rev. Cancer 2015, 15, 3. [Google Scholar] [CrossRef]
- Park, J.; Kwon, M.; Shin, E.-C. Immune checkpoint inhibitors for cancer treatment. Arch. Pharmacal Res. 2016, 39, 1577–1587. [Google Scholar] [CrossRef] [PubMed]
- Pennock, N.; White, J.T.; Cross, E.W.; Cheney, E.E.; Tamburini, B.A.; Kedl, R.M. T cell responses: Naïve to memory and everything in between. Adv. Physiol. Educ. 2013, 37, 273–283. [Google Scholar] [CrossRef] [Green Version]
- Smyth, M.J.; Ngiow, S.F.; Ribas, A.; Teng, M.W.L. Combination cancer immunotherapies tailored to the tumour microenvironment. Nat. Rev. Clin. Oncol. 2016, 13, 143–158. [Google Scholar] [CrossRef] [PubMed]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef]
- Postow, M.A.; Callahan, M.K.; Wolchok, J.D. Immune Checkpoint Blockade in Cancer Therapy. J. Clin. Oncol. 2015, 33, 1974–1982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diesendruck, Y.; Benhar, I. Novel immune check point inhibiting antibodies in cancer therapy—Opportunities and challenges. Drug Resist. Updates 2017, 30, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Vreeland, T.J.; Clifton, G.T.; Herbert, G.S.; Hale, D.F.; Jackson, D.O.; Berry, J.S.; Peoples, G.E. Gaining ground on a cure through synergy: Combining checkpoint inhibitors with cancer vaccines. Expert Rev. Clin. Immunol. 2016, 12, 1347–1357. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, L.; Velcheti, V. Checkpoint immunotherapy: Good for cancer therapy, bad for rheumatic diseases. Ann. Rheum. Dis. 2017, 76, 1–3. [Google Scholar] [CrossRef] [Green Version]
- Alekseenko, I.V.; Snezhkov, E.V.; Chernov, I.P.; Pleshkan, V.V.; Potapov, V.K.; Sass, A.V.; Monastyrskaya, G.S.; Kopantzev, E.P.; Vinogradova, T.V.; Khramtsov, Y.V.; et al. Therapeutic properties of a vector carrying the HSV thymidine kinase and GM-CSF genes and delivered as a complex with a cationic copolymer. J. Transl. Med. 2015, 13, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Duarte, S.; Carle, G.; Faneca, H.; de Lima, M.C.P.; Pierrefite-Carle, V. Suicide gene therapy in cancer: Where do we stand now? Cancer Lett. 2012, 324, 160–170. [Google Scholar] [CrossRef]
- Portsmouth, D.; Hlavaty, J.; Renner, M. Suicide genes for cancer therapy. Mol. Asp. Med. 2007, 28, 4–41. [Google Scholar] [CrossRef]
- Altaner, C. Prodrug cancer gene therapy. Cancer Lett. 2008, 270, 191–201. [Google Scholar] [CrossRef]
- Zhang, J.; Kale, V.; Chen, M. Gene-Directed Enzyme Prodrug Therapy. AAPS J. 2015, 17, 102–110. [Google Scholar] [CrossRef] [Green Version]
- Karjoo, Z.; Chen, X.; Hatefi, A. Progress and problems with the use of suicide genes for targeted cancer therapy. Adv. Drug Deliv. Rev. 2016, 99, 113–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, Y.; Fan, J.; Deng, L.; Huang, B.; Zhou, B. Antitumor efficacy of cytosine deaminase-armed vaccinia virus plus 5-fluorocytosine in colorectal cancers. Cancer Cell Int. 2020, 20, 243. [Google Scholar] [CrossRef]
- Mishra, A.P.; Chandra, S.; Tiwari, R.; Srivastava, A.; Tiwari, G. Therapeutic Potential of Prodrugs Towards Targeted Drug Delivery. Open Med. Chem. J. 2018, 12, 111–123. [Google Scholar] [CrossRef] [Green Version]
- Souza, C.; Pellosi, D.; Tedesco, A.C. Prodrugs for targeted cancer therapy. Expert Rev. Anticancer Ther. 2019, 19, 483–502. [Google Scholar] [CrossRef] [PubMed]
- Franzyk, H.; Christensen, S. Targeting Toxins toward Tumors. Molecules 2021, 26, 1292. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.-P.; Seo, H.-H.; Shin, J.-H.; Park, H.-B.; Lim, D.-P.; Eom, H.-S.; Bae, Y.-S.; Kim, I.-H.; Choi, K.; Lee, S.-J. Adenovirus Expressing Both Thymidine Kinase and Soluble PD1 Enhances Antitumor Immunity by Strengthening CD8 T-cell Response. Mol. Ther. 2013, 21, 688–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barba, D.; Hardin, J.; Sadelain, M.; Gage, F.H. Development of anti-tumor immunity following thymidine kinase-mediated killing of experimental brain tumors. Proc. Natl. Acad. Sci. USA 1994, 91, 4348–4352. [Google Scholar] [CrossRef] [Green Version]
- Kuriyama, S.; Kikukawa, M.; Masui, K.; Okuda, H.; Nakatani, T.; Akahane, T.; Mitoro, A.; Tominaga, K.; Tsujinoue, H.; Yoshiji, H.; et al. Cancer gene therapy with HSV-tk/GCV system depends on t-cell-mediated immune responses and causes apoptotic death of tumor cellsIn vivo. Int. J. Cancer 1999, 83, 374–380. [Google Scholar] [CrossRef]
- Haack, K.; Linnebacher, M.; Eisold, S.; Zöller, M.; von Knebel Doeberitz, M.; Gebert, J. Induction of protective immunity against syngeneic rat cancer cells by expression of the cytosine deaminase suicide gene. Cancer Gene Ther. 2000, 7, 1357–1364. [Google Scholar] [CrossRef] [PubMed]
- Touati, W.; Tran, T.; Seguin, J.; Diry, M.; Flinois, J.-P.; Baillou, C.; Lescaille, G.; Andre, F.; Tartour, E.; Lemoine, F.; et al. A Suicide Gene Therapy Combining the Improvement of Cyclophosphamide Tumor Cytotoxicity and the Development of an Anti-Tumor Immune Response. Curr. Gene Ther. 2014, 14, 236–246. [Google Scholar] [CrossRef]
- Nayagom, B.; Amara, I.; Habiballah, M.; Amrouche, F.; Beaune, P.; De Waziers, I. Immunogenic cell death in a combined synergic gene- and immune-therapy against cancer. OncoImmunology 2019, 8, e1667743. [Google Scholar] [CrossRef] [PubMed]
- Greco, O.; Dachs, G.U. Gene directed enzyme/prodrug therapy of cancer: Historical appraisal and future prospectives. J. Cell. Physiol. 2001, 187, 22–36. [Google Scholar] [CrossRef]
- Van Putten, E.H.; Dirven, C.M.; van den Bent, M.J.; Lamfers, M.L. Sitimagene ceradenovec: A gene-based drug for the treatment of operable high-grade glioma. Future Oncol. 2010, 6, 1691–1710. [Google Scholar] [CrossRef] [PubMed]
- Westphal, M.; Ylä-Herttuala, S.; Martin, J.; Warnke, P.; Menei, P.; Eckland, D.; Kinley, J.; Kay, R.; Ram, Z. Adenovirus-mediated gene therapy with sitimagene ceradenovec followed by intravenous ganciclovir for patients with operable high-grade glioma (ASPECT): A randomised, open-label, phase 3 trial. Lancet Oncol. 2013, 14, 823–833. [Google Scholar] [CrossRef]
- Collins, S.A.; Shah, A.H.; Ostertag, D.; Kasahara, N.; Jolly, D.J. Clinical development of retroviral replicating vector Toca 511 for gene therapy of cancer. Expert Opin. Biol. Ther. 2021, 1–16. [Google Scholar] [CrossRef]
- Culver, K.; Ram, Z.; Wallbridge, S.; Ishii, H.; Oldfield, E.; Blaese, R. In vivo gene transfer with retroviral vector-producer cells for treatment of experimental brain tumors. Science 1992, 256, 1550–1552. [Google Scholar] [CrossRef]
- Connors, T.A.; Knox, R.J. Prodrugs in cancer chemotherapy. Stem Cells 1995, 13, 501–511. [Google Scholar] [CrossRef]
- Rangel-Sosa, M.M.; Aguilar-Córdova, E.; Rojas-Martinez, A. Immunotherapy and gene therapy as novel treatments for cancer. Colomb. Med. 2017, 48, 138–147. [Google Scholar] [CrossRef] [Green Version]
- Malekshah, O.M.; Chen, X.; Nomani, A.; Sarkar, S.; Hatefi, A. Enzyme/Prodrug Systems for Cancer Gene Therapy. Curr. Pharmacol. Rep. 2016, 2, 299–308. [Google Scholar] [CrossRef] [Green Version]
- Slavcev, R.A.; Wettig, S.; Kaur, T. Nanomedicine Based Approaches to Cancer Diagonsis and Therapy, Non-Viral Gene Therapy; Yuan, P.X., Ed.; InTech: Rijeka, Croatia, 2011. [Google Scholar]
- Brockstedt, D.G.; Diagana, M.; Zhang, Y.; Tran, K.; Belmar, N.; Meier, M.; Yang, A.; Boissiere, F.; Lin, A.; Chiang, Y. Development of anti-tumor immunity against a non-immunogenic mammary carcinoma through in vivo somatic GM-CSF, IL-2, and HSVtk combination gene therapy. Mol. Ther. 2002, 6, 627–636. [Google Scholar] [CrossRef]
- Majumdar, A.S.; Zolotorev, A.; Samuel, S.; Tran, K.; Vertin, B.; Hall-Meier, M.; Antoni, B.-A.; Adeline, E.; Philip, M.; Philip, R. Efficacy of herpes simplex virus thymidine kinase in combination with cytokine gene therapy in an experimental metastatic breast cancer model. Cancer Gene Ther. 2000, 7, 1086–1099. [Google Scholar] [CrossRef] [Green Version]
- Jones, R.K.; Pope, I.M.; Kinsella, A.R.; Watson, A.J.; Christmas, S. Combined suicide and granulocyte–macrophage colony-stimulating factor gene therapy induces complete tumor regression and generates antitumor immunity. Cancer Gene Ther. 2000, 7, 1519–1528. [Google Scholar] [CrossRef] [Green Version]
- Pesonen, S.; Kangasniemi, L.; Hemminki, A. Oncolytic Adenoviruses for the Treatment of Human Cancer: Focus on Translational and Clinical Data. Mol. Pharm. 2011, 8, 12–28. [Google Scholar] [CrossRef] [PubMed]
- Bayne, L.J.; Beatty, G.; Jhala, N.; Clark, C.E.; Rhim, A.D.; Stanger, B.Z.; Vonderheide, R.H. Tumor-Derived Granulocyte-Macrophage Colony-Stimulating Factor Regulates Myeloid Inflammation and T Cell Immunity in Pancreatic Cancer. Cancer Cell 2012, 21, 822–835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutschalk, C.M.; Yanamandra, A.K.; Linde, N.; Meides, A.; Depner, S.; Mueller, M.M. GM-CSF enhances tumor invasion by elevated MMP-2, -9, and -26 expression. Cancer Med. 2013, 2, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Ostrand-Rosenberg, S.; Sinha, P.; Beury, D.W.; Clements, V.K. Cross-talk between myeloid-derived suppressor cells (MDSC), macrophages, and dendritic cells enhances tumor-induced immune suppression. Semin. Cancer Biol. 2012, 22, 275–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metcalf, D.; Nicola, N. The Hemopoietic Colony-Stimulating Factors: From Biology to Clinical Applications; Cambridge University Press: Cambridge, MA, USA, 1995. [Google Scholar]
- Bhattacharya, P.; Thiruppathi, M.; Elshabrawy, H.A.; Alharshawi, K.; Kumar, P.; Prabhakar, B.S. GM-CSF: An immune modulatory cytokine that can suppress autoimmunity. Cytokine 2015, 75, 261–271. [Google Scholar] [CrossRef] [Green Version]
- Naik, S.H. Demystifying the development of dendritic cell subtypes, a little. Immunol. Cell Biol. 2008, 86, 439–452. [Google Scholar] [CrossRef] [PubMed]
- Van De Laar, L.; Coffer, P.; Woltman, A. Regulation of dendritic cell development by GM-CSF: Molecular control and implications for immune homeostasis and therapy. Blood 2012, 119, 3383–3393. [Google Scholar] [CrossRef] [PubMed]
- Zhan, Y.; Lew, A.M.; Chopin, M. The Pleiotropic Effects of the GM-CSF Rheostat on Myeloid Cell Differentiation and Function: More Than a Numbers Game. Front. Immunol. 2019, 10, 2679. [Google Scholar] [CrossRef] [PubMed]
- Willart, M.A.; Deswarte, K.; Pouliot, P.; Braun, H.; Beyaert, R.; Lambrecht, B.N.; Hammad, H. Interleukin-1α controls allergic sensitization to inhaled house dust mite via the epithelial release of GM-CSF and IL-33. J. Exp. Med. 2012, 209, 1505–1517. [Google Scholar] [CrossRef] [Green Version]
- Kanerva, A.; Nokisalmi, P.; Diaconu, I.; Koski-Palkén, A.; Cerullo, V.; Liikanen, I.; Tähtinen, S.; Oksanen, M.; Heiskanen, R.; Pesonen, S.; et al. Antiviral and Antitumor T-cell Immunity in Patients Treated with GM-CSF–Coding Oncolytic Adenovirus. Clin. Cancer Res. 2013, 19, 2734–2744. [Google Scholar] [CrossRef] [Green Version]
- Herndler-Brandstetter, D.; Flavell, R.A. Producing GM-CSF: A unique T helper subset? Cell Res. 2014, 24, 1379–1380. [Google Scholar] [CrossRef] [Green Version]
- Sheng, W.; Yang, F.; Zhou, Y.; Yang, H.; Low, P.Y.; Kemeny, D.M.; Tan, P.; Moh, A.; Kaplan, M.H.; Zhang, Y.; et al. STAT5 programs a distinct subset of GM-CSF-producing T helper cells that is essential for autoimmune neuroinflammation. Cell Res. 2014, 24, 1387–1402. [Google Scholar] [CrossRef] [Green Version]
- Rosas, M.; Gordon, S.; Taylor, P.R. Characterisation of the expression and function of the GM-CSF receptor α-chain in mice. Eur. J. Immunol. 2007, 37, 2518–2528. [Google Scholar] [CrossRef] [Green Version]
- Clive, K.S.; Tyler, J.A.; Clifton, G.T.; Holmes, J.P.; Mittendorf, E.A.; Ponniah, S.; Peoples, G.E. Use of GM-CSF as an adjuvant with cancer vaccines: Beneficial or detrimental? Expert Rev. Vaccines 2010, 9, 519–525. [Google Scholar] [CrossRef]
- Nebiker, C.; Han, J.; Eppenberger-Castori, S.; Iezzi, G.; Hirt, C.; Amicarella, F.; Cremonesi, E.; Huber, X.; Padovan, E.; Angrisani, B.; et al. GM-CSF Production by Tumor Cells Is Associated with Improved Survival in Colorectal Cancer. Clin. Cancer Res. 2014, 20, 3094–3106. [Google Scholar] [CrossRef] [Green Version]
- Hong, I.-S. Stimulatory versus suppressive effects of GM-CSF on tumor progression in multiple cancer types. Exp. Mol. Med. 2016, 48, e242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aliper, A.M.; Frieden-Korovkina, V.P.; Buzdin, A.; Roumiantsev, S.A.; Zhavoronkov, A. A role for G-CSF and GM-CSF in nonmyeloid cancers. Cancer Med. 2014, 3, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Hoeller, C.; Michielin, O.; Ascierto, P.A.; Szabo, Z.; Blank, C.U. Systematic review of the use of granulocyte–macrophage colony-stimulating factor in patients with advanced melanoma. Cancer Immunol. Immunother. 2016, 65, 1015–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sverdlov, E.D. Systems biology and personalized medicine: To be or not to be? Ross. Fiziol. Zh. Im. I. M. Sechenova 2014, 100, 505–541. [Google Scholar]
- Sverdlov, E.D. Multidimensional complexity of cancer. Simple solutions are needed. Biochemistry 2016, 81, 731–738. [Google Scholar] [CrossRef]
- Matzinger, P. An Innate Sense of Danger. Ann. N. Y. Acad. Sci. 2002, 961, 341–342. [Google Scholar] [CrossRef]
- Matzinger, P. The Danger Model: A Renewed Sense of Self. Science 2002, 296, 301–305. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Evans, J.E.; Rock, K.L. Molecular identification of a danger signal that alerts the immune system to dying cells. Nature 2003, 425, 516–521. [Google Scholar] [CrossRef]
- Galluzzi, L.; Buqué, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunogenic cell death in cancer and infectious disease. Nat. Rev. Immunol. 2017, 17, 97–111. [Google Scholar] [CrossRef]
- Heil, M.; Land, W.G. Danger signals—Damaged-self recognition across the tree of life. Front. Plant Sci. 2014, 5, 578. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.S.; Liu, Z.; Bartlett, D.L. Oncolytic Immunotherapy: Dying the Right Way is a Key to Eliciting Potent Antitumor Immunity. Front. Oncol. 2014, 4, 74. [Google Scholar] [CrossRef] [Green Version]
- Wicks, I.P.; Roberts, A.W. Targeting GM-CSF in inflammatory diseases. Nat. Rev. Rheumatol. 2016, 12, 37–48. [Google Scholar] [CrossRef]
- Koyama, Y.; Yoshihara, C.; Ito, T. Novel Antitumor Strategy Utilizing a Plasmid Expressing a Mycobacterium tuberculosis Antigen as a “Danger Signal” to Block Immune Escape of Tumor Cells. Pharmaceutics 2015, 7, 165–174. [Google Scholar] [CrossRef]
- Kroemer, G.; Galluzzi, L.; Kepp, O.; Zitvogel, L. Immunogenic Cell Death in Cancer Therapy. Annu. Rev. Immunol. 2013, 31, 51–72. [Google Scholar] [CrossRef]
- Golden, E.B.; Epellicciotta, I.; Edemaria, S.; Ebarcellos-Hoff, M.H.; Formenti, S.C. The convergence of radiation and immunogenic cell death signaling pathways. Front. Oncol. 2012, 2, 88. [Google Scholar] [CrossRef] [Green Version]
- Woller, N.; Gürlevik, E.; Ureche, C.-I.; Schumacher, A.; Kühnel, F. Oncolytic Viruses as Anticancer Vaccines. Front. Oncol. 2014, 4, 188. [Google Scholar] [CrossRef] [Green Version]
- Uusi-Kerttula, H.; Hulin-Curtis, S.; Davies, J.; Parker, A.L. Oncolytic Adenovirus: Strategies and Insights for Vector Design and Immuno-Oncolytic Applications. Viruses 2015, 7, 6009–6042. [Google Scholar] [CrossRef] [Green Version]
- Hojeij, R.; Domingos-Pereira, S.; Nkosi, M.; Gharbi, D.; Derré, L.; Schiller, J.T.; Jichlinski, P.; Nardelli-Haefliger, D. Immunogenic Human Papillomavirus Pseudovirus-Mediated Suicide-Gene Therapy for Bladder Cancer. Int. J. Mol. Sci. 2016, 17, 1125. [Google Scholar] [CrossRef] [PubMed]
- Walters, J.N.; Ferraro, B.; Duperret, E.K.; Kraynyak, K.A.; Chu, J.; Saint-Fleur, A.; Yan, J.; Levitsky, H.; Khan, A.S.; Sardesai, N.; et al. A Novel DNA Vaccine Platform Enhances Neo-antigen-like T Cell Responses against WT1 to Break Tolerance and Induce Anti-tumor Immunity. Mol. Ther. 2017, 25, 976–988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [Green Version]
- Palucka, A.K.; Coussens, L.M. The basis of oncoimmunology. Cell 2016, 164, 1233–1247. [Google Scholar] [CrossRef] [Green Version]
- Kohlhapp, F.J.; Kaufman, H.L. Molecular Pathways: Mechanism of Action for Talimogene Laherparepvec, a New Oncolytic Virus Immunotherapy. Clin. Cancer Res. 2016, 22, 1048–1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, J.M.; Andrei, A.-C.; McNeel, D.G. Prioritization of cancer antigens: Keeping the target in sight. Expert Rev. Vaccines 2009, 8, 1657–1661. [Google Scholar] [CrossRef] [PubMed]
- Cheever, M.A.; Allison, J.; Ferris, A.S.; Finn, O.J.; Hastings, B.M.; Hecht, T.T.; Mellman, I.; Prindiville, S.A.; Viner, J.L.; Weiner, L.M.; et al. The Prioritization of Cancer Antigens: A National Cancer Institute Pilot Project for the Acceleration of Translational Research. Clin. Cancer Res. 2009, 15, 5323–5337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eike, L.-M.; Yang, N.; Rekdal, Ø.; Sveinbjørnsson, B. The oncolytic peptide LTX-315 induces cell death and DAMP release by mitochondria distortion in human melanoma cells. Oncotarget 2015, 6, 34910–34923. [Google Scholar] [CrossRef] [Green Version]
- Camilio, K.A.; Rekdal, O.; Sveinbjornsson, B. LTX-315 (Oncopore): A short synthetic anticancer peptide and novel immunotherapeutic agent. Oncoimmunology 2014, 3, e29181. [Google Scholar] [CrossRef] [Green Version]
- Luchner, M.; Reinke, S.; Milicic, A. TLR Agonists as Vaccine Adjuvants Targeting Cancer and Infectious Diseases. Pharmaceutics 2021, 13, 142. [Google Scholar] [CrossRef]
- Vijayan, A.; Rumbo, M.; Carnoy, C.; Sirard, J.-C. Compartmentalized Antimicrobial Defenses in Response to Flagellin. Trends Microbiol. 2018, 26, 423–435. [Google Scholar] [CrossRef] [Green Version]
- Leigh, N.D.; Bian, G.; Ding, X.; Liu, H.; Aygun-Sunar, S.; Burdelya, L.G.; Gudkov, A.V.; Cao, X. A Flagellin-Derived Toll-Like Receptor 5 Agonist Stimulates Cytotoxic Lymphocyte-Mediated Tumor Immunity. PLoS ONE 2014, 9, e85587. [Google Scholar] [CrossRef]
- Sfondrini, L.; Rossini, A.; Besusso, D.; Merlo, A.; Tagliabue, E.; Nard, S.M.; Balsari, A. Antitumor Activity of the TLR-5 Ligand Flagellin in Mouse Models of Cancer. J. Immunol. 2006, 176, 6624–6630. [Google Scholar] [CrossRef]
- Geng, D.; Kaczanowska, S.; Tsai, A.; Younger, K.; Ochoa, A.; Rapoport, A.P.; Ostrand-Rosenberg, S.; Davila, E. TLR5 Ligand–Secreting T Cells Reshape the Tumor Microenvironment and Enhance Antitumor Activity. Cancer Res. 2015, 75, 1959–1971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, X.; Guo, C.; Yi, H.; Qian, J.; Fisher, P.B.; Subjeck, J.R.; Wang, X.-Y. A Multifunctional Chimeric Chaperone Serves as a Novel Immune Modulator Inducing Therapeutic Antitumor Immunity. Cancer Res. 2013, 73, 2093–2103. [Google Scholar] [CrossRef] [Green Version]
- Fan, R.; Wang, C.; Wang, Y.; Ren, P.; Gan, P.; Ji, H.; Xia, Z.; Hu, S.; Zeng, Q.; Huang, W.; et al. Enhanced antitumoral efficacy and immune response following conditionally replicative adenovirus containing constitutive HSF1 delivery to rodent tumors. J. Transl. Med. 2012, 10, 101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, X.F.; Ren, W.; Rollins, L.; Pittman, P.; Shah, M.; Shen, L.; Gu, Q.; Strube, R.; Hu, F.; Chen, S.-Y. A broadly applicable, personalized heat shock protein-mediated oncolytic tumor vaccine. Cancer Res. 2003, 63, 7321–7329. [Google Scholar] [PubMed]
- Li, J.-L.; Liu, H.-L.; Zhang, X.-R.; Xu, J.-P.; Hu, W.-K.; Liang, M.; Chen, S.-Y.; Hu, F.; Chu, D.-T. A phase I trial of intratumoral administration of recombinant oncolytic adenovirus overexpressing HSP70 in advanced solid tumor patients. Gene Ther. 2009, 16, 376–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montico, B.; Nigro, A.; Casolaro, V.; Col, J.D. Immunogenic Apoptosis as a Novel Tool for Anticancer Vaccine Development. Int. J. Mol. Sci. 2018, 19, 594. [Google Scholar] [CrossRef] [Green Version]
- Showalter, A.; Limaye, A.; Oyer, J.L.; Igarashi, R.; Kittipatarin, C.; Copik, A.; Khaled, A.R. Cytokines in immunogenic cell death: Applications for cancer immunotherapy. Cytokine 2017, 97, 123–132. [Google Scholar] [CrossRef]
- Hernandez, C.; Huebener, P.; Schwabe, R.F. Damage-associated molecular patterns in cancer: A double-edged sword. Oncogene 2016, 35, 5931–5941. [Google Scholar] [CrossRef]
- Locy, H.; De Mey, S.L.; De Mey, W.; De Ridder, M.; Thielemans, K.; Maenhout, S.K. Immunomodulation of the Tumor Microenvironment: Turn Foe Into Friend. Front. Immunol. 2018, 9, 2909. [Google Scholar] [CrossRef]
- Dillman, R.O. An update on GM-CSF and its potential role in melanoma management. Melanoma Manag. 2020, 7, MMT49. [Google Scholar] [CrossRef]
- Emens, L.A.; Middleton, G. The Interplay of Immunotherapy and Chemotherapy: Harnessing Potential Synergies. Cancer Immunol. Res. 2015, 3, 436–443. [Google Scholar] [CrossRef] [Green Version]
- Vanmeerbeek, I.; Sprooten, J.; De Ruysscher, D.; Tejpar, S.; Vandenberghe, P.; Fucikova, J.; Spisek, R.; Zitvogel, L.; Kroemer, G.; Galluzzi, L.; et al. Trial watch: Chemotherapy-induced immunogenic cell death in immuno-oncology. OncoImmunology 2020, 9, 1703449. [Google Scholar] [CrossRef] [Green Version]
- Wargo, J.A.; Reuben, A.; Cooper, Z.; Oh, K.S.; Sullivan, R.J. Immune Effects of Chemotherapy, Radiation, and Targeted Therapy and Opportunities for Combination With Immunotherapy. Semin. Oncol. 2015, 42, 601–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, W.-L.; Hua, Z.-C. Evaluation of effectiveness of granulocyte-macrophage colony-stimulating factor therapy to cancer patients after chemotherapy: A meta-analysis. Oncotarget 2018, 9, 28226–28239. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, H.A.; Villar, R.C. Radiotherapy and immune response: The systemic effects of a local treatment. Clinics 2018, 73 (Suppl. S1), e557s. [Google Scholar] [CrossRef]
- Reynders, K.; De Ruysscher, D. Radiotherapy and Immunotherapy: Improving Cancer Treatment through Synergy. Prog. Tumor Res. 2015, 42, 67–78. [Google Scholar] [CrossRef]
- Golden, E.B.; Chhabra, A.; Chachoua, A.; Adams, S.; Donach, M.; Fenton-Kerimian, M.; Friedman, K.; Ponzo, F.; Babb, J.S.; Goldberg, J.; et al. Local radiotherapy and granulocyte-macrophage colony-stimulating factor to generate abscopal responses in patients with metastatic solid tumours: A proof-of-principle trial. Lancet Oncol. 2015, 16, 795–803. [Google Scholar] [CrossRef]
- Formenti, S.C.; Golden, E.B.; DeMaria, S. Local radiotherapy and GM-CSF in metastatic cancer: Lessons from a proof of principle trial. OncoImmunology 2016. [Google Scholar] [CrossRef]
- Hu, Z.I.; McArthur, H.L.; Ho, A.Y. The Abscopal Effect of Radiation Therapy: What Is It and How Can We Use It in Breast Cancer? Curr. Breast Cancer Rep. 2017, 9, 45–51. [Google Scholar] [CrossRef] [Green Version]
- Anderson, C.C. Application of central immunologic concepts to cancer: Helping T cells and B cells become intolerant of tumors. Eur. J. Immunol. 2014, 44, 1921–1924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Burg, S.H.; Arens, R.; Ossendorp, F.; van Hall, T.; Melief, C.J. Vaccines for established cancer: Overcoming the challenges posed by immune evasion. Nat. Rev. Cancer 2016, 16, 219–233. [Google Scholar] [CrossRef] [PubMed]
- Tagliamonte, M.; Petrizzo, A.; Tornesello, M.L.; Buonaguro, F.M.; Buonaguro, L. Antigen-specific vaccines for cancer treatment. Hum. Vaccines Immunother. 2014, 10, 3332–3346. [Google Scholar] [CrossRef] [PubMed]
- Miguel, A.; Sendra, L.; Noé, V.; Ciudad, C.; Dasí, F.; Hervas, D.; Herrero, M.J.; Aliño, S.F. Silencing of Foxp3 enhances the antitumor efficacy of GM-CSF genetically modified tumor cell vaccine against B16 melanoma. OncoTargets Ther. 2017, 10, 503–514. [Google Scholar] [CrossRef] [Green Version]
- Yan, W.-L.; Shen, K.-Y.; Tien, C.-Y.; Chen, Y.-A.; Liu, S.-J. Recent progress in GM-CSF-based cancer immunotherapy. Immunotherapy 2017, 9, 347–360. [Google Scholar] [CrossRef]
- Ungerechts, G.; Bossow, S.; Leuchs, B.; Holm, P.S.; Rommelaere, J.; Coffey, M.; Coffin, R.; Bell, J.; Nettelbeck, D.M. Moving oncolytic viruses into the clinic: Clinical-grade production, purification, and characterization of diverse oncolytic viruses. Mol. Ther. Methods Clin. Dev. 2016, 3, 16018. [Google Scholar] [CrossRef] [Green Version]
- Fukuhara, H.; Ino, Y.; Todo, T. Oncolytic virus therapy: A new era of cancer treatment at dawn. Cancer Sci. 2016, 107, 1373–1379. [Google Scholar] [CrossRef]
- Aurelian, L. Oncolytic viruses as immunotherapy: Progress and remaining challenges. OncoTargets Ther. 2016, 9, 2627–2637. [Google Scholar] [CrossRef] [Green Version]
- De Gruijl, T.D.; Janssen, A.B.; van Beusechem, V.W. Arming oncolytic viruses to leverage antitumor immunity. Expert Opin. Biol. Ther. 2015, 15, 959–971. [Google Scholar] [CrossRef]
- Melcher, A.; Parato, K.; Rooney, C.M.; Bell, J.C. Thunder and Lightning: Immunotherapy and Oncolytic Viruses Collide. Mol. Ther. 2011, 19, 1008–1016. [Google Scholar] [CrossRef]
- Prestwich, R.J.; Errington, F.; Harrington, K.; Pandha, H.S.; Selby, P.; Melcher, A. Oncolytic Viruses: Do They Have a Role in Anti-Cancer Therapy? Clin. Med. Oncol. 2008, 2, 83–96. [Google Scholar] [CrossRef] [Green Version]
- Prestwich, R.J.; Errington, F.; Hatfield, P.; Merrick, A.E.; Ilett, E.J.; Selby, P.J.; Melcher, A.A. The immune system—Is it relevant to cancer development, progression and treatment? Clin. Oncol. 2008, 20, 101–112. [Google Scholar] [CrossRef]
- Prestwich, R.J.; Harrington, K.J.; Pandha, H.S.; Vile, R.G.; Melcher, A.A.; Errington, F. Oncolytic viruses: A novel form of immunotherapy. Expert Rev. Anticancer Ther. 2008, 8, 1581–1588. [Google Scholar] [CrossRef]
- Rojas, J.J.; Sampath, P.; Hou, W.; Thorne, S.H. Defining Effective Combinations of Immune Checkpoint Blockade and Oncolytic Virotherapy. Clin. Cancer Res. 2015, 21, 5543–5551. [Google Scholar] [CrossRef] [Green Version]
- Pearl, T.M.; Markert, J.M.; Cassady, K.A.; Ghonime, M.G. Oncolytic Virus-Based Cytokine Expression to Improve Immune Activity in Brain and Solid Tumors. Mol. Ther. Oncolytics 2019, 13, 14–21. [Google Scholar] [CrossRef] [Green Version]
- Bommareddy, P.K.; Patel, A.; Hossain, S.; Kaufman, H.L. Talimogene Laherparepvec (T-VEC) and Other Oncolytic Viruses for the Treatment of Melanoma. Am. J. Clin. Dermatol. 2017, 18, 1–15. [Google Scholar] [CrossRef]
- Farkona, S.; Diamandis, E.; Blasutig, I.M. Cancer immunotherapy: The beginning of the end of cancer? BMC Med. 2016, 14, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corrigan, P.A.; Beaulieu, C.; Patel, R.B.; Lowe, D.K. Talimogene Laherparepvec. Ann. Pharmacother. 2017, 51, 675–681. [Google Scholar] [CrossRef] [PubMed]
- Herter-Sprie, G.S.; Koyama, S.; Korideck, H.; Hai, J.; Deng, J.; Li, Y.Y.; Buczkowski, K.A.; Grant, A.K.; Ullas, S.; Rhee, K.; et al. Synergy of radiotherapy and PD-1 blockade in Kras-mutant lung cancer. JCI Insight 2016, 1, e87415. [Google Scholar] [CrossRef] [PubMed]
- Bezborodova, O.A.; Nemtsova, E.R.; Gevorkov, A.R.; Boyko, A.V.; Venediktova, J.B.; Alekseenko, I.V.; Kostina, M.B.; Monastyrskaya, G.S.; Sverdlov, E.D.; Khmelevskiy, E.V.; et al. Antitumor efficacy of combined gene and radiotherapy in animals. Dokl. Biochem. Biophys. 2016, 470, 345–348. [Google Scholar] [CrossRef]
- Kaliberov, S.A.; Buchsbaum, D.J. Chapter seven—Cancer treatment with gene therapy and radiation therapy. Adv. Cancer Res. 2012, 115, 221–263. [Google Scholar] [CrossRef] [Green Version]
- Alekseenko, I.V.; Pleshkan, V.V.; Kopantzev, E.P.; Stukacheva, E.A.; Chernov, I.P.; Vinogradova, T.V.; Sverdlov, E.D. Activity of the Upstream Component of Tandem TERT/Survivin Promoters Depends on Features of the Downstream Component. PLoS ONE 2012, 7, e46474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondratyeva, L.G.; Kashkin, K.N.; Chernov, I.; Stukacheva, E.A.; Dydich, D.A.; Kopantzev, E.P.; Sverdlov, E.D. PCNA: A Constitutive Human Promoter for Gene Expression for Functional Studies and Therapeutic Applications. Mol. Genet. Microbiol. Virol. 2017, 32, 137–140. [Google Scholar] [CrossRef]
- Antonova, D.V.; Alekseenko, I.V.; Siniushina, A.K.; Kuzmich, A.I.; Pleshkan, V.V. Searching for Promoters to Drive Stable and Long-Term Transgene Expression in Fibroblasts for Syngeneic Mouse Tumor Models. Int. J. Mol. Sci. 2020, 21, 6098. [Google Scholar] [CrossRef]
- Shepelev, M.V.; Korobko, E.V.; Georgiev, G.P.; Sverdlov, E.D.; Korobko, I.V. Application of mRNA regulatory regions to improve tumor specificity of transgene expression. Cancer Gene Ther. 2011, 18, 682–684. [Google Scholar] [CrossRef] [PubMed]
- Ohlfest, J.R.; Freese, A.B.; Largaespada, D.A. Nonviral vectors for cancer gene therapy: Prospects for integrating vectors and combination therapies. Curr. Gene Ther. 2005, 5, 629–641. [Google Scholar] [CrossRef] [PubMed]
- Lam, P.; Khan, G.; Stripecke, R.; Hui, K.M.; Kasahara, N.; Peng, K.-W.; Guinn, B.-A. The innovative evolution of cancer gene and cellular therapies. Cancer Gene Ther. 2013, 20, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Halama, A.; Kuliński, M.; Librowski, T.; Lochyński, S. Polymer-based non-viral gene delivery as a concept for the treatment of cancer. Pharmacol. Rep. 2009, 61, 993–999. [Google Scholar] [CrossRef]
- Malecki, M. Frontiers in Suicide Gene Therapy of Cancer. J. Genet. Syndr. Gene Ther. 2012, 2012, e114. [Google Scholar] [CrossRef]
- Sung, Y.K.; Kim, S.W. Recent advances in the development of gene delivery systems. Biomater. Res. 2019, 23, 1–7. [Google Scholar] [CrossRef]
- Dolgin, E. How COVID unlocked the power of RNA vaccines. Nature 2021, 589, 189–191. [Google Scholar] [CrossRef]
- Park, K.S.; Sun, X.; Aikins, M.E.; Moon, J.J. Non-viral COVID-19 vaccine delivery systems. Adv. Drug Deliv. Rev. 2021, 169, 137–151. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Van Der Meel, R.; Chen, X.; Lammers, T. The EPR effect and beyond: Strategies to improve tumor targeting and cancer nanomedicine treatment efficacy. Theranostics 2020, 10, 7921–7924. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Long, L.; Maeda, H. Enhancement of Tumor-Targeted Delivery of Bacteria with Nitroglycerin Involving Augmentation of the EPR Effect. Methods Mol. Biol. 2016, 1409, 9–23. [Google Scholar] [PubMed]
- Chernyavska, M.; Schmid, M.; Freitag, P.; Palacio-Castañeda, V.; Piruska, A.; Huck, W.T.S.; Plückthun, A.; Verdurmen, W.P.R. Unravelling Receptor and RGD Motif Dependence of Retargeted Adenoviral Vectors using Advanced Tumor Model Systems. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef]
- Chawla, S.P.; Bruckner, H.; Morse, M.A.; Assudani, N.; Hall, F.L.; Gordon, E.M. A Phase I-II Study Using Rexin-G Tumor-Targeted Retrovector Encoding a Dominant-Negative Cyclin G1 Inhibitor for Advanced Pancreatic Cancer. Mol. Ther. Oncolytics 2019, 12, 56–67. [Google Scholar] [CrossRef] [Green Version]
- Kuzmich, A.; Rakitina, O.; Didych, D.; Potapov, V.; Zinovyeva, M.; Alekseenko, I.; Sverdlov, E. Novel Histone-Based DNA Carrier Targeting Cancer-Associated Fibroblasts. Polymers 2020, 12, 1695. [Google Scholar] [CrossRef]
- Durymanov, M.O.; Slastnikova, T.A.; Kuzmich, A.I.; Khramtsov, Y.V.; Ulasov, A.V.; Rosenkranz, A.A.; Egorov, S.Y.; Sverdlov, E.D.; Sobolev, A.S. Microdistribution of MC1R-targeted polyplexes in murine melanoma tumor tissue. Biomaterials 2013, 34, 10209–10216. [Google Scholar] [CrossRef] [Green Version]
- Williams, E.M.; Little, R.F.; Mowday, A.; Rich, M.; Chan-Hyams, J.V.; Copp, J.N.; Smaill, J.B.; Patterson, A.; Ackerley, D.F. Nitroreductase gene-directed enzyme prodrug therapy: Insights and advances toward clinical utility. Biochem. J. 2015, 471, 131–153. [Google Scholar] [CrossRef]
- Montfoort, N.E.; Der Aa, E.E.; Woltman, A.M. Understanding MHC Class I Presentation of Viral Antigens by Human Dendritic Cells as a Basis for Rational Design of Therapeutic Vaccines. Front. Immunol. 2014, 5, 182. [Google Scholar] [CrossRef] [Green Version]
- Aznar, M.A.; Tinari, N.; Rullán, A.J.; Sánchez-Paulete, A.R.; Rodriguez-Ruiz, M.E.; Melero, I. Intratumoral Delivery of Immunotherapy—Act Locally, Think Globally. J. Immunol. 2017, 198, 31–39. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alekseenko, I.; Kuzmich, A.; Kondratyeva, L.; Kondratieva, S.; Pleshkan, V.; Sverdlov, E. Step-by-Step Immune Activation for Suicide Gene Therapy Reinforcement. Int. J. Mol. Sci. 2021, 22, 9376. https://doi.org/10.3390/ijms22179376
Alekseenko I, Kuzmich A, Kondratyeva L, Kondratieva S, Pleshkan V, Sverdlov E. Step-by-Step Immune Activation for Suicide Gene Therapy Reinforcement. International Journal of Molecular Sciences. 2021; 22(17):9376. https://doi.org/10.3390/ijms22179376
Chicago/Turabian StyleAlekseenko, Irina, Alexey Kuzmich, Liya Kondratyeva, Sofia Kondratieva, Victor Pleshkan, and Eugene Sverdlov. 2021. "Step-by-Step Immune Activation for Suicide Gene Therapy Reinforcement" International Journal of Molecular Sciences 22, no. 17: 9376. https://doi.org/10.3390/ijms22179376