
Regulation of ADAM10 by the TspanC8 Family of Tetraspanins and Their Therapeutic Potential


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Importance of the Molecular Scissor ADAM10 in Health and Disease
3. Tetraspanins as Regulators of Other Membrane Proteins
3.1. Tetraspanin Overview
3.2. Tetraspanins Interact with and Regulate Other Membrane Proteins
3.3. Insights from Recent Tetraspanin Structural Studies
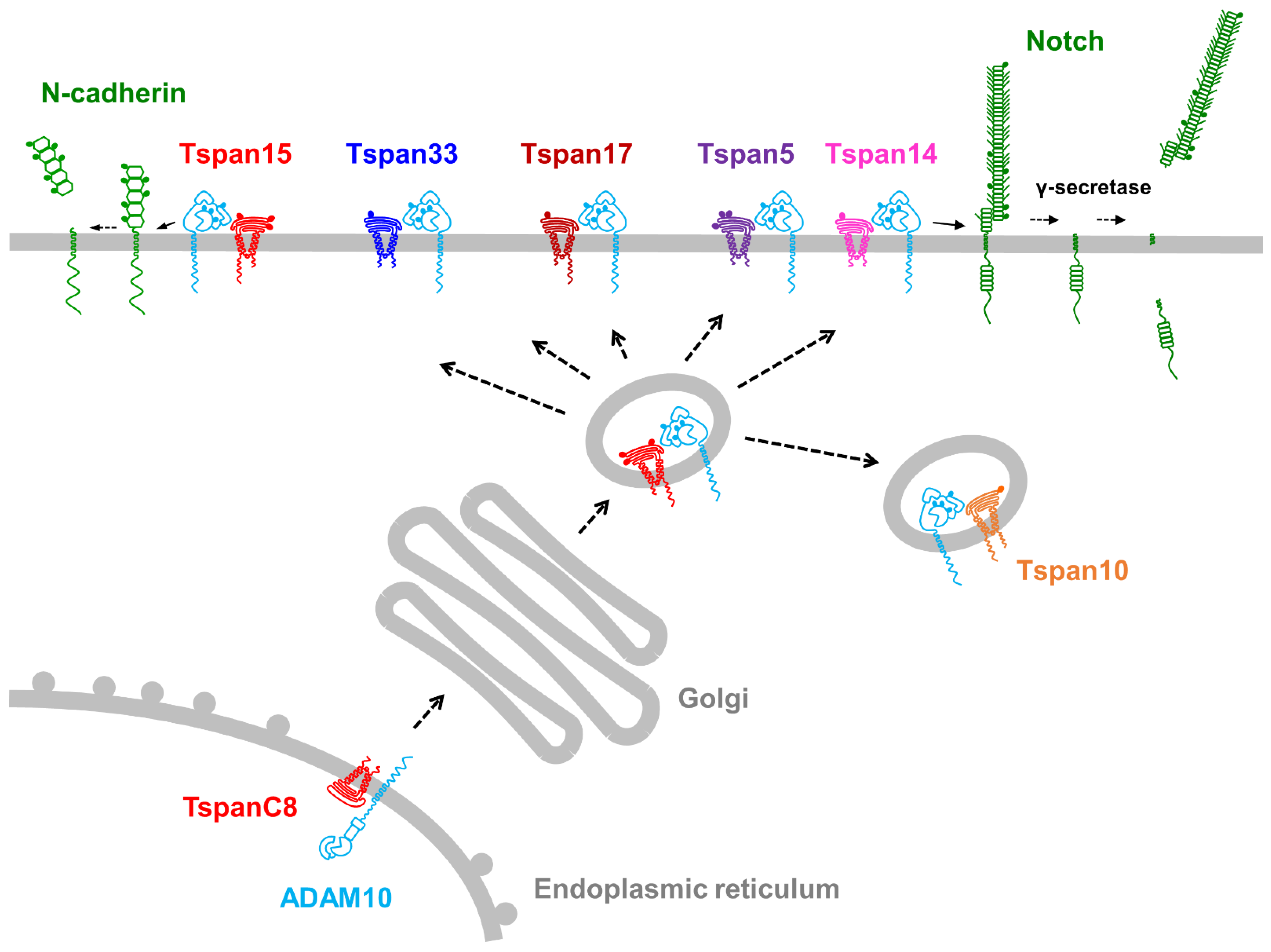
4. The TspanC8 Subgroup of Tetraspanins Regulate ADAM10
4.1. TspanC8 Overview
4.2. Tspan15/ADAM10 Promotes Cell Invasion and Cleaves N-Cadherin
4.3. Tspan5/ADAM10 and Tspan14/ADAM10 Cleave and Activate Notch
4.4. Additional Substrate Specificities for TspanC8/ADAM10 Complexes
4.5. Potential Mechanisms for TspanC8/ADAM10 Substrate Specificity
5. Concluding Remarks: Therapeutic Potential of TspanC8 Targeting
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Khokha, R.; Murthy, A.; Weiss, A. Metalloproteinases and their natural inhibitors in inflammation and immunity. Nat. Rev. Immunol. 2013, 13, 649–665. [Google Scholar] [CrossRef] [PubMed]
- Wong, E.; Maretzky, T.; Peleg, Y.; Blobel, C.P.; Sagi, I. The Functional Maturation of A Disintegrin and Metalloproteinase (ADAM) 9, 10, and 17 Requires Processing at a Newly Identified Proprotein Convertase (PC) Cleavage Site. J. Biol. Chem. 2015, 290, 12135–12146. [Google Scholar] [CrossRef] [Green Version]
- Klein, T.; Bischoff, R. Active Metalloproteases of the A Disintegrin And Metalloprotease (ADAM) Family: Biological Function and Structure. J. Proteome Res. 2011, 10, 17–33. [Google Scholar] [CrossRef]
- Groot, A.J.; Habets, R.; Yahyanejad, S.; Hodin, C.M.; Reiss, K.; Saftig, P.; Theys, J.; Vooijs, M. Regulated Proteolysis of NOTCH2 and NOTCH3 Receptors by ADAM10 and Presenilins. Mol. Cell. Biol. 2014, 34, 2822–2832. [Google Scholar] [CrossRef] [Green Version]
- Hartmann, D.; De Strooper, B.; Serneels, L.; Craessaerts, K.; Herreman, A.; Annaert, W.; Umans, L.; Lübke, T.; Illert, A.L.; Von Figura, K.; et al. The disintegrin/metalloprotease ADAM 10 is essential for Notch signalling but not for alpha-secretase activity in fibroblasts. Hum. Mol. Genet. 2002, 11, 2615–2624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Tetering, G.; van Diest, P.; Verlaan, I.; van der Wall, E.; Kopan, R.; Vooijs, M. Metalloprotease ADAM10 is required for Notch1 site 2 cleavage. J. Biol. Chem. 2009, 284, 31018–31027. [Google Scholar] [CrossRef] [Green Version]
- Zunke, F.; Rose-John, S. The shedding protease ADAM17: Physiology and pathophysiology. Biochim. Biophys. Acta (BBA) Bioenerg. 2017, 1864, 2059–2070. [Google Scholar] [CrossRef]
- Dulloo, I.; Muliyil, S.; Freeman, M. The molecular, cellular and pathophysiological roles of iRhom pseudoproteases. Open Biol. 2019, 9, 190003. [Google Scholar] [CrossRef] [Green Version]
- Geesala, R.; Issuree, P.D.; Maretzky, T. Novel functions of inactive rhomboid proteins in immunity and disease. J. Leukoc. Biol. 2019, 106, 823–835. [Google Scholar] [CrossRef] [PubMed]
- Matthews, A.L.; Noy, P.J.; Reyat, J.S.; Tomlinson, M.G. Regulation of A disintegrin and metalloproteinase (ADAM) family sheddases ADAM10 and ADAM17: The emerging role of tetraspanins and rhomboids. Platelets 2017, 28, 333–341. [Google Scholar] [CrossRef] [Green Version]
- Alabi, R.O.; Farber, G.; Blobel, C.P. Intriguing Roles for Endothelial ADAM10/Notch Signaling in the Development of Organ-Specific Vascular Beds. Physiol. Rev. 2018, 98, 2025–2061. [Google Scholar] [CrossRef] [PubMed]
- Saftig, P.; Lichtenthaler, S.F. The alpha secretase ADAM10: A metalloprotease with multiple functions in the brain. Prog. Neurobiol. 2015, 135, 1–20. [Google Scholar] [CrossRef]
- Weber, S.; Niessen, M.T.; Prox, J.; Lüllmann-Rauch, R.; Schmitz, A.; Schwanbeck, R.; Blobel, C.P.; Jorissen, E.; de Strooper, B.; Niessen, C.M.; et al. The disintegrin/metalloproteinase Adam10 is essential for epidermal integrity and Notch-mediated signaling. Development 2011, 138, 495–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sprinzak, D.; Blacklow, S.C. Biophysics of Notch Signaling. Annu. Rev. Biophys. 2021, 50, 157–189. [Google Scholar] [CrossRef]
- Kuhn, P.-H.; Colombo, A.V.; Schusser, B.; Dreymueller, D.; Wetzel, S.; Schepers, U.; Herber, J.; Ludwig, A.; Kremmer, E.; Montag, D.; et al. Systematic substrate identification indicates a central role for the metalloprotease ADAM10 in axon targeting and synapse function. eLife 2016, 5, 5. [Google Scholar] [CrossRef]
- Maretzky, T.; Reiss, K.; Ludwig, A.; Buchholz, J.; Scholz, F.; Proksch, E.; de Strooper, B.; Hartmann, D.; Saftig, P. ADAM10 mediates E-cadherin shedding and regulates epithelial cell-cell adhesion, migration, and -catenin translocation. Proc. Natl. Acad. Sci. USA 2005, 102, 9182–9187. [Google Scholar] [CrossRef] [Green Version]
- Reiss, K.; Maretzky, T.; Ludwig, A.; Tousseyn, T.; de Strooper, B.; Hartmann, D.; Saftig, P. ADAM10 cleavage of N-cadherin and regulation of cell-cell adhesion and beta-catenin nuclear signalling. EMBO J. 2005, 24, 742–752. [Google Scholar] [CrossRef] [PubMed]
- Schulz, B.; Pruessmeyer, J.; Maretzky, T.; Ludwig, A.; Blobel, C.P.; Saftig, P.; Reiss, K. ADAM10 Regulates Endothelial Permeability and T-Cell Transmigration by Proteolysis of Vascular Endothelial Cadherin. Circ. Res. 2008, 102, 1192–1201. [Google Scholar] [CrossRef] [Green Version]
- Sahin, U.; Weskamp, G.; Kelly, K.; Zhou, H.-M.; Higashiyama, S.; Peschon, J.; Hartmann, D.; Saftig, P.; Blobel, C.P. Distinct roles for ADAM10 and ADAM17 in ectodomain shedding of six EGFR ligands. J. Cell Biol. 2004, 164, 769–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, T.M., Jr.; Tharakan, A.; Martin, R.K. Targeting ADAM10 in Cancer and Autoimmunity. Front. Immunol. 2020, 11, 499. [Google Scholar] [CrossRef] [Green Version]
- Wetzel, S.; Seipold, L.; Saftig, P. The metalloproteinase ADAM10: A useful therapeutic target? Biochim. Biophys. Acta (BBA) Bioenerg. 2017, 1864, 2071–2081. [Google Scholar] [CrossRef]
- Postina, R.; Schroeder, A.; Dewachter, I.; Bohl, J.; Schmitt, U.; Kojro, E.; Prinzen, C.; Endres, K.; Hiemke, C.; Blessing, M.; et al. A disintegrin-metalloproteinase prevents amyloid plaque formation and hippocampal defects in an Alzheimer disease mouse model. J. Clin. Investig. 2004, 113, 1456–1464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bender, M.; Hagedorn, I.; Nieswandt, B. Genetic and antibody-induced glycoprotein VI deficiency equally protects mice from mechanically and FeCl3-induced thrombosis. J. Thromb. Haemost. 2011, 9, 1423–1426. [Google Scholar] [CrossRef] [PubMed]
- Gardiner, E.E.; Karunakaran, D.; Shen, Y.; Arthur, J.F.; Andrews, R.K.; Berndt, M.C. Controlled shedding of platelet glycoprotein (GP)VI and GPIb?IX?V by ADAM family metalloproteinases. J. Thromb. Haemost. 2007, 5, 1530–1537. [Google Scholar] [CrossRef]
- Ahmed, M.U.; Kaneva, V.; Loyau, S.; Nechipurenko, D.; Receveur, N.; Le Bris, M.; Janus-Bell, E.; Didelot, M.; Rauch, A.; Susen, S.; et al. Pharmacological Blockade of Glycoprotein VI Promotes Thrombus Disaggregation in the Absence of Thrombin. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 2127–2142. [Google Scholar] [CrossRef] [PubMed]
- Sulis, M.L.; Saftig, P.; Ferrando, A.A. Redundancy and Specificity of the Metalloprotease System Mediating Oncogenic NOTCH1 Activation in T-ALL. Leukemia 2011, 25, 1564–1569. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.C.; Liu, X.; Li, Y.; Covington, M.; Wynn, R.; Huber, R.; Hillman, M.; Yang, G.; Ellis, D.; Marando, C.; et al. Identification of ADAM10 as a major source of HER2 ectodomain sheddase activity in HER2 overexpressing breast cancer cells. Cancer Biol. Ther. 2006, 5, 657–664. [Google Scholar] [CrossRef] [Green Version]
- Zheng, H.; Zhong, A.; Xie, S.; Wang, Y.; Sun, J.; Zhang, J.; Tong, Y.; Chen, M.; Zhang, G.; Ma, Q.; et al. Elevated serum HER-2 predicts poor prognosis in breast cancer and is correlated to ADAM10 expression. Cancer Med. 2019, 8, 679–685. [Google Scholar] [CrossRef]
- Feldinger, K.; Generali, D.; Kramer-Marek, G.; Gijsen, M.; Ng, T.B.; Wong, J.H.; Strina, C.; Cappelletti, M.; Andreis, D.; Li, J.-L.; et al. ADAM10 mediates trastuzumab resistance and is correlated with survival in HER2 positive breast cancer. Oncotarget 2014, 5, 6633–6646. [Google Scholar] [CrossRef] [Green Version]
- Mathews, J.A.; Ford, J.; Norton, S.; Kang, D.-J.; Dellinger, A.; Gibb, D.R.; Ford, A.Q.; Massay, H.; Kepley, C.L.; Scherle, P.; et al. A potential new target for asthma therapy: A Disintegrin and Metalloprotease 10 (ADAM10) involvement in murine experimental asthma. Allergy 2011, 66, 1193–1200. [Google Scholar] [CrossRef] [Green Version]
- Ludwig, A.; Hundhausen, C.; Lambert, M.H.; Broadway, N.; Andrews, R.C.; Bickett, D.M.; Leesnitzer, M.A.; Becherer, J.D. Metalloproteinase Inhibitors for the Disintegrin-Like Metalloproteinases ADAM10 and ADAM17 that Differentially Block Constitutive and Phorbol Ester-Inducible Shedding of Cell Surface Molecules. Comb. Chem. High Throughput Screen. 2005, 8, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Witters, L.; Scherle, P.; Friedman, S.; Fridman, J.; Caulder, E.; Newton, R.; Lipton, A. Synergistic Inhibition with a Dual Epidermal Growth Factor Receptor/HER-2/neu Tyrosine Kinase Inhibitor and a Disintegrin and Metalloprotease Inhibitor. Cancer Res. 2008, 68, 7083–7089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Deventer, S.; Arp, A.B.; van Spriel, A.B. Dynamic Plasma Membrane Organization: A Complex Symphony. Trends Cell Biol. 2021, 31, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Charrin, S.; Jouannet, S.; Boucheix, C.; Rubinstein, E. Tetraspanins at a glance. J. Cell Sci. 2014, 127, 3641–3648. [Google Scholar] [CrossRef] [Green Version]
- Dahmane, S.; Doucet, C.; Le Gall, A.; Chamontin, C.; Dosset, P.; Murcy, F.; Fernandez, L.; Salas, D.; Rubinstein, E.; Mougel, M.; et al. Nanoscale organization of tetraspanins during HIV-1 budding by correlative dSTORM/AFM. Nanoscale 2019, 11, 6036–6044. [Google Scholar] [CrossRef]
- Marjon, K.D.; Termini, C.; Karlen, K.L.; Saito-Reis, C.; Soria, C.E.; Lidke, K.A.; Gillette, J.M. Tetraspanin CD82 regulates bone marrow homing of acute myeloid leukemia by modulating the molecular organization of N-cadherin. Oncogene 2016, 35, 4132–4140. [Google Scholar] [CrossRef] [Green Version]
- Zuidscherwoude, M.; Göttfert, F.; Dunlock, V.M.E.; Figdor, C.; Bogaart, G.V.D.; Van Spriel, A.B. The tetraspanin web revisited by super-resolution microscopy. Sci. Rep. 2015, 5, 12201. [Google Scholar] [CrossRef] [Green Version]
- Zimmerman, B.; Kelly, B.; McMillan, B.J.; Seegar, T.C.; Dror, R.O.; Kruse, A.C.; Blacklow, S.C. Crystal Structure of a Full-Length Human Tetraspanin Reveals a Cholesterol-Binding Pocket. Cell 2016, 167, 1041–1051.e11. [Google Scholar] [CrossRef] [Green Version]
- Susa, K.J.; Rawson, S.; Kruse, A.C.; Blacklow, S.C. Cryo-EM structure of the B cell co-receptor CD19 bound to the tetraspanin CD81. Science 2021, 371, 300–305. [Google Scholar] [CrossRef]
- Umeda, R.; Satouh, Y.; Takemoto, M.; Nakada-Nakura, Y.; Liu, K.; Yokoyama, T.; Shirouzu, M.; Iwata, S.; Nomura, N.; Sato, K.; et al. Structural insights into tetraspanin CD9 function. Nat. Commun. 2020, 11, 1606. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Liu, X.R.; Greenberg, Z.J.; Zhou, F.; He, P.; Fan, L.; Liu, S.; Shen, G.; Egawa, T.; Gross, M.L.; et al. Open conformation of tetraspanins shapes interaction partner networks on cell membranes. EMBO J. 2020, 39. [Google Scholar] [CrossRef] [PubMed]
- Hemler, M.E. Tetraspanin proteins promote multiple cancer stages. Nat. Rev. Cancer 2014, 14, 49–60. [Google Scholar] [CrossRef]
- Junge, H.J.; Yang, S.; Burton, J.B.; Paes, K.; Shu, X.; French, D.M.; Costa, M.; Rice, D.S.; Ye, W. TSPAN12 regulates retinal vascular development by promoting Norrin- but not Wnt-induced FZD4/beta-catenin signaling. Cell 2009, 139, 299–311. [Google Scholar] [CrossRef] [Green Version]
- Gavin, R.L.; Koo, C.Z.; Tomlinson, M.G. Tspan18 is a novel regulator of thrombo-inflammation. Med. Microbiol. Immunol. 2020, 209, 553–564. [Google Scholar] [CrossRef] [PubMed]
- Noy, P.J.; Gavin, R.L.; Colombo, D.; Haining, E.J.; Reyat, J.S.; Payne, H.; Thielmann, I.; Lokman, A.B.; Neag, G.; Yang, J.; et al. Tspan18 is a novel regulator of the Ca2+ channel Orai1 and von Willebrand factor release in endothelial cells. Haematologica 2018, 104, 1892–1905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oosterheert, W.; Xenaki, K.T.; Neviani, V.; Pos, W.; Doulkeridou, S.; Manshande, J.; Pearce, N.M.; Kroon-Batenburg, L.M.; Lutz, M.; Henegouwen, P.M.V.B.E.; et al. Implications for tetraspanin-enriched microdomain assembly based on structures of CD9 with EWI-F. Life Sci. Alliance 2020, 3, e202000883. [Google Scholar] [CrossRef]
- Susa, K.J.; Seegar, T.C.; Blacklow, S.C.; Kruse, A.C. A dynamic interaction between CD19 and the tetraspanin CD81 controls B cell co-receptor trafficking. eLife 2020, 9, 9. [Google Scholar] [CrossRef]
- Kitadokoro, K.; Bordo, D.; Galli, G.; Petracca, R.; Falugi, F.; Abrignani, S.; Grandi, G.; Bolognesi, M. CD81 extracellular domain 3D structure: Insight into the tetraspanin superfamily structural motifs. EMBO J. 2001, 20, 12–18. [Google Scholar] [CrossRef] [Green Version]
- Charrin, S.; Yalaoui, S.; Bartosch, B.; Cocquerel, L.; Franetich, J.F.; Boucheix, C.; Mazier, D.; Rubinstein, E.; Silvie, O. The Ig domain protein CD9P-1 down-regulates CD81 ability to support Plasmodium yoelii infection. J. Biol. Chem. 2009, 284, 31572–31578. [Google Scholar] [CrossRef] [Green Version]
- Hochheimer, N.; Sies, R.; Aschenbrenner, A.C.; Schneider, D.; Lang, T. Classes of non-conventional tetraspanins defined by alternative splicing. Sci. Rep. 2019, 9, 14075. [Google Scholar] [CrossRef] [Green Version]
- Dornier, E.; Coumailleau, F.; Ottavi, J.F.; Moretti, J.; Boucheix, C.; Mauduit, P.; Schweisguth, F.; Rubinstein, E. TspanC8 tetraspanins regulate ADAM10/Kuzbanian trafficking and promote Notch activation in flies and mammals. J. Cell Biol. 2012, 199, 481–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haining, E.J.; Yang, J.; Bailey, R.L.; Khan, K.; Collier, R.; Tsai, S.; Watson, S.; Frampton, J.; Garcia, P.; Tomlinson, M.G. The TspanC8 Subgroup of Tetraspanins Interacts with A Disintegrin and Metalloprotease 10 (ADAM10) and Regulates Its Maturation and Cell Surface Expression. J. Biol. Chem. 2012, 287, 39753–39765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prox, J.; Willenbrock, M.; Weber, S.; Lehmann, T.; Schmidt-Arras, D.; Schwanbeck, R.; Saftig, P.; Schwake, M. Tetraspanin15 regulates cellular trafficking and activity of the ectodomain sheddase ADAM10. Cell. Mol. Life Sci. 2012, 69, 2919–2932. [Google Scholar] [CrossRef]
- Jouannet, S.; Saint-Pol, J.; Fernandez, L.; Nguyen, V.; Charrin, S.; Boucheix, C.; Brou, C.; Milhiet, P.-E.; Rubinstein, E. TspanC8 tetraspanins differentially regulate the cleavage of ADAM10 substrates, Notch activation and ADAM10 membrane compartmentalization. Cell. Mol. Life Sci. 2016, 73, 1895–1915. [Google Scholar] [CrossRef] [Green Version]
- Noy, P.J.; Yang, J.; Reyat, J.S.; Matthews, A.L.; Charlton, A.E.; Furmston, J.; Rogers, D.A.; Rainger, G.E.; Tomlinson, M.G. TspanC8 Tetraspanins and A Disintegrin and Metalloprotease 10 (ADAM10) Interact via Their Extracellular Regions: Evidence for distinct binding mechanisms for different TspanC8 proteins. J. Biol. Chem. 2016, 291, 3145–3157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seipold, L.; Altmeppen, H.; Koudelka, T.; Tholey, A.; Kasparek, P.; Sedlacek, R.; Schweizer, M.; Bär, J.; Mikhaylova, M.; Glatzel, M.; et al. In vivo regulation of the A disintegrin and metalloproteinase 10 (ADAM10) by the tetraspanin 15. Cell. Mol. Life Sci. 2018, 75, 3251–3267. [Google Scholar] [CrossRef]
- Eschenbrenner, E.; Jouannet, S.; Clay, D.; Chaker, J.; Boucheix, C.; Brou, C.; Tomlinson, M.G.; Charrin, S.; Rubinstein, E. TspanC8 tetraspanins differentially regulate ADAM10 endocytosis and half-life. Life Sci. Alliance 2019, 3, e201900444. [Google Scholar] [CrossRef] [Green Version]
- Koo, C.Z.; Harrison, N.; Noy, P.J.; Szyroka, J.; Matthews, A.L.; Hsia, H.-E.; Müller, S.A.; Tüshaus, J.; Goulding, J.; Willis, K.; et al. The tetraspanin Tspan15 is an essential subunit of an ADAM10 scissor complex. J. Biol. Chem. 2020, 295, 12822–12839. [Google Scholar] [CrossRef] [Green Version]
- Saint-Pol, J.; Billard, M.; Dornier, E.; Eschenbrenner, E.; Danglot, L.; Boucheix, C.; Charrin, S.; Rubinstein, E. New insights into the tetraspanin Tspan5 using novel monoclonal antibodies. J. Biol. Chem. 2017, 292, 9551–9566. [Google Scholar] [CrossRef] [Green Version]
- Matthews, A.L.; Koo, C.Z.; Szyroka, J.; Harrison, N.; Kanhere, A.; Tomlinson, M.G. Regulation of Leukocytes by TspanC8 Tetraspanins and the “Molecular Scissor” ADAM10. Front. Immunol. 2018, 9, 9. [Google Scholar] [CrossRef]
- Matthews, A.L.; Szyroka, J.; Collier, R.; Noy, P.J.; Tomlinson, M.G. Scissor sisters: Regulation of ADAM10 by the TspanC8 tetraspanins. Biochem. Soc. Trans. 2017, 45, 719–730. [Google Scholar] [CrossRef] [Green Version]
- Saint-Pol, J.; Eschenbrenner, E.; Dornier, E.; Boucheix, C.; Charrin, S.; Rubinstein, E. Regulation of the trafficking and the function of the metalloprotease ADAM10 by tetraspanins. Biochem. Soc. Trans. 2017, 45, 937–944. [Google Scholar] [CrossRef]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Hiroshima, K.; Shiiba, M.; Oka, N.; Hayashi, F.; Ishida, S.; Fukushima, R.; Koike, K.; Iyoda, M.; Nakashima, D.; Tanzawa, H.; et al. Tspan15 plays a crucial role in metastasis in oral squamous cell carcinoma. Exp. Cell Res. 2019, 384, 111622. [Google Scholar] [CrossRef] [PubMed]
- Sidahmed-Adrar, N.; Ottavi, J.; Benzoubir, N.; Saadi, T.A.; Saleh, M.B.; Mauduit, P.; Guettier, C.; Desterke, C.; Le Naour, F. Tspan15 Is a New Stemness-Related Marker in Hepatocellular Carcinoma. Proteomics 2019, 19, e1900025. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Zhang, Z.; Li, L.; Qin, Y.R.; Liu, H.; Jiang, C.; Zeng, T.T.; Li, M.Q.; Xie, D.; Li, Y.; et al. TSPAN15 interacts with BTRC to promote oesophageal squamous cell carcinoma metastasis via activating NF-kappaB signaling. Nat. Commun. 2018, 9, 1423. [Google Scholar] [CrossRef] [PubMed]
- Sépult, C.; Bellefroid, M.; Rocks, N.; Donati, K.; Gérard, C.; Gilles, C.; Ludwig, A.; Duysinx, B.; Noel, A.; Cataldo, D. ADAM10 mediates malignant pleural mesothelioma invasiveness. Oncogene 2019, 38, 3521–3534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suyama, K.; Shapiro, I.; Guttman, M.; Hazan, R.B. A signaling pathway leading to metastasis is controlled by N-cadherin and the FGF receptor. Cancer Cell 2002, 2, 301–314. [Google Scholar] [CrossRef] [Green Version]
- Xie, Q.; Guo, H.; He, P.; Deng, H.; Gao, Y.; Dong, N.; Niu, W.; Liu, T.; Li, M.; Wang, S.; et al. Tspan5 promotes epithelial-mesenchymal transition and tumour metastasis of hepatocellular carcinoma by activating Notch signalling. Mol. Oncol. 2021. [Google Scholar] [CrossRef]
- Zhou, J.; Fujiwara, T.; Ye, S.; Li, X.; Zhao, H. Downregulation of Notch Modulators, Tetraspanin 5 and 10, Inhibits Osteoclastogenesis in Vitro. Calcif. Tissue Int. 2014, 95, 209–217. [Google Scholar] [CrossRef]
- Chastagner, P.; Rubinstein, E.; Brou, C. Ligand-activated Notch undergoes DTX4-mediated ubiquitylation and bilateral endocytosis before ADAM10 processing. Sci. Signal. 2017, 10, eaag2989. [Google Scholar] [CrossRef] [Green Version]
- Reyat, J.S.; Chimen, M.; Noy, P.J.; Szyroka, J.; Rainger, G.E.; Tomlinson, M.G. ADAM10-Interacting Tetraspanins Tspan5 and Tspan17 Regulate VE-Cadherin Expression and Promote T Lymphocyte Transmigration. J. Immunol. 2017, 199, 666–676. [Google Scholar] [CrossRef] [Green Version]
- Brummer, T.; Muller, S.A.; Pan-Montojo, F.; Yoshida, F.; Fellgiebel, A.; Tomita, T.; Endres, K.; Lichtenthaler, S.F. NrCAM is a marker for substrate-selective activation of ADAM10 in Alzheimer’s disease. EMBO Mol. Med. 2019, 11, e9695. [Google Scholar] [CrossRef] [PubMed]
- Shah, J.; Rouaud, F.; Guerrera, D.; Vasileva, E.; Popov, L.M.; Kelley, W.L.; Rubinstein, E.; Carette, J.; Amieva, M.; Citi, S. A Dock-and-Lock Mechanism Clusters ADAM10 at Cell-Cell Junctions to Promote α-Toxin Cytotoxicity. Cell Rep. 2018, 25, 2132–2147.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seegar, T.C.; Killingsworth, L.B.; Saha, N.; Meyer, P.A.; Patra, D.; Zimmerman, B.; Janes, P.; Rubinstein, E.; Nikolov, D.B.; Skiniotis, G.; et al. Structural Basis for Regulated Proteolysis by the α-Secretase ADAM10. Cell 2017, 171, 1638–1648.e7. [Google Scholar] [CrossRef] [Green Version]
- Uemura, K.; Kihara, T.; Kuzuya, A.; Okawa, K.; Nishimoto, T.; Ninomiya, H.; Sugimoto, H.; Kinoshita, A.; Shimohama, S. Characterization of sequential N-cadherin cleavage by ADAM10 and PS1. Neurosci. Lett. 2006, 402, 278–283. [Google Scholar] [CrossRef] [PubMed]
- Marambaud, P.; Shioi, J.; Serban, G.; Georgakopoulos, A.; Sarner, S.; Nagy, V.; Baki, L.; Wen, P.; Efthimiopoulos, S.; Shao, Z.; et al. A presenilin-1/gamma-secretase cleavage releases the E-cadherin intracellular domain and regulates disassembly of adherens junctions. EMBO J. 2002, 21, 1948–1956. [Google Scholar] [CrossRef] [Green Version]
- Sunnarborg, S.W.; Hinkle, C.L.; Stevenson, M.; Russell, W.E.; Raska, C.S.; Peschon, J.J.; Castner, B.J.; Gerhart, M.J.; Paxton, R.J.; Black, R.A.; et al. Tumor Necrosis Factor-α Converting Enzyme (TACE) Regulates Epidermal Growth Factor Receptor Ligand Availability. J. Biol. Chem. 2002, 277, 12838–12845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robert, J.-M.H.; Amoussou, N.G.; Le Mai, H.; Logé, C.; Brouard, S. Tetraspanins: Useful multifunction proteins for the possible design and development of small-molecule therapeutic tools. Drug Discov. Today 2021, 26, 56–68. [Google Scholar] [CrossRef]
- van der Horst, H.J.; Oostindie, S.C.; Cillessen, S.; Gelderloos, A.T.; Overdijk, M.B.; Nijhof, I.S.; Zweegman, S.; Chamuleau, M.E.D.; Breij, E.C.W.; Mutis, T. Potent Preclinical Efficacy of DuoHexaBody-CD37 in B-Cell Malignancies. Hemasphere 2021, 5, e504. [Google Scholar] [CrossRef] [PubMed]
- Oostindie, S.C.; Van Der Horst, H.J.; Kil, L.P.; Strumane, K.; Overdijk, M.B.; Brink, E.N.V.D.; Brakel, J.H.N.V.D.; Rademaker, H.J.; Van Kessel, B.; Noort, J.V.D.; et al. DuoHexaBody-CD37®, a novel biparatopic CD37 antibody with enhanced Fc-mediated hexamerization as a potential therapy for B-cell malignancies. Blood Cancer J. 2020, 10, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Scarfò, I.; Ormhøj, M.; Frigault, M.J.; Castano, A.P.; Lorrey, S.; Bouffard, A.A.; Van Scoyk, A.; Rodig, S.J.; Shay, A.J.; Aster, J.C.; et al. Anti-CD37 chimeric antigen receptor T cells are active against B- and T-cell lymphomas. Blood 2018, 132, 1495–1506. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, A.; Aster, J.C. Notch signaling in cancer: Complexity and challenges on the path to clinical translation. Semin. Cancer Biol. 2021. [Google Scholar] [CrossRef]
- Ventress, J.K.; Partridge, L.J.; Read, R.C.; Cozens, D.; MacNeil, S.; Monk, P.N. Peptides from Tetraspanin CD9 Are Potent Inhibitors of Staphylococcus Aureus Adherence to Keratinocytes. PLoS ONE 2016, 11, e0160387. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Harrison, N.; Koo, C.Z.; Tomlinson, M.G. Regulation of ADAM10 by the TspanC8 Family of Tetraspanins and Their Therapeutic Potential. Int. J. Mol. Sci. 2021, 22, 6707. https://doi.org/10.3390/ijms22136707
Harrison N, Koo CZ, Tomlinson MG. Regulation of ADAM10 by the TspanC8 Family of Tetraspanins and Their Therapeutic Potential. International Journal of Molecular Sciences. 2021; 22(13):6707. https://doi.org/10.3390/ijms22136707
Chicago/Turabian StyleHarrison, Neale, Chek Ziu Koo, and Michael G. Tomlinson. 2021. "Regulation of ADAM10 by the TspanC8 Family of Tetraspanins and Their Therapeutic Potential" International Journal of Molecular Sciences 22, no. 13: 6707. https://doi.org/10.3390/ijms22136707