
Macrophages Compensate for Loss of Protein Tyrosine Phosphatase N2 in Dendritic Cells to Protect from Elevated Colitis

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
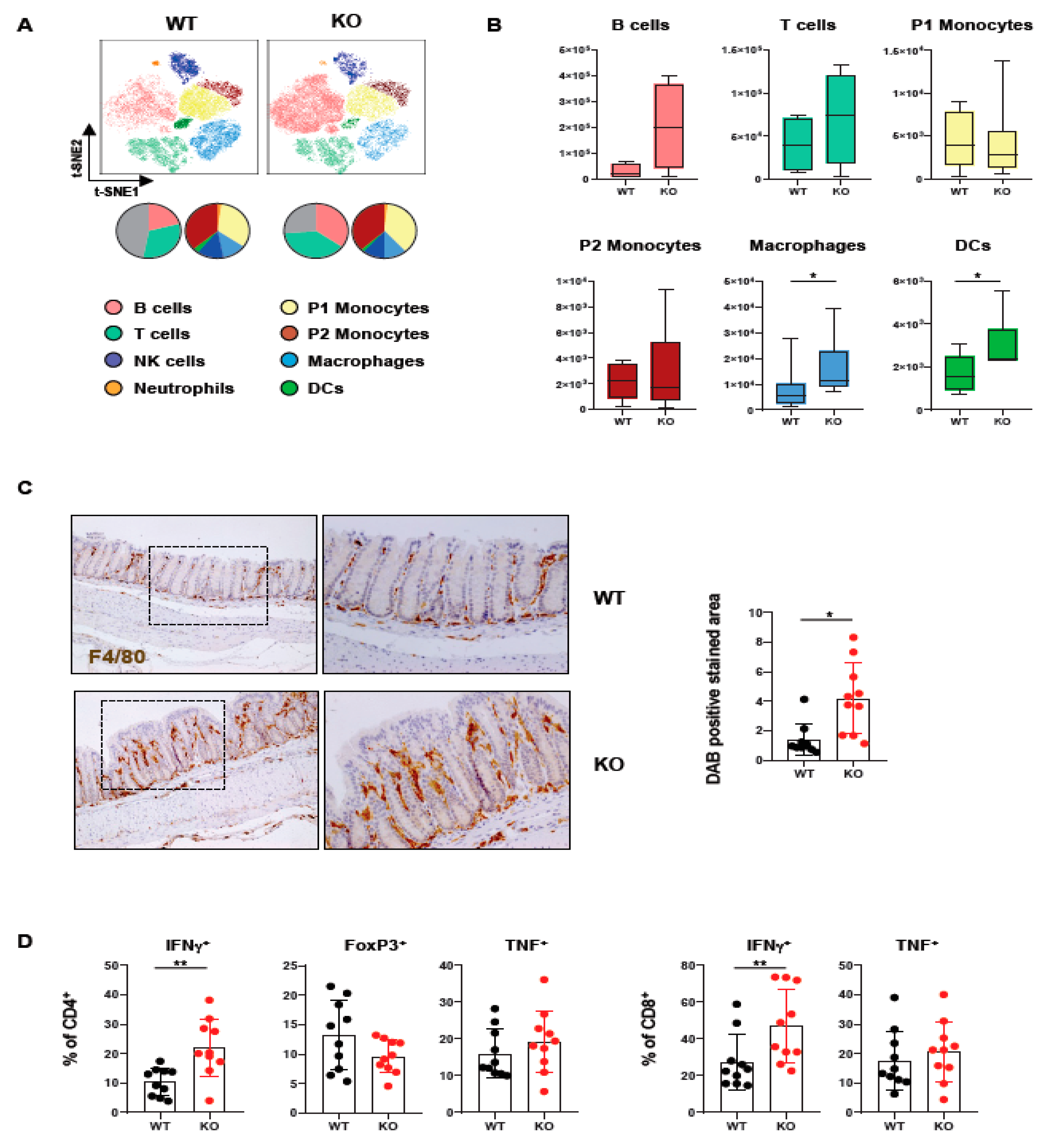
2.1. Loss of PTPN2 in DCs Affects Immune Cell Infiltrations in Lamina Propria
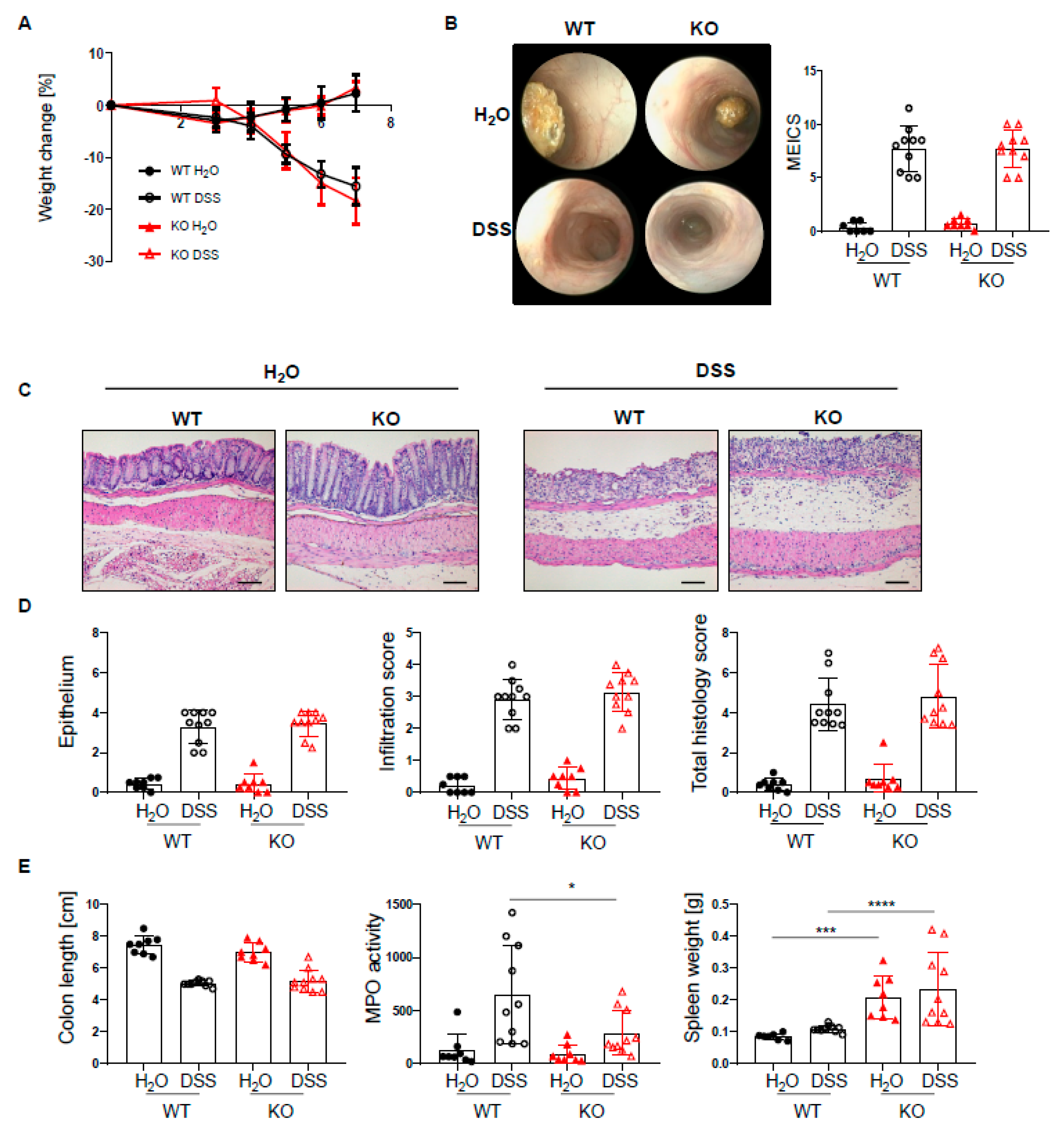
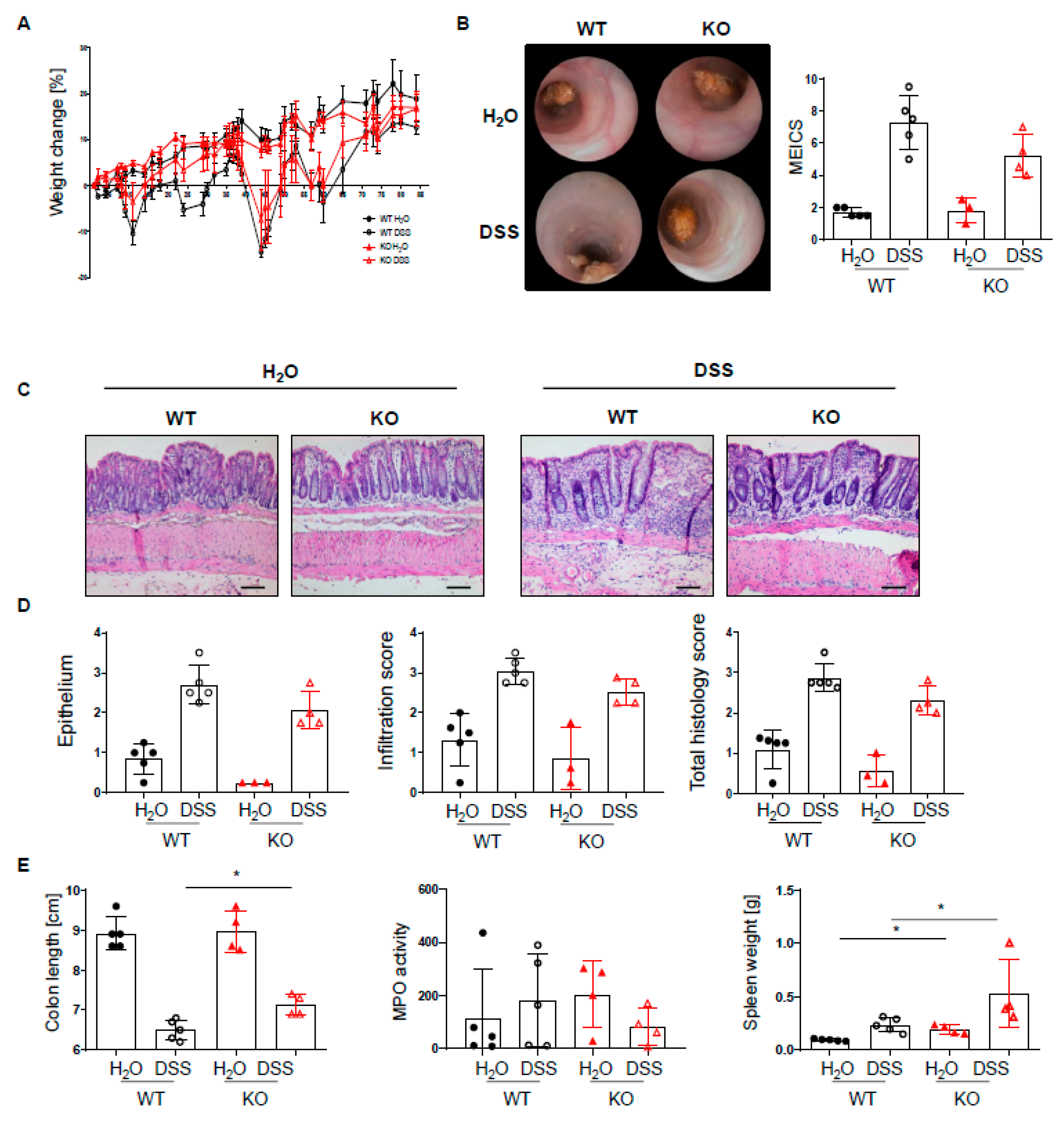
2.2. PTPN2 Deficiency in DCs Has No Impact on Intestinal Inflammation in Acute DSS Colitis
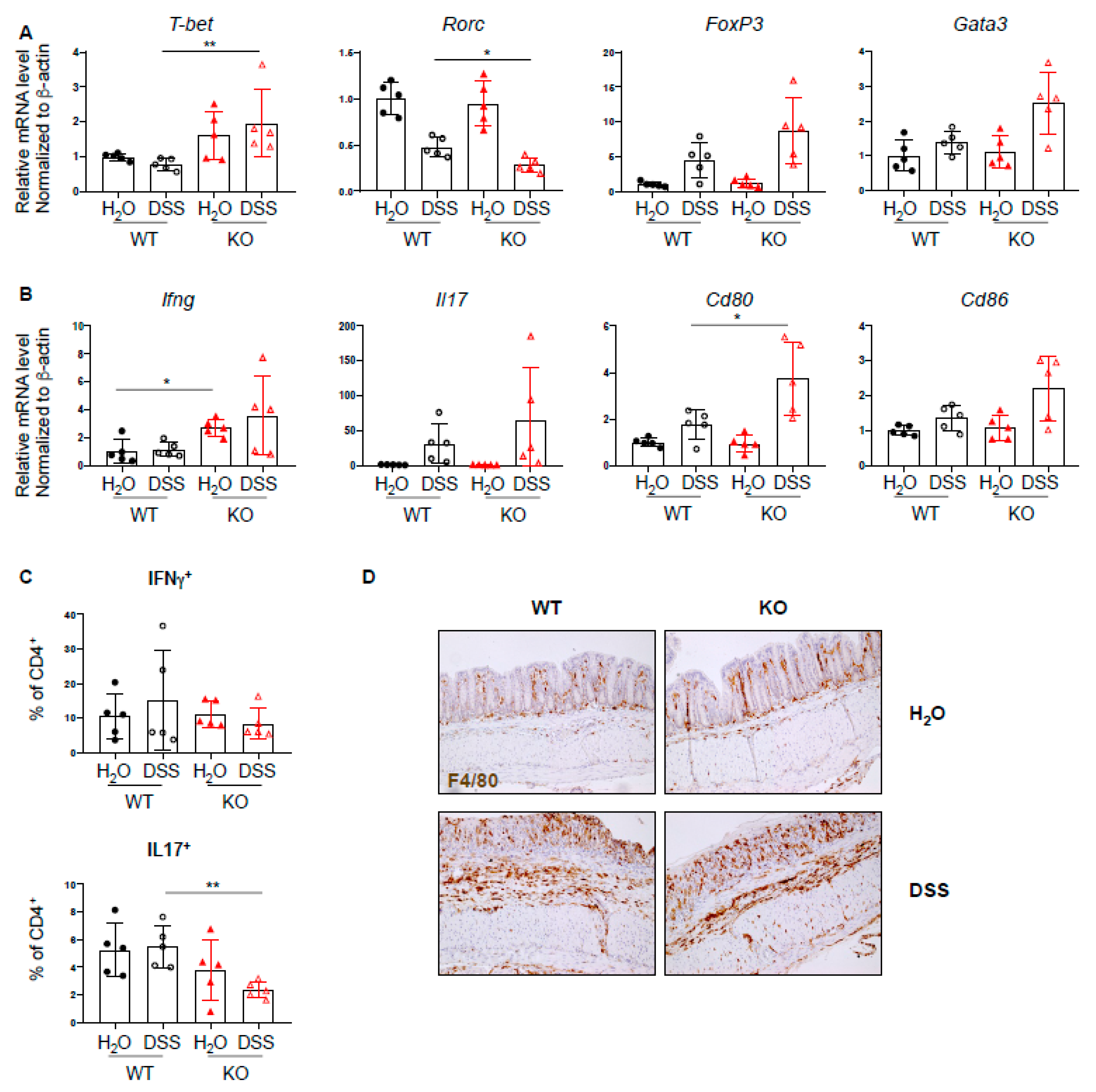
2.3. Increased Th1 Response in PTPN2fl/flxCD11cCre Mice in Acute Colitis
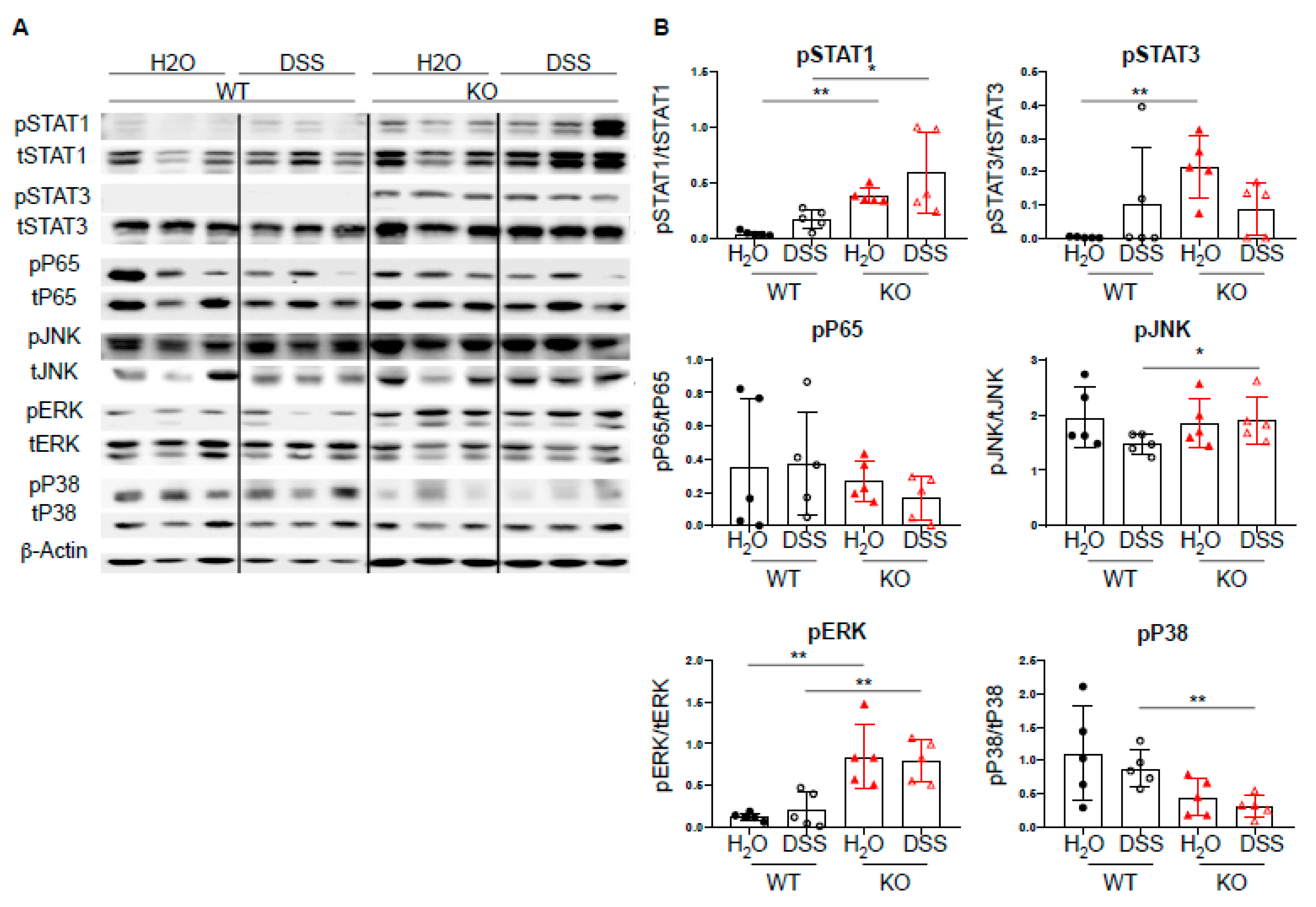
2.4. DC-Specific PTPN2 Regulates Phosphorylation of STAT1 in Acute DSS Colitis
2.5. No Changes in PTPN2fl/flxCD11cCre Mice upon Induction of Chronic Inflammation
2.6. Loss of PTPN2 in DCs Has No Effect on Colitis Severity in a T Cell Transfer Colitis Model
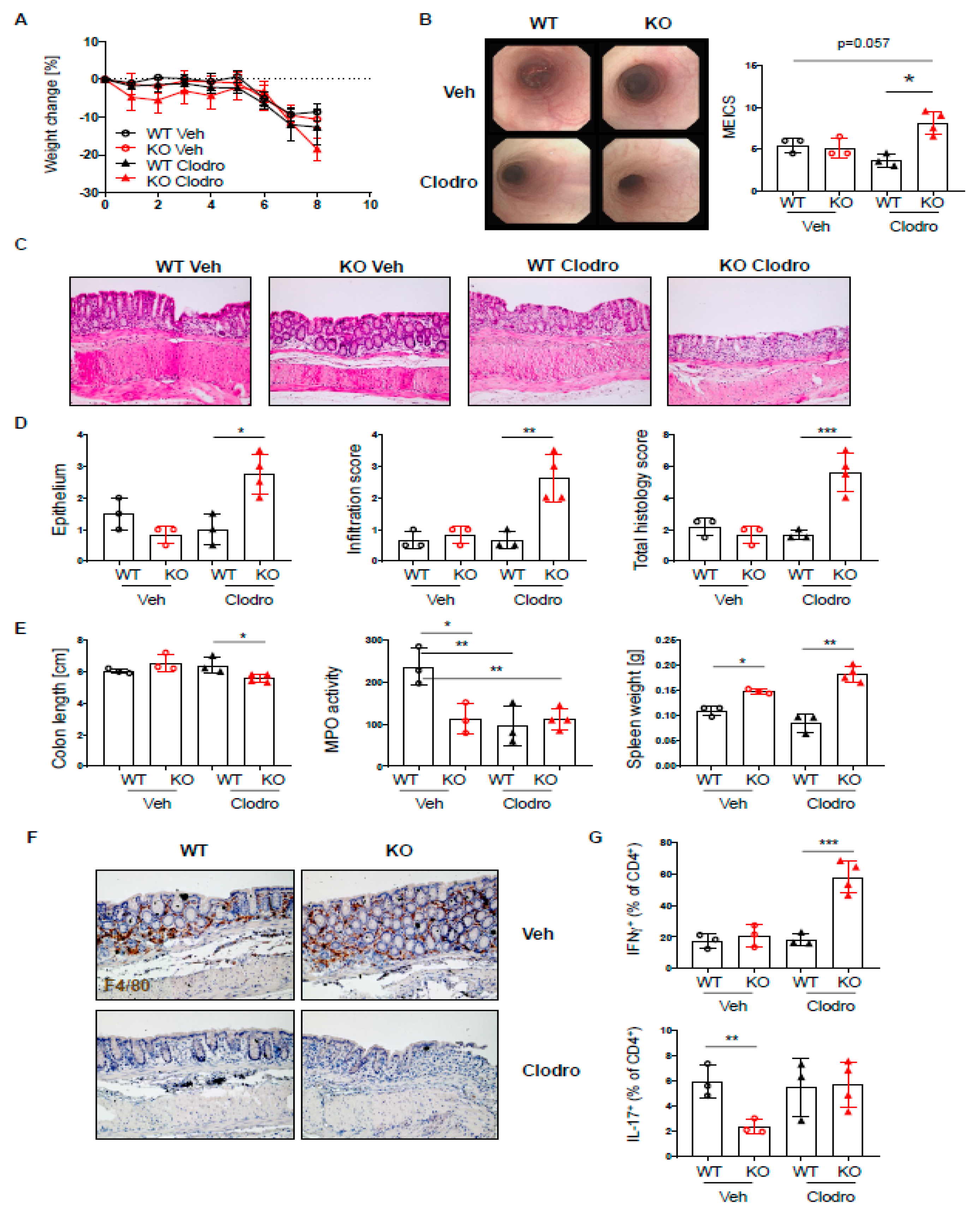
2.7. Macrophage Depletion Results in Elevated Acute DSS Colitis in PTPN2-CD11cCre Mice
3. Discussion
4. Methods
4.1. Mice, Colitis Induction, and Macrophage Depletion
4.2. Endoscopic Assessment of Colitis Severity and Histological Score
4.3. RNA Isolation, RT-PCR, and Quantitative PCR
4.4. Protein Isolation and Western Blotting
4.5. Myeloperoxidase (MPO) Activity Assay
4.6. Immunohistochemistry
4.7. Isolation of Lamina Propria Lymphocytes
4.8. Flow Cytometry
4.9. Cell Sorting
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Podolsky, D.K. Inflammatory Bowel Disease. N. Engl. J. Med. 2002, 347, 417–429. [Google Scholar] [CrossRef]
- Burton, P.R.; Clayton, D.G.; Cardon, L.R.; Craddock, N.; Deloukas, P.; Duncanson, A.; Kwiatrowski, D.P.; McCarthy, M.I.; Ouwehand, W.H.; Samani, N.J.; et al. Genome-wide association study of 14,000 cases of seven common diseases and 3000 shared controls. Nature 2007, 447, 661–678. [Google Scholar]
- Khor, B.; Gardet, A.; Xavier, R.J. Genetics and pathogenesis of inflammatory bowel disease. Nature 2011, 474, 307–317. [Google Scholar] [CrossRef] [Green Version]
- Todd, J.A.; Walker, N.M.; Cooper, J.D.; Smyth, D.J.; Downes, K.; Plagnol, V.; Bailey, R.; Nejentsev, S.; Field, S.F.; Payne, F. Robust associations of four new chromosome regions from genome-wide analyses of type 1 diabetes. Nat. Genet. 2007, 39, 857–864. [Google Scholar] [CrossRef] [PubMed]
- Aradi, B.; Kato, M.; Filkova, M.; Karouzakis, E.; Klein, K.; Scharl, M.; Kolling, C.; Michel, B.A.; Gay, R.E.; Buzas, E.I. Protein Tyrosine Phosphatase Nonreceptor Type 2: An Important Regulator of lnterleukin-6 Production in Rheumatoid Arthritis Synovial Fibroblasts. Arthritis Rheumatol. 2015, 67, 2624–2633. [Google Scholar] [CrossRef] [PubMed]
- Scharl, M.; Hruz, P.; McCole, D.F. Protein Tyrosine Phosphatase non-Receptor Type 2 regulates IFN-γ-induced cytokine signaling in THP-1 monocytes. Inflamm. Bowel Dis. 2010, 16, 2055–2064. [Google Scholar] [CrossRef]
- Scharl, M.; McCole, D.F.; Weber, A.; Vavricka, S.R.; Frei, P.; Kellermeier, S.; Pesch, T.; Fried, M.; Rogler, G. Protein tyrosine phosphatase N2 regulates TNFα-induced signalling and cytokine secretion in human intestinal epithelial cells. Gut 2011, 60, 189–197. [Google Scholar] [CrossRef] [Green Version]
- Scharl, M.; Mwinyi, J.; Fischbeck, A.; Leucht, K.; Eloranta, J.J.; Arikkat, J.; Pesch, T.; Kellermeier, S.; Mair, A.; Kullak-Ublick, G.A. Crohn’s Disease-associated Polymorphism Within the PTPN2 Gene Affects Muramyl-Dipeptide-induced Cytokine Secretion and Autophagy. Inflamm. Bowel Dis. 2011, 18, 900–912. [Google Scholar] [CrossRef]
- Van Vliet, C.; Bukczynska, P.E.; Puryer, M.A.; Sadek, C.M.; Shields, B.J.; Tremblay, M.L.; Tiganis, T. Selective regulation of tumor necrosis factor–induced Erk signaling by Src family kinases and the T cell protein tyrosine phosphatase. Nat. Immunol. 2005, 6, 253–260. [Google Scholar] [CrossRef]
- Ten Hoeve, J.; de Jesus Ibarra-Sanchez, M.; Fu, Y.; Zhu, W.; Tremblay, M.; David, M.; Shuai, K. Identification of a Nuclear Stat1 Protein Tyrosine Phosphatase. Mol. Cell. Biol. 2002, 22, 5662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simoncic, P.D.; Lee-Loy, A.; Barber, D.L.; Tremblay, M.L.; McGlade, C.J. The T Cell Protein Tyrosine Phosphatase Is a Negative Regulator of Janus Family Kinases 1 and 3. Curr. Biol. 2002, 12, 446–453. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, T.; Sekine, Y.; Kashima, K.; Kubota, A.; Sato, N.; Aoki, N.; Matsuda, T. The nuclear isoform of protein-tyrosine phosphatase TC-PTP regulates interleukin-6-mediated signaling pathway through STAT3 dephosphorylation. Biochem. Biophys. Res. Commun. 2002, 297, 811–817. [Google Scholar] [CrossRef] [Green Version]
- You-Ten, K.E.; Muise, E.S.; Itié, A.; Michaliszyn, E.; Wagner, J.; Jothy, S.; Lapp, W.S.; Tremblay, M.L. Impaired Bone Marrow Microenvironment and Immune Function in T Cell Protein Tyrosine Phosphatase–deficient Mice. J. Exp. Med. 1997, 186, 683. [Google Scholar] [CrossRef] [Green Version]
- Heinonen, K.M.; Nestel, F.P.; Newell, E.W.; Charette, G.; Seemayer, T.A.; Tremblay, M.L.; Lapp, W.S. T-cell protein tyrosine phosphatase deletion results in progressive systemic inflammatory disease. Blood 2004, 103, 3457–3464. [Google Scholar] [CrossRef] [PubMed]
- Doody, K.M.; Bourdeau, A.; Tremblay, M.L. T-cell protein tyrosine phosphatase is a key regulator in immune cell signaling: Lessons from the knockout mouse model and implications in human disease. Immunol. Rev. 2009, 228, 325–341. [Google Scholar] [CrossRef] [PubMed]
- Wiede, F.; Shields, B.J.; Chew, S.H.; Kyparissoudis, K.; van Vliet, C.; Galic, S.; Tremblay, M.L.; Russell, S.M.; Godfrey, D.I.; Tiganis, T. T cell protein tyrosine phosphatase attenuates T cell signaling to maintain tolerance in mice. J. Clin. Investig. 2011, 121, 4758–4774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spalinger, M.R.; Kasper, S.; Chassard, C.; Raselli, T.; Frey-Wagner, I.; Gottier, C.; Lang, S.; Atrott, K.; Vavricka, S.R.; Mair, F. PTPN2 controls differentiation of CD4(+) T cells and limits intestinal inflammation and intestinal dysbiosis. Mucosal Immunol. 2015, 8, 918–929. [Google Scholar] [CrossRef] [Green Version]
- Kasper, S.H.; Spalinger, M.R.; Leonardi, I.; Gerstgrasser, A.; Raselli, T.; Gottier, C.; Atrott, K.; Frey-Wagner, I.; Fischbeck-Terhalle, A.; Rogler, G. Deficiency of Protein Tyrosine Phosphatase Non-Receptor Type 2 in Intestinal Epithelial Cells Has No Appreciable Impact on Dextran Sulphate Sodium Colitis Severity But Promotes Wound Healing. Digestion 2016, 93, 249–259. [Google Scholar] [CrossRef] [Green Version]
- Spalinger, M.R.; Manzini, R.; Hering, L.; Riggs, J.B.; Gottier, C.; Lang, S.; Atrott, K.; Fettelschoss, A.; Olomski, F.; Kündig, T.M. PTPN2 Regulates Inflammasome Activation and Controls Onset of Intestinal Inflammation and Colon Cancer. Cell Rep. 2018, 22, 1835–1848. [Google Scholar] [CrossRef] [Green Version]
- Hering, L.; Katkeviciute, E.; Schwarzfischer, M.; Busenhart, P.; Gottier, C.; Mrdjen, D.; Komuczki, J.; Wawrzyniak, M.; Lang, S.; Atrott, K. Protein Tyrosine Phosphatase Non-Receptor Type 2 Function in Dendritic Cells Is Crucial to Maintain Tissue Tolerance. Front. Immunol. 2020, 11, 1856. [Google Scholar] [CrossRef]
- Cerovic, V.; Bain, C.C.; Mowat, A.M.; Milling, S.W. Intestinal macrophages and dendritic cells: What’s the difference? Trends Immunol 2014, 35, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Coombes, J.L.; Powrie, F. Dendritic cells in intestinal immune regulation. Nat. Rev. Immunol. 2008, 8, 435–446. [Google Scholar] [CrossRef] [PubMed]
- Abe, K.; Nguyen, K.P.; Fine, S.D.; Mo, J.-H.; Shen, C.; Shenouda, S.; Corr, M.; Jung, S.; Lee, J.; Eckmann, L. Conventional dendritic cells regulate the outcome of colonic inflammation independently of T cells. Proc. Natl. Acad. Sci. USA 2007, 104, 17022. [Google Scholar] [CrossRef] [Green Version]
- Bain, C.C.; Mowat, A.M. Intestinal macrophages—Specialised adaptation to a unique environment. Eur. J. Immunol. 2011, 41, 2494–2498. [Google Scholar] [CrossRef] [PubMed]
- Krajina, T.; Leithäuser, F.; Möller, P.; Trobonjaca, Z.; Reimann, J. Colonic lamina propria dendritic cells in mice with CD4+ T cell-induced colitis. Eur. J. Immunol. 2003, 33, 1073–1083. [Google Scholar] [CrossRef]
- Vuckovic, S.; Florin, T.H.; Khalil, D.; Zhang, M.F.; Patel, K.; Hamilton, I.; Hart, D.N. CD40 and CD86 upregulation with divergent CMRF44 expression on blood dendritic cells in inflammatory bowel diseases. Am. J. Gastroenterol. 2001, 96, 2946–2956. [Google Scholar] [CrossRef]
- Velde, A.A.; van Kooyk, Y.; Braat, H.; Hommes, D.W.; Dellemijn, T.A.M.; Slors, J.F.M.; van Deventer, S.J.H.; Vyth-Dreese, F.A. Increased expression of DC-SIGN+IL-12+IL-18+ and CD83+IL-12–IL-18– dendritic cell populations in the colonic mucosa of patients with Crohn’s disease. Eur. J. Immunol. 2003, 33, 143–151. [Google Scholar] [CrossRef]
- Van Gassen, S.; Callebaut, B.; Van Helden, M.J.; Lambrecht, B.N.; Demeester, P.; Dhaene, T.; Saeys, Y. FlowSOM: Using self-organizing maps for visualization and interpretation of cytometry data. Cytom. A 2015, 87, 636–645. [Google Scholar] [CrossRef]
- Na, Y.R.; Stakenborg, M.; Seok, S.H.; Matteoli, G. Macrophages in intestinal inflammation and resolution: A potential therapeutic target in IBD. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 531–543. [Google Scholar] [CrossRef]
- Bader, J.E.; Enos, R.T.; Velázquez, K.T.; Carson, M.S.; Nagarkatti, M.; Nagarkatti, P.S.; Chatzistamou, I.; Davis, J.M.; Carson, J.A.; Robinson, C.M. Macrophage depletion using clodronate liposomes decreases tumorigenesis and alters gut microbiota in the AOM/DSS mouse model of colon cancer. Am. J. Physiol. Gastrointest. Liver Physiol. 2018, 314, G22–G31. [Google Scholar] [CrossRef]
- Wirtz, S.; Neufert, C.; Weigmann, B.; Neurath, M.F. Chemically induced mouse models of intestinal inflammation. Nat. Protoc. 2007, 2, 541–546. [Google Scholar] [CrossRef]
- Strober, W.; Fuss, I.J.; Blumberg, R.S. The Immunology of Mucosal Models of Inflammation. Annu. Rev. Immunol. 2002, 20, 495–549. [Google Scholar] [CrossRef]
- Mowat, A.M.; Agace, W.W. Regional specialization within the intestinal immune system. Nat. Rev. Immunol. 2014, 14, 667–685. [Google Scholar] [CrossRef]
- Wang, S.; Ye, Q.; Zeng, X.; Qiao, S. Functions of Macrophages in the Maintenance of Intestinal Homeostasis. J. Immunol. Res. 2019, 2019, 1512969. [Google Scholar] [CrossRef] [Green Version]
- Araki, Y.; Mukaisyo, K.; Sugihara, H.; Hattori, T. Decomposition of dextran sulfate sodium under alkaline, acidic, high temperature and high pressure conditions. Oncol. Rep. 2008, 20, 147–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medani, M.; Collins, D.; Docherty, N.G.; Baird, A.W.; O’Connell, P.R.; Winter, D.C. Emerging role of hydrogen sulfide in colonic physiology and pathophysiology. Inflamm. Bowel Dis. 2011, 17, 1620–1625. [Google Scholar] [CrossRef] [PubMed]
- McConnell, E.L.; Basit, A.W.; Murdan, S. Measurements of rat and mouse gastrointestinal pH, fluid and lymphoid tissue, and implications for in-vivo experiments. J. Pharm. Pharmacol. 2008, 60, 63–70. [Google Scholar] [CrossRef]
- Laroui, H.; Ingersoll, S.A.; Liu, H.C.; Baker, M.T.; Ayyadurai, S.; Charania, M.A.; Laroui, F.; Yan, Y.; Sitaraman, S.V.; Merlin, D. Dextran sodium sulfate (DSS) induces colitis in mice by forming nano-lipocomplexes with medium-chain-length fatty acids in the colon. PLoS ONE 2012, 7, e32084. [Google Scholar]
- Okeke, E.B.; Uzonna, J.E. The Pivotal Role of Regulatory T Cells in the Regulation of Innate Immune Cells. Front. Immunol. 2019, 10, 680. [Google Scholar] [CrossRef] [Green Version]
- Becker, C.; Fantini, M.C.; Neurath, M.F. High resolution colonoscopy in live mice. Nat. Protoc. 2006, 1, 2900–2904. [Google Scholar] [CrossRef]
- Obermeier, F.; Kojouharoff, G.; Hans, W.; Schölmerich, J.; Gross, V.; Falk, W. Interferon-gamma (IFN-gamma)- and tumour necrosis factor (TNF)-induced nitric oxide as toxic effector molecule in chronic dextran sulphate sodium (DSS)-induced colitis in mice. Clin. Exp. Immunol. 1999, 116, 238–245. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hering, L.; Katkeviciute, E.; Schwarzfischer, M.; Niechcial, A.; Riggs, J.B.; Wawrzyniak, M.; Atrott, K.; van de Sande, M.; Lang, S.; Becher, B.; et al. Macrophages Compensate for Loss of Protein Tyrosine Phosphatase N2 in Dendritic Cells to Protect from Elevated Colitis. Int. J. Mol. Sci. 2021, 22, 6820. https://doi.org/10.3390/ijms22136820
Hering L, Katkeviciute E, Schwarzfischer M, Niechcial A, Riggs JB, Wawrzyniak M, Atrott K, van de Sande M, Lang S, Becher B, et al. Macrophages Compensate for Loss of Protein Tyrosine Phosphatase N2 in Dendritic Cells to Protect from Elevated Colitis. International Journal of Molecular Sciences. 2021; 22(13):6820. https://doi.org/10.3390/ijms22136820
Chicago/Turabian StyleHering, Larissa, Egle Katkeviciute, Marlene Schwarzfischer, Anna Niechcial, Julianne B. Riggs, Marcin Wawrzyniak, Kirstin Atrott, Marnix van de Sande, Silvia Lang, Burkhard Becher, and et al. 2021. "Macrophages Compensate for Loss of Protein Tyrosine Phosphatase N2 in Dendritic Cells to Protect from Elevated Colitis" International Journal of Molecular Sciences 22, no. 13: 6820. https://doi.org/10.3390/ijms22136820