
Exogenous Hydrogen Sulfide Plays an Important Role by Regulating Autophagy in Diabetic-Related Diseases


Abstract
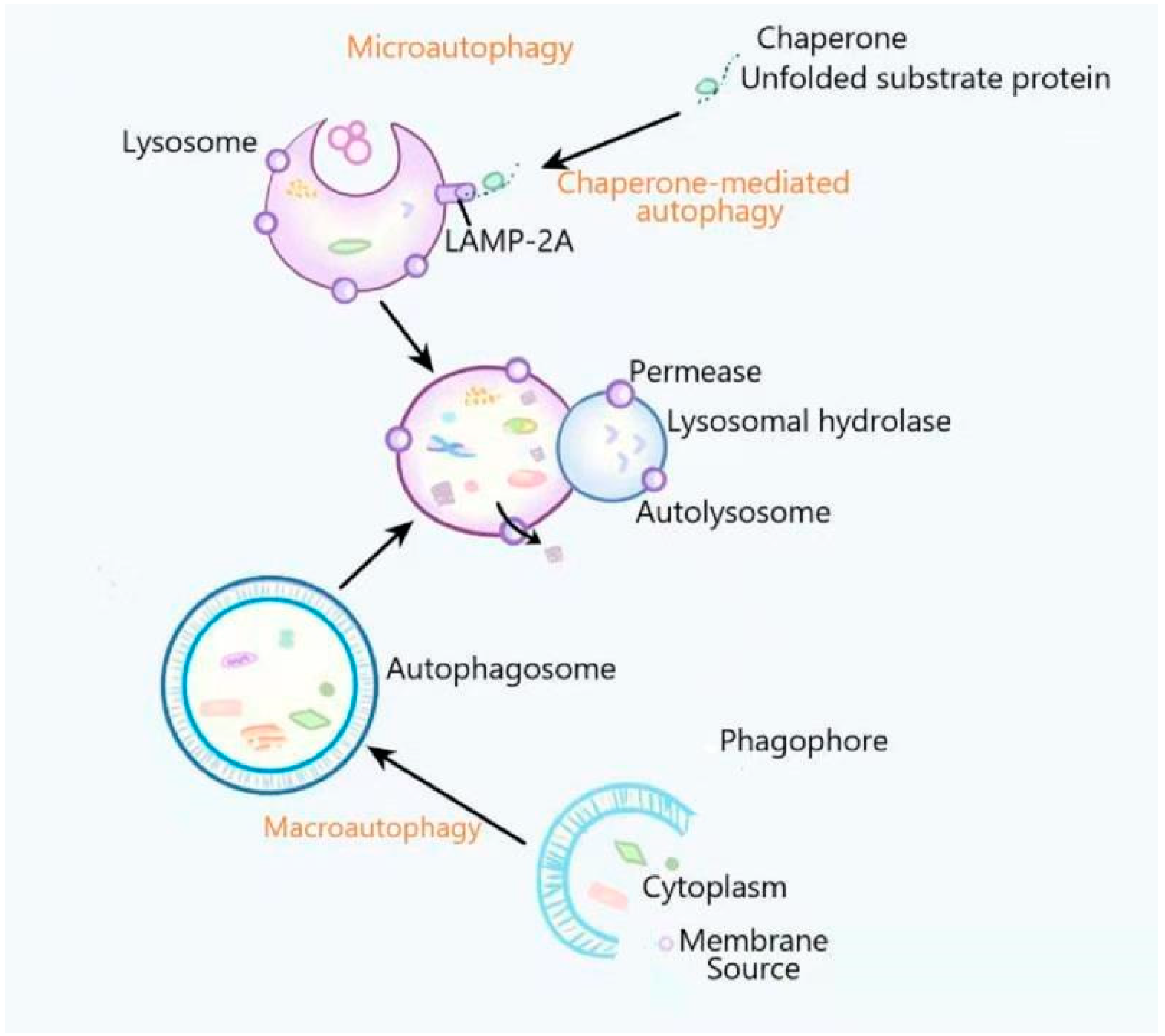
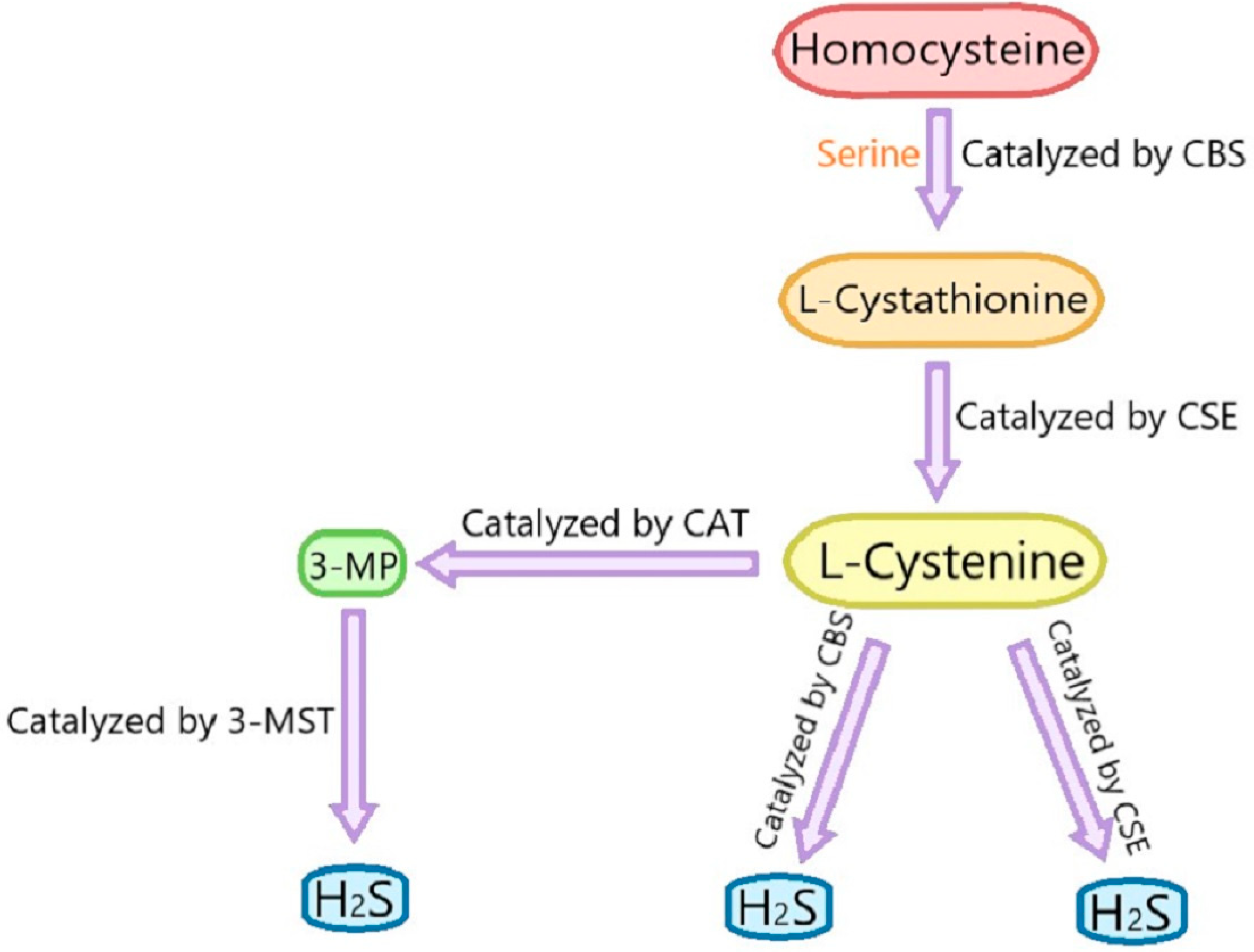
:1. Introduction
2. Exogenous H2S Plays an Important Role by Regulating Autophagy in Diabetic Cardiomyopathy
3. Exogenous H2S Plays an Important Role by Regulating Autophagy in Diabetic Vascular Endothelial Cell Dysfunction
4. Exogenous H2S Plays an Important Role by Regulating Autophagy in Diabetic Renal Fibrosis
5. Exogenous H2S Plays an Important Role by Regulating Autophagy in Diabetic Macroangiopathy
6. Exogenous H2S Plays an Important Role by Regulating Autophagy in Diabetic Depression
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Behrends, C.; Sowa, M.E.; Gygi, S.P.; Harper, J.W. Network organization of the human autophagy system. Nature 2010, 466, 68–76. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Zhang, X.; Wu, T.; Hu, X.; Su, J.; Chen, X. Autophagy in Skin Diseases. Dermatology 2019, 235, 380–389. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular definitions of autophagy and related processes. EMBO J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef] [PubMed]
- Antunes, F.; Erustes, A.G.; Costa, A.J.; Nascimento, A.C.; Bincoletto, C.; Ureshino, R.P.; Pereira, G.J.S.; Smaili, S.S. Autophagy and intermittent fasting: The connection for cancer therapy? Clinics (Sao Paulo) 2018, 73, e814s. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Cuervo, A.M. The coming of age of chaperone-mediated autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 365–381. [Google Scholar] [CrossRef]
- Fujiwara, Y.; Wada, K.; Kabuta, T. Lysosomal degradation of intracellular nucleic acids-multiple autophagic pathways. J. Biochem. 2017, 161, 145–154. [Google Scholar] [CrossRef] [Green Version]
- Pohl, C.; Dikic, I. Cellular quality control by the ubiquitin-proteasome system and autophagy. Science 2019, 366, 818–822. [Google Scholar] [CrossRef]
- Pyo, J.O.; Nah, J.; Jung, Y.K. Molecules and their functions in autophagy. Exp. Mol. Med. 2012, 44, 73–80. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wu, D.; Wang, H. Hydrogen sulfide plays an important protective role by influencing autophagy in diseases. Physiol. Res. 2019, 68, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Fujita, N.; Itoh, T.; Omori, H.; Fukuda, M.; Noda, T.; Yoshimori, T. The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol. Biol. Cell 2008, 19, 2092–2100. [Google Scholar] [CrossRef] [Green Version]
- Ichimiya, T.; Yamakawa, T.; Hirano, T.; Yokoyama, Y.; Hayashi, Y.; Hirayama, D.; Wagatsuma, K.; Itoi, T.; Nakase, H. Autophagy and Autophagy-Related Diseases: A Review. Int. J. Mol. Sci. 2020, 21, 8974. [Google Scholar] [CrossRef]
- Tooze, S.A.; Yoshimori, T. The origin of the autophagosomal membrane. Nat. Cell Biol. 2010, 12, 831–835. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Yoshimori, T.; Ohsumi, Y. The role of Atg proteins in autophagosome formation. Annu. Rev. Cell Dev. Biol. 2011, 27, 107–132. [Google Scholar] [CrossRef] [PubMed]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Levine, B. Autosis and autophagic cell death: The dark side of autophagy. Cell Death Differ. 2015, 22, 367–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Huerta, P.; Troncoso-Escudero, P.; Jerez, C.; Hetz, C.; Vidal, R.L. The intersection between growth factors, autophagy and ER stress: A new target to treat neurodegenerative diseases? Brain Res. 2016, 1649, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Deretic, V.; Saitoh, T.; Akira, S. Autophagy in infection, inflammation and immunity. Nat. Rev. Immunol. 2013, 13, 722–737. [Google Scholar] [CrossRef]
- Kaushik, S.; Cuervo, A.M. Proteostasis and aging. Nat. Med. 2015, 21, 1406–1415. [Google Scholar] [CrossRef] [PubMed]
- Kuma, A.; Komatsu, M.; Mizushima, N. Autophagy-monitoring and autophagy-deficient mice. Autophagy 2017, 13, 1619–1628. [Google Scholar] [CrossRef] [Green Version]
- Shirakabe, A.; Ikeda, Y.; Sciarretta, S.; Zablocki, D.K.; Sadoshima, J. Aging and Autophagy in the Heart. Circ. Res. 2016, 118, 1563–1576. [Google Scholar] [CrossRef] [Green Version]
- Menzies, F.M.; Fleming, A.; Rubinsztein, D.C. Compromised autophagy and neurodegenerative diseases. Nat. Rev. Neurosci. 2015, 16, 345–357. [Google Scholar] [CrossRef]
- Zhao, S.; Li, X.; Li, X.; Wei, X.; Wang, H. Hydrogen Sulfide Plays an Important Role in Diabetic Cardiomyopathy. Front. Cell Dev. Biol. 2021, 9, 627336. [Google Scholar] [CrossRef]
- Nie, X.; Xia, F.; Liu, Y.; Zhou, Y.; Ye, W.; Hean, P.; Meng, J.; Liu, H.; Liu, L.; Wen, J.; et al. Downregulation of Wnt3 Suppresses Colorectal Cancer Development Through Inhibiting Cell Proliferation and Migration. Front. Pharmacol. 2019, 10. [Google Scholar] [CrossRef]
- Nie, X.; Liu, H.; Liu, L.; Wang, Y.-D.; Chen, W.-D. Emerging Roles of Wnt Ligands in Human Colorectal Cancer. Front. Oncol. 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.D.; Wang, H.; Zhu, Y.Z. The Drug Developments of Hydrogen Sulfide on Cardiovascular Disease. Oxid. Med. Cell. Longev. 2018, 2018, 4010395. [Google Scholar] [CrossRef] [Green Version]
- Beltowski, J. Synthesis, Metabolism, and Signaling Mechanisms of Hydrogen Sulfide: An Overview. Methods Mol. Biol. 2019, 2007, 1–8. [Google Scholar] [CrossRef]
- Wang, H.; Shi, X.; Qiu, M.; Lv, S.; Liu, H. Hydrogen Sulfide Plays an Important Protective Role through Influencing Endoplasmic Reticulum Stress in Diseases. Int. J. Biol. Sci. 2020, 16, 264–271. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Liang, F.; Shah Masood, W.; Yan, X. Hydrogen sulfide protected gastric epithelial cell from ischemia/reperfusion injury by Keap1 s-sulfhydration, MAPK dependent anti-apoptosis and NF-kappaB dependent anti-inflammation pathway. Eur. J. Pharmacol. 2014, 725, 70–78. [Google Scholar] [CrossRef]
- Zheng, J.; Zhao, T.; Yuan, Y.; Hu, N.; Tang, X. Hydrogen sulfide (H2S) attenuates uranium-induced acute nephrotoxicity through oxidative stress and inflammatory response via Nrf2-NF-kappaB pathways. Chem. Biol. Interact. 2015, 242, 353–362. [Google Scholar] [CrossRef]
- Yang, G.; Wu, L.; Jiang, B.; Yang, W.; Qi, J.; Cao, K.; Meng, Q.; Mustafa, A.K.; Mu, W.; Zhang, S.; et al. H2S as a physiologic vasorelaxant: Hypertension in mice with deletion of cystathionine gamma-lyase. Science 2008, 322, 587–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Huang, Y.; Zhang, R.; Chen, Q.; Chen, J.; Zong, Y.; Liu, J.; Feng, S.; Liu, A.D.; Holmberg, L.; et al. Correction to: Hydrogen sulfide upregulates KATP channel expression in vascular smooth muscle cells of spontaneously hypertensive rats. J. Mol. Med. 2021, 99, 439–440. [Google Scholar] [CrossRef]
- Du, J.; Huang, Y.; Yan, H.; Zhang, Q.; Zhao, M.; Zhu, M.; Liu, J.; Chen, S.X.; Bu, D.; Tang, C.; et al. Hydrogen sulfide suppresses oxidized low-density lipoprotein (ox-LDL)-stimulated monocyte chemoattractant protein 1 generation from macrophages via the nuclear factor kappaB (NF-kappaB) pathway. J. Biol. Chem. 2014, 289, 9741–9753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calabrese, V.; Scuto, M.; Salinaro, A.T.; Dionisio, G.; Modafferi, S.; Ontario, M.L.; Greco, V.; Sciuto, S.; Schmitt, C.P.; Calabrese, E.J.; et al. Hydrogen Sulfide and Carnosine: Modulation of Oxidative Stress and Inflammation in Kidney and Brain Axis. Antioxidants 2020, 9, 1303. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Gu, Y.; Wen, M.; Zhao, S.; Wang, W.; Ma, Y.; Meng, G.; Han, Y.; Wang, Y.; Liu, G.; et al. Hydrogen Sulfide Induces Keap1 S-sulfhydration and Suppresses Diabetes-Accelerated Atherosclerosis via Nrf2 Activation. Diabetes 2016, 65, 3171–3184. [Google Scholar] [CrossRef] [Green Version]
- Xue, X.; Ling, X.; Xi, W.; Wang, P.; Sun, J.; Yang, Q.; Xiao, J. Exogenous hydrogen sulfide reduces atrial remodeling and atrial fibrillation induced by diabetes mellitus via activation of the PI3K/Akt/eNOS pathway. Mol. Med. Rep. 2020, 22, 1759–1766. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, M.; Ishizaki, T.; Kimura, T. Protective effect of hydrogen sulfide on pancreatic beta-cells. Nitric Oxide 2015, 46, 32–36. [Google Scholar] [CrossRef]
- Yang, G.; Tang, G.; Zhang, L.; Wu, L.; Wang, R. The pathogenic role of cystathionine gamma-lyase/hydrogen sulfide in streptozotocin-induced diabetes in mice. Am. J. Pathol. 2011, 179, 869–879. [Google Scholar] [CrossRef]
- Jacobsen, L.V.; Flint, A.; Olsen, A.K.; Ingwersen, S.H. Liraglutide in Type 2 Diabetes Mellitus: Clinical Pharmacokinetics and Pharmacodynamics. Clin. Pharm. 2016, 55, 657–672. [Google Scholar] [CrossRef] [Green Version]
- Fan, M.; Jiang, H.; Zhang, Y.; Ma, Y.; Li, L.; Wu, J. Liraglutide Enhances Autophagy and Promotes Pancreatic beta Cell Proliferation to Ameliorate Type 2 Diabetes in High-Fat-Fed and Streptozotocin-Treated Mice. Med. Sci. Monit. 2018, 24, 2310–2316. [Google Scholar] [CrossRef]
- Marwick, T.H.; Ritchie, R.; Shaw, J.E.; Kaye, D. Implications of Underlying Mechanisms for the Recognition and Management of Diabetic Cardiomyopathy. J. Am. Coll. Cardiol. 2018, 71, 339–351. [Google Scholar] [CrossRef]
- Pant, T.; Dhanasekaran, A.; Fang, J.; Bai, X.; Bosnjak, Z.J.; Liang, M.; Ge, Z.D. Current status and strategies of long noncoding RNA research for diabetic cardiomyopathy. BMC Cardiovasc. Disord. 2018, 18, 197. [Google Scholar] [CrossRef] [Green Version]
- Ohno, S.; Kohjitani, A.; Miyata, M.; Tohya, A.; Yamashita, K.; Hashiguchi, T.; Ohishi, M.; Sugimura, M. Recovery of Endothelial Function after Minor-to-Moderate Surgery Is Impaired by Diabetes Mellitus, Obesity, Hyperuricemia and Sevoflurane-Based Anesthesia. Int. Heart J. 2018, 59, 559–565. [Google Scholar] [CrossRef] [Green Version]
- Baumgardt, S.L.; Paterson, M.; Leucker, T.M.; Fang, J.; Zhang, D.X.; Bosnjak, Z.J.; Warltier, D.C.; Kersten, J.R.; Ge, Z.D. Chronic Co-Administration of Sepiapterin and L-Citrulline Ameliorates Diabetic Cardiomyopathy and Myocardial Ischemia/Reperfusion Injury in Obese Type 2 Diabetic Mice. Circ. Heart Fail. 2016, 9, e002424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.E.; Baumgardt, S.L.; Fang, J.; Paterson, M.; Liu, Y.; Du, J.; Shi, Y.; Qiao, S.; Bosnjak, Z.J.; Warltier, D.C.; et al. Cardiomyocyte GTP Cyclohydrolase 1 Protects the Heart Against Diabetic Cardiomyopathy. Sci. Rep. 2016, 6, 27925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Funabashi, N.; Takaoka, H.; Ozawa, K.; Kobayashi, Y. Endocardial Fibrotic Lesions Have a Greater Effect on Peak Longitudinal Strain than Epicardial Fibrotic Lesions in Hypertrophic Cardiomyopathy Patients. Int. Heart J. 2018, 59, 347–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takaoka, H.; Funabashi, N.; Ozawa, K.; Uehara, M.; Sano, K.; Komuro, I.; Kobayashi, Y. Improved Diagnosis of Detection of Late Enhancement in Left Ventricular Myocardium Using 2nd Generation 320-Slice CT Reconstructed with FIRST in Non-Ischemic Cardiomyopathy. Int. Heart J. 2018, 59, 542–549. [Google Scholar] [CrossRef] [Green Version]
- Karbasforooshan, H.; Karimi, G. The role of SIRT1 in diabetic cardiomyopathy. Biomed. Pharm. 2017, 90, 386–392. [Google Scholar] [CrossRef] [PubMed]
- Yao, Q.; Ke, Z.Q.; Guo, S.; Yang, X.S.; Zhang, F.X.; Liu, X.F.; Chen, X.; Chen, H.G.; Ke, H.Y.; Liu, C. Curcumin protects against diabetic cardiomyopathy by promoting autophagy and alleviating apoptosis. J. Mol. Cell. Cardiol. 2018, 124, 26–34. [Google Scholar] [CrossRef]
- Sato, A.; Asano, T.; Isono, M.; Ito, K.; Asano, T. Panobinostat synergizes with bortezomib to induce endoplasmic reticulum stress and ubiquitinated protein accumulation in renal cancer cells. BMC Urol. 2014, 14, 71. [Google Scholar] [CrossRef]
- Shang, F.; Taylor, A. Ubiquitin-proteasome pathway and cellular responses to oxidative stress. Free Radic. Biol. Med. 2011, 51, 5–16. [Google Scholar] [CrossRef] [Green Version]
- An, Y.; Zhang, H.; Wang, C.; Jiao, F.; Xu, H.; Wang, X.; Luan, W.; Ma, F.; Ni, L.; Tang, X.; et al. Activation of ROS/MAPKs/NF-kappaB/NLRP3 and inhibition of efferocytosis in osteoclast-mediated diabetic osteoporosis. FASEB J. 2019, 33, 12515–12527. [Google Scholar] [CrossRef] [Green Version]
- Grumati, P.; Dikic, I. Ubiquitin signaling and autophagy. J. Biol. Chem. 2018, 293, 5404–5413. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Chen, X.; Zhao, Y.; Ponnusamy, M.; Liu, Y. The role of ubiquitin proteasomal system and autophagy-lysosome pathway in Alzheimer’s disease. Rev. Neurosci. 2017, 28, 861–868. [Google Scholar] [CrossRef]
- Wu, J.; Tian, Z.; Sun, Y.; Lu, C.; Liu, N.; Gao, Z.; Zhang, L.; Dong, S.; Yang, F.; Zhong, X.; et al. Exogenous H2S facilitating ubiquitin aggregates clearance via autophagy attenuates type 2 diabetes-induced cardiomyopathy. Cell Death Dis. 2017, 8, e2992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Tang, Y.; Wen, L.; Kong, X.; Chen, X.; Liu, P.; Zhou, Z.; Chen, W.; Xiao, C.; Xiao, P.; et al. Neferine reduces cisplatin-induced nephrotoxicity by enhancing autophagy via the AMPK/mTOR signaling pathway. Biochem. Biophys. Res. Commun. 2017, 484, 694–701. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.; Sun, J.; Fei, A.; Gao, C.; Pan, S.; Wu, Z. Hydrogen sulfide treatment alleviated ventilator-induced lung injury through regulation of autophagy and endoplasmic reticulum stress. Int. J. Biol. Sci. 2019, 15, 2872–2884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, S.C.; Rabinovitch, P.S.; Kaeberlein, M. mTOR is a key modulator of ageing and age-related disease. Nature 2013, 493, 338–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Zhong, P.; Sun, L. Exogenous hydrogen sulfide mitigates NLRP3 inflammasome-mediated inflammation through promoting autophagy via the AMPK-mTOR pathway. Biol. Open 2019, 8. [Google Scholar] [CrossRef] [Green Version]
- Yang, F.; Zhang, L.; Gao, Z.; Sun, X.; Yu, M.; Dong, S.; Wu, J.; Zhao, Y.; Xu, C.; Zhang, W.; et al. Exogenous H2S Protects Against Diabetic Cardiomyopathy by Activating Autophagy via the AMPK/mTOR Pathway. Cell. Physiol. Biochem. 2017, 43, 1168–1187. [Google Scholar] [CrossRef] [Green Version]
- Asbun, J.; Villarreal, F.J. The pathogenesis of myocardial fibrosis in the setting of diabetic cardiomyopathy. J. Am. Coll. Cardiol. 2006, 47, 693–700. [Google Scholar] [CrossRef] [Green Version]
- Falcao-Pires, I.; Leite-Moreira, A.F. Diabetic cardiomyopathy: Understanding the molecular and cellular basis to progress in diagnosis and treatment. Heart Fail. Rev. 2012, 17, 325–344. [Google Scholar] [CrossRef] [PubMed]
- Xiao, T.; Luo, J.; Wu, Z.; Li, F.; Zeng, O.; Yang, J. Effects of hydrogen sulfide on myocardial fibrosis and PI3K/AKT1-regulated autophagy in diabetic rats. Mol. Med. Rep. 2016, 13, 1765–1773. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Guo, R.; Yu, L.; Zhang, Y.; Ren, J. Aldehyde dehydrogenase 2 (ALDH2) rescues myocardial ischaemia/reperfusion injury: Role of autophagy paradox and toxic aldehyde. Eur. Heart J. 2011, 32, 1025–1038. [Google Scholar] [CrossRef] [Green Version]
- Glance, L.G.; Dick, A.W.; Glantz, J.C.; Wissler, R.N.; Qian, F.; Marroquin, B.M.; Mukamel, D.B.; Kellermann, A.L. Rates of major obstetrical complications vary almost five-fold among US hospitals. Health Aff. 2014, 33, 1330–1336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwak, H.B.; Lee, Y.; Kim, J.H.; Van Remmen, H.; Richardson, A.G.; Lawler, J.M. MnSOD overexpression reduces fibrosis and pro-apoptotic signaling in the aging mouse heart. J. Gerontol. A Biol. Sci. Med. Sci. 2015, 70, 533–544. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Liu, M.; Song, X.; Zheng, X.; Yi, J.; Liu, D.; Wang, S.; Chu, C.; Yang, J. Exogenous Hydrogen Sulfide Ameliorates Diabetic Myocardial Fibrosis by Inhibiting Cell Aging Through SIRT6/AMPK Autophagy. Front. Pharmacol. 2020, 11, 1150. [Google Scholar] [CrossRef]
- Wang, S.; Ge, W.; Harns, C.; Meng, X.; Zhang, Y.; Ren, J. Ablation of toll-like receptor 4 attenuates aging-induced myocardial remodeling and contractile dysfunction through NCoRI-HDAC1-mediated regulation of autophagy. J. Mol. Cell. Cardiol. 2018, 119, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Kida, Y.; Goligorsky, M.S. Sirtuins, Cell Senescence, and Vascular Aging. Can. J. Cardiol. 2016, 32, 634–641. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Chen, Z.; Jiang, Z.; Luo, P.; Liu, L.; Huang, Y.; Wang, H.; Wang, Y.; Long, L.; Tan, X.; et al. Cordycepin prevents radiation ulcer by inhibiting cell senescence via NRF2 and AMPK in rodents. Nat. Commun. 2019, 10, 2538. [Google Scholar] [CrossRef] [PubMed]
- Legeay, S.; Fautrat, P.; Norman, J.B.; Antonova, G.; Kennard, S.; Bruder-Nascimento, T.; Patel, V.S.; Faure, S.; Belin de Chantemele, E.J. Selective deficiency in endothelial PTP1B protects from diabetes and endoplasmic reticulum stress-associated endothelial dysfunction via preventing endothelial cell apoptosis. Biomed. Pharm. 2020, 127, 110200. [Google Scholar] [CrossRef]
- Cheang, W.S.; Wong, W.T.; Wang, L.; Cheng, C.K.; Lau, C.W.; Ma, R.C.W.; Xu, A.; Wang, N.; Huang, Y.; Tian, X.Y. Resveratrol ameliorates endothelial dysfunction in diabetic and obese mice through sirtuin 1 and peroxisome proliferator-activated receptor delta. Pharmacol. Res. 2019, 139, 384–394. [Google Scholar] [CrossRef]
- Hemling, P.; Zibrova, D.; Strutz, J.; Sohrabi, Y.; Desoye, G.; Schulten, H.; Findeisen, H.; Heller, R.; Godfrey, R.; Waltenberger, J. Hyperglycemia-induced endothelial dysfunction is alleviated by thioredoxin mimetic peptides through the restoration of VEGFR-2-induced responses and improved cell survival. Int. J. Cardiol. 2020, 308, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Pallichankandy, S.; Rahman, A.; Thayyullathil, F.; Galadari, S. ROS-dependent activation of autophagy is a critical mechanism for the induction of anti-glioma effect of sanguinarine. Free Radic. Biol. Med. 2015, 89, 708–720. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Tan, J.; Miao, Y.; Lei, P.; Zhang, Q. ROS and Autophagy: Interactions and Molecular Regulatory Mechanisms. Cell. Mol. Neurobiol. 2015, 35, 615–621. [Google Scholar] [CrossRef]
- Liu, J.; Wu, J.; Sun, A.; Sun, Y.; Yu, X.; Liu, N.; Dong, S.; Yang, F.; Zhang, L.; Zhong, X.; et al. Hydrogen sulfide decreases high glucose/palmitate-induced autophagy in endothelial cells by the Nrf2-ROS-AMPK signaling pathway. Cell Biosci. 2016, 6, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ha, J.; Guan, K.L.; Kim, J. AMPK and autophagy in glucose/glycogen metabolism. Mol. Asp. Med. 2015, 46, 46–62. [Google Scholar] [CrossRef]
- Muller, S.G.; Jardim, N.S.; Quines, C.B.; Nogueira, C.W. Diphenyl diselenide regulates Nrf2/Keap-1 signaling pathway and counteracts hepatic oxidative stress induced by bisphenol A in male mice. Environ. Res. 2018, 164, 280–287. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Wang, L.; Zhang, G.; Zhang, Z.; Guo, W. Oxidative stress activated by Keap-1/Nrf2 signaling pathway in pathogenesis of preeclampsia. Int. J. Clin. Exp. Pathol. 2020, 13, 382–392. [Google Scholar]
- Wu, M.; Han, W.; Song, S.; Du, Y.; Liu, C.; Chen, N.; Wu, H.; Shi, Y.; Duan, H. NLRP3 deficiency ameliorates renal inflammation and fibrosis in diabetic mice. Mol. Cell. Endocrinol. 2018, 478, 115–125. [Google Scholar] [CrossRef]
- Liu, L.; Wang, Y.; Yan, R.; Liang, L.; Zhou, X.; Liu, H.; Zhang, X.; Mao, Y.; Peng, W.; Xiao, Y.; et al. BMP-7 inhibits renal fibrosis in diabetic nephropathy via miR-21 downregulation. Life Sci. 2019, 238, 116957. [Google Scholar] [CrossRef]
- Duffield, J.S. Cellular and molecular mechanisms in kidney fibrosis. J. Clin. Investig. 2014, 124, 2299–2306. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y. Cellular and molecular mechanisms of renal fibrosis. Nat. Rev. Nephrol. 2011, 7, 684–696. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Xiao, T.; Li, F.; Li, Y.; Zeng, O.; Liu, M.; Liang, B.; Li, Z.; Chu, C.; Yang, J. Hydrogen sulfide reduced renal tissue fibrosis by regulating autophagy in diabetic rats. Mol. Med. Rep. 2017, 16, 1715–1722. [Google Scholar] [CrossRef] [Green Version]
- Lu, M.; Li, H.; Liu, W.; Zhang, X.; Li, L.; Zhou, H. Curcumin attenuates renal interstitial fibrosis by regulating autophagy and retaining mitochondrial function in unilateral ureteral obstruction rats. Basic Clin. Pharmacol. Toxicol. 2021, 128, 594–604. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Tan, K.; Luo, Q.; Bai, X. Dihydromyricetin promotes autophagy and attenuates renal interstitial fibrosis by regulating miR-155-5p/PTEN signaling in diabetic nephropathy. Bosn. J. Basic Med. Sci. 2020, 20, 372–380. [Google Scholar] [CrossRef]
- Katakami, N. Mechanism of Development of Atherosclerosis and Cardiovascular Disease in Diabetes Mellitus. J. Atheroscler. Thromb. 2018, 25, 27–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montero, D.; Walther, G.; Perez-Martin, A.; Vicente-Salar, N.; Roche, E.; Vinet, A. Vascular smooth muscle function in type 2 diabetes mellitus: A systematic review and meta-analysis. Diabetologia 2013, 56, 2122–2133. [Google Scholar] [CrossRef] [Green Version]
- Di Tucci, C.; Di Feliciantonio, M.; Vena, F.; Capone, C.; Schiavi, M.C.; Pietrangeli, D.; Muzii, L.; Benedetti Panici, P. Alpha lipoic acid in obstetrics and gynecology. Gynecol. Endocrinol. 2018, 34, 729–733. [Google Scholar] [CrossRef]
- Kim, H.J.; Kim, J.Y.; Lee, S.J.; Kim, H.J.; Oh, C.J.; Choi, Y.K.; Lee, H.J.; Do, J.Y.; Kim, S.Y.; Kwon, T.K.; et al. Alpha-Lipoic acid prevents neointimal hyperplasia via induction of p38 mitogen-activated protein kinase/Nur77-mediated apoptosis of vascular smooth muscle cells and accelerates postinjury reendothelialization. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 2164–2172. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.R.; Kim, A.; Kim, K.S.; Park, Y.Y.; Park, J.H.; Kim, K.H.; Kim, S.J.; Park, K.K. Alpha-lipoic acid attenuates atherosclerotic lesions and inhibits proliferation of vascular smooth muscle cells through targeting of the Ras/MEK/ERK signaling pathway. Mol. Biol. Rep. 2012, 39, 6857–6866. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Kim, H.J.; Lee, K.; Kim, J.M.; Kim, H.S.; Kim, J.R.; Ha, C.M.; Choi, Y.K.; Lee, S.J.; Kim, J.Y.; et al. alpha-Lipoic acid attenuates vascular calcification via reversal of mitochondrial function and restoration of Gas6/Axl/Akt survival pathway. J. Cell. Mol. Med. 2012, 16, 273–286. [Google Scholar] [CrossRef]
- Qiu, X.; Liu, K.; Xiao, L.; Jin, S.; Dong, J.; Teng, X.; Guo, Q.; Chen, Y.; Wu, Y. Alpha-lipoic acid regulates the autophagy of vascular smooth muscle cells in diabetes by elevating hydrogen sulfide level. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3723–3738. [Google Scholar] [CrossRef]
- Fisher, L.; Hessler, D.M.; Polonsky, W.H.; Masharani, U.; Peters, A.L.; Blumer, I.; Strycker, L.A. Prevalence of depression in Type 1 diabetes and the problem of over-diagnosis. Diabet. Med. 2016, 33, 1590–1597. [Google Scholar] [CrossRef]
- Trief, P.M.; Foster, N.C.; Chaytor, N.; Hilliard, M.E.; Kittelsrud, J.M.; Jaser, S.S.; Majidi, S.; Corathers, S.D.; Bzdick, S.; Adkins, D.W.; et al. Longitudinal Changes in Depression Symptoms and Glycemia in Adults With Type 1 Diabetes. Diabetes Care 2019, 42, 1194–1201. [Google Scholar] [CrossRef]
- Tang, Z.J.; Zou, W.; Yuan, J.; Zhang, P.; Tian, Y.; Xiao, Z.F.; Li, M.H.; Wei, H.J.; Tang, X.Q. Antidepressant-like and anxiolytic-like effects of hydrogen sulfide in streptozotocin-induced diabetic rats through inhibition of hippocampal oxidative stress. Behav. Pharmacol. 2015, 26, 427–435. [Google Scholar] [CrossRef]
- Mariga, A.; Mitre, M.; Chao, M.V. Consequences of brain-derived neurotrophic factor withdrawal in CNS neurons and implications in disease. Neurobiol. Dis. 2017, 97, 73–79. [Google Scholar] [CrossRef] [Green Version]
- Chao, M.V.; Hempstead, B.L. p75 and Trk: A two-receptor system. Trends Neurosci. 1995, 18, 321–326. [Google Scholar] [CrossRef]
- Smith, E.D.; Prieto, G.A.; Tong, L.; Sears-Kraxberger, I.; Rice, J.D.; Steward, O.; Cotman, C.W. Rapamycin and interleukin-1beta impair brain-derived neurotrophic factor-dependent neuron survival by modulating autophagy. J. Biol. Chem. 2014, 289, 20615–20629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.Y.; Wei, H.J.; Wu, L.; Liu, S.M.; Tang, Y.Y.; Zou, W.; Wang, C.Y.; Zhang, P.; Tang, X.Q. BDNF-TrkB pathway mediates antidepressant-like roles of H2 S in diabetic rats via promoting hippocampal autophagy. Clin. Exp. Pharmacol. Physiol. 2020, 47, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Lin, S.; Qiu, J.; Sun, W.; Dong, M.; Xiang, Y.; Wang, L.; Du, P. NLRP3 inflammasome negatively regulates podocyte autophagy in diabetic nephropathy. Biochem. Biophys. Res. Commun. 2020, 521, 791–798. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| The Name of Diabetic-Related Disease | Mechanism | Reference |
|---|---|---|
| Diabetic cardiomyopathy | Promoting ubiquitin aggregation clearance through promoting autophagy via ubiquitylation of Keap-1 | [54] |
| Diabetic cardiomyopathy | Activating autophagy through activating AMPK/mTOR signaling pathway | [59] |
| Diabetes-induced myocardial fibrosis | Inhibiting autophagy via activating PI3K/AKT1 signaling pathway | [63] |
| Diabetes-induced myocardial fibrosis | Suppressing myocardial cell senescence via activating autophagy through activating SIRT6/AMPK signaling pathway | [66] |
| High glucose-induced endothelial cell dysfunction | Inhibiting autophagy via the Nrf2/ROS/AMPK signaling pathway | [75] |
| Diabetes-induced renal fibrosis | Activating autophagy via inhibiting TGFβ1/NF-κB/AKT pathways | [83] |
| Diabetic dysfunctional vascular smooth muscle | Inhibiting autophagy via suppressing the AMPK/mTOR pathway | [92] |
| Diabetic depression | Promoting autophagy via activating BDNF/TrkB pathway | [99] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lv, S.; Liu, H.; Wang, H. Exogenous Hydrogen Sulfide Plays an Important Role by Regulating Autophagy in Diabetic-Related Diseases. Int. J. Mol. Sci. 2021, 22, 6715. https://doi.org/10.3390/ijms22136715
Lv S, Liu H, Wang H. Exogenous Hydrogen Sulfide Plays an Important Role by Regulating Autophagy in Diabetic-Related Diseases. International Journal of Molecular Sciences. 2021; 22(13):6715. https://doi.org/10.3390/ijms22136715
Chicago/Turabian StyleLv, Shuangyu, Huiyang Liu, and Honggang Wang. 2021. "Exogenous Hydrogen Sulfide Plays an Important Role by Regulating Autophagy in Diabetic-Related Diseases" International Journal of Molecular Sciences 22, no. 13: 6715. https://doi.org/10.3390/ijms22136715