
Small RNA, Transcriptome and Degradome Analysis of the Transgenerational Heat Stress Response Network in Durum Wheat




Abstract
:1. Introduction
2. Results
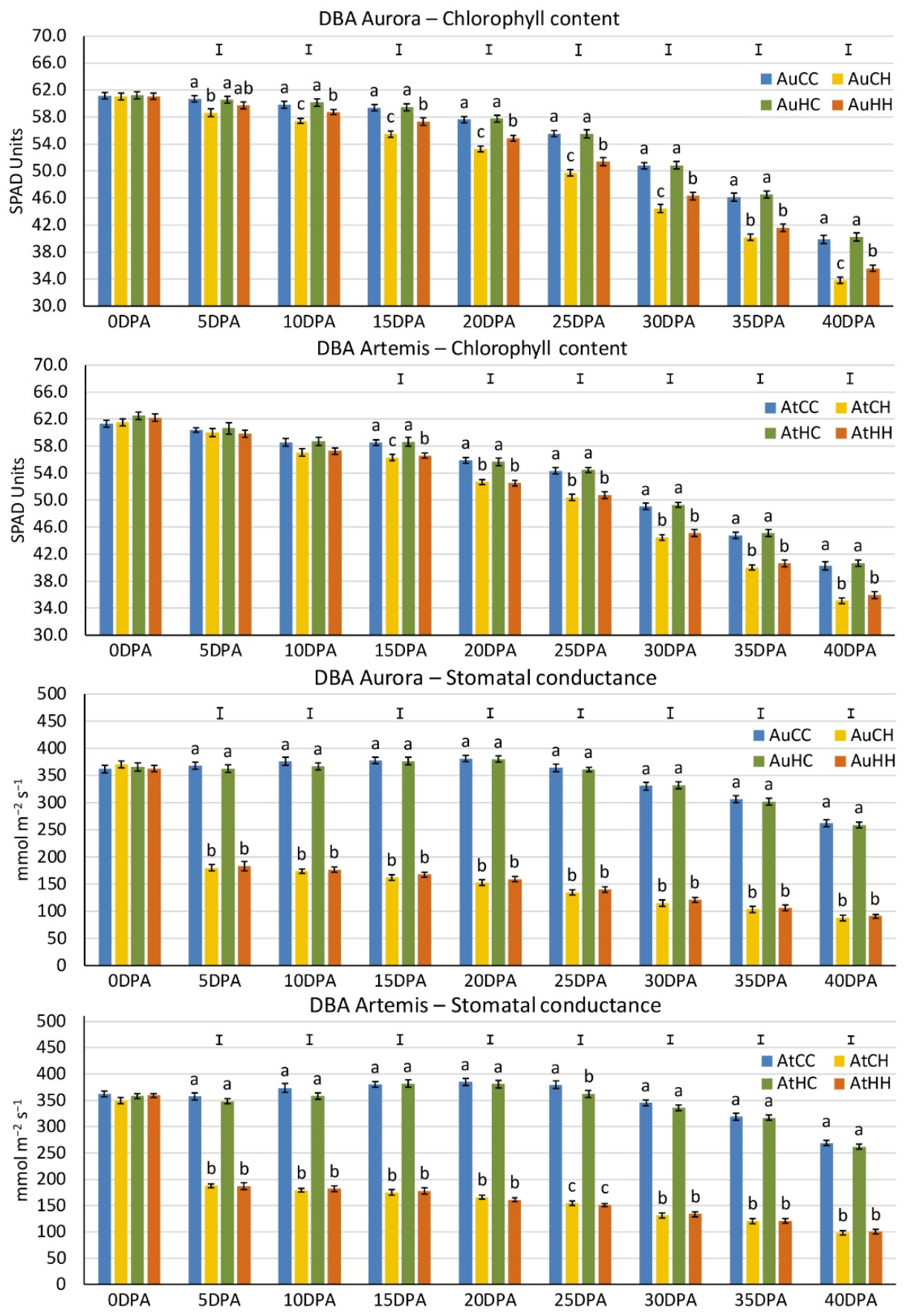
2.1. Physiological, Grain Quality and Yield Performance of Two Durum Wheat Varieties under the Effects of Transgenerational and Progeny Heat Stress
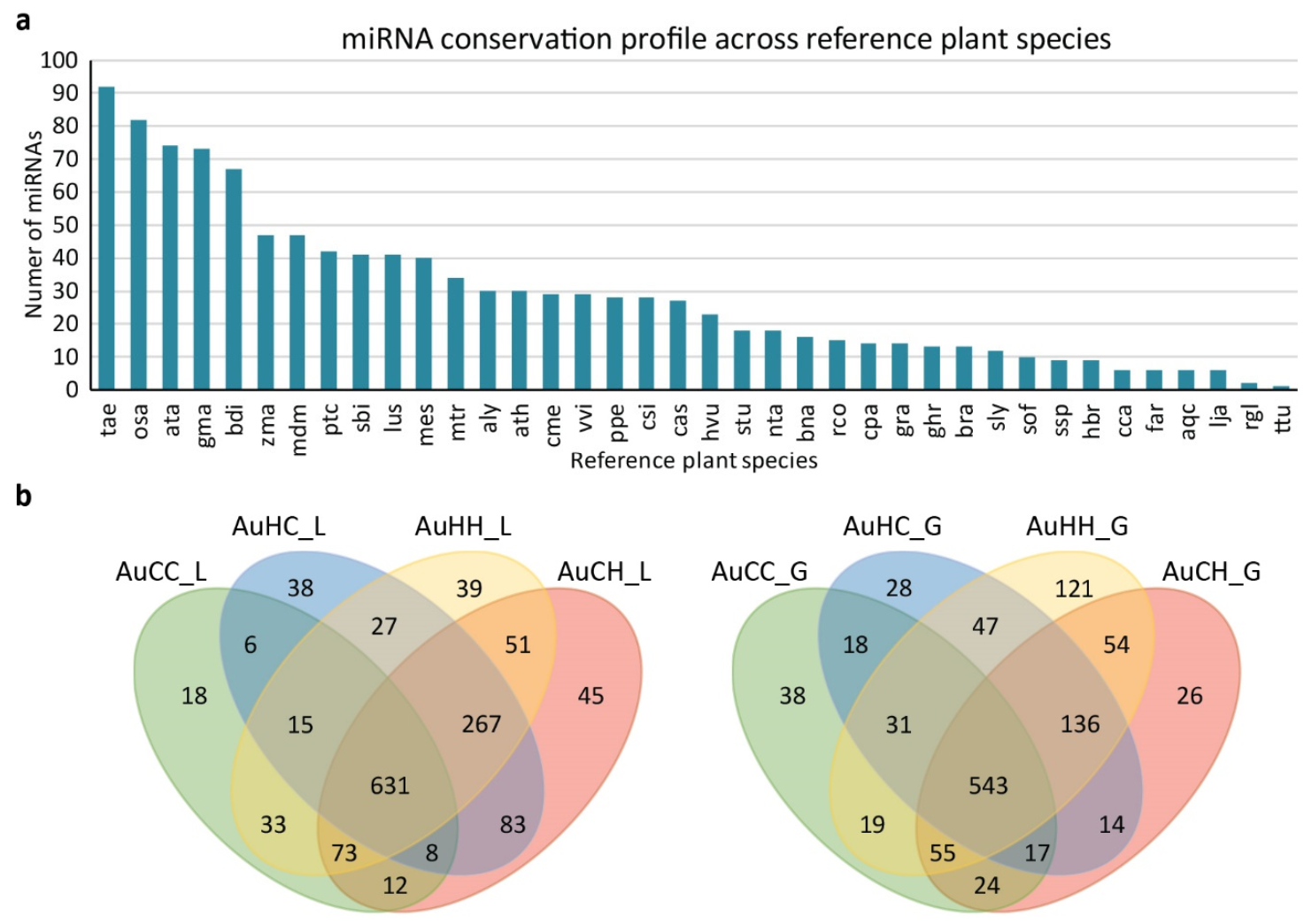
2.2. Analysis of the miRNAome and Differentially Expressed miRNAs (DEMs)
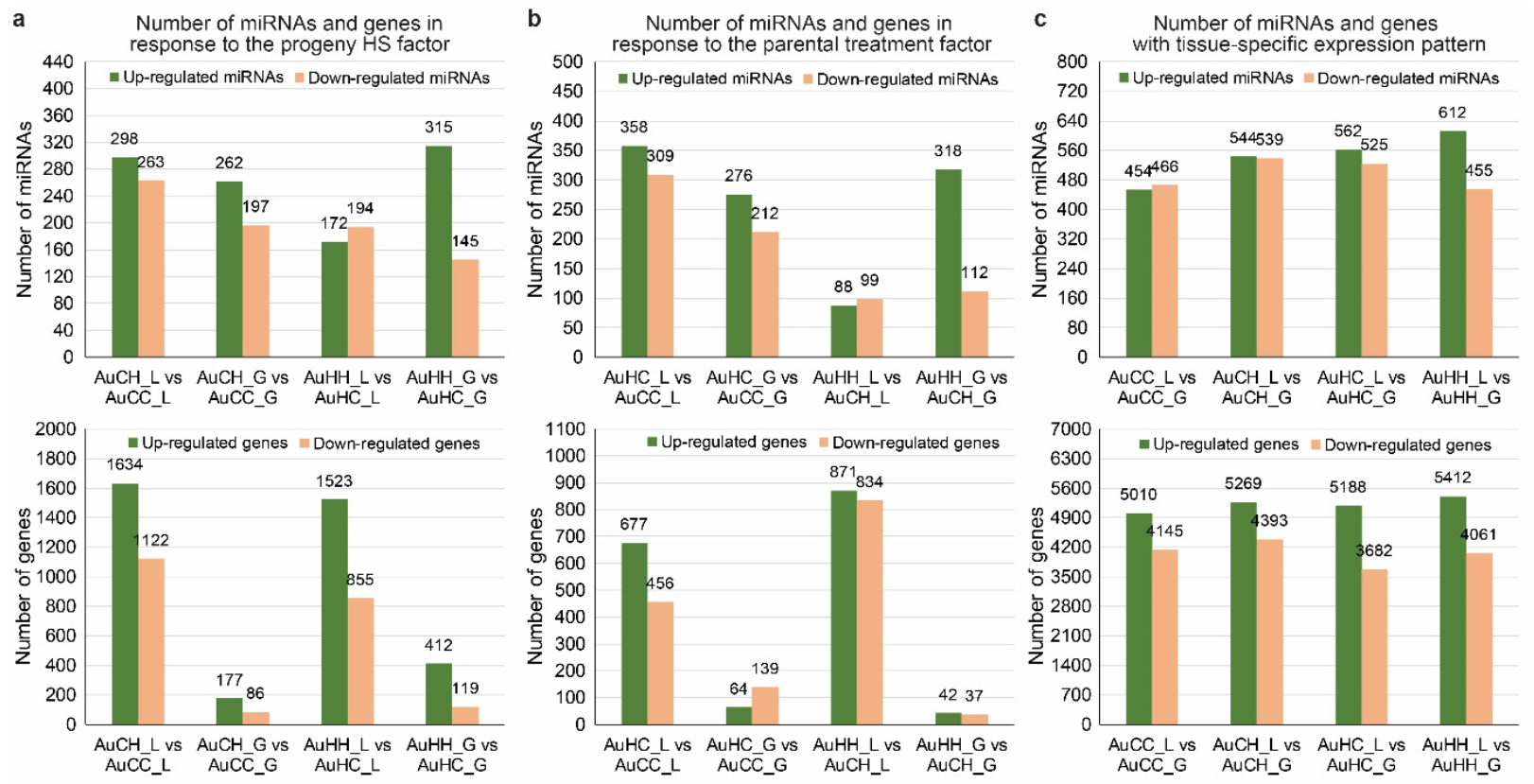
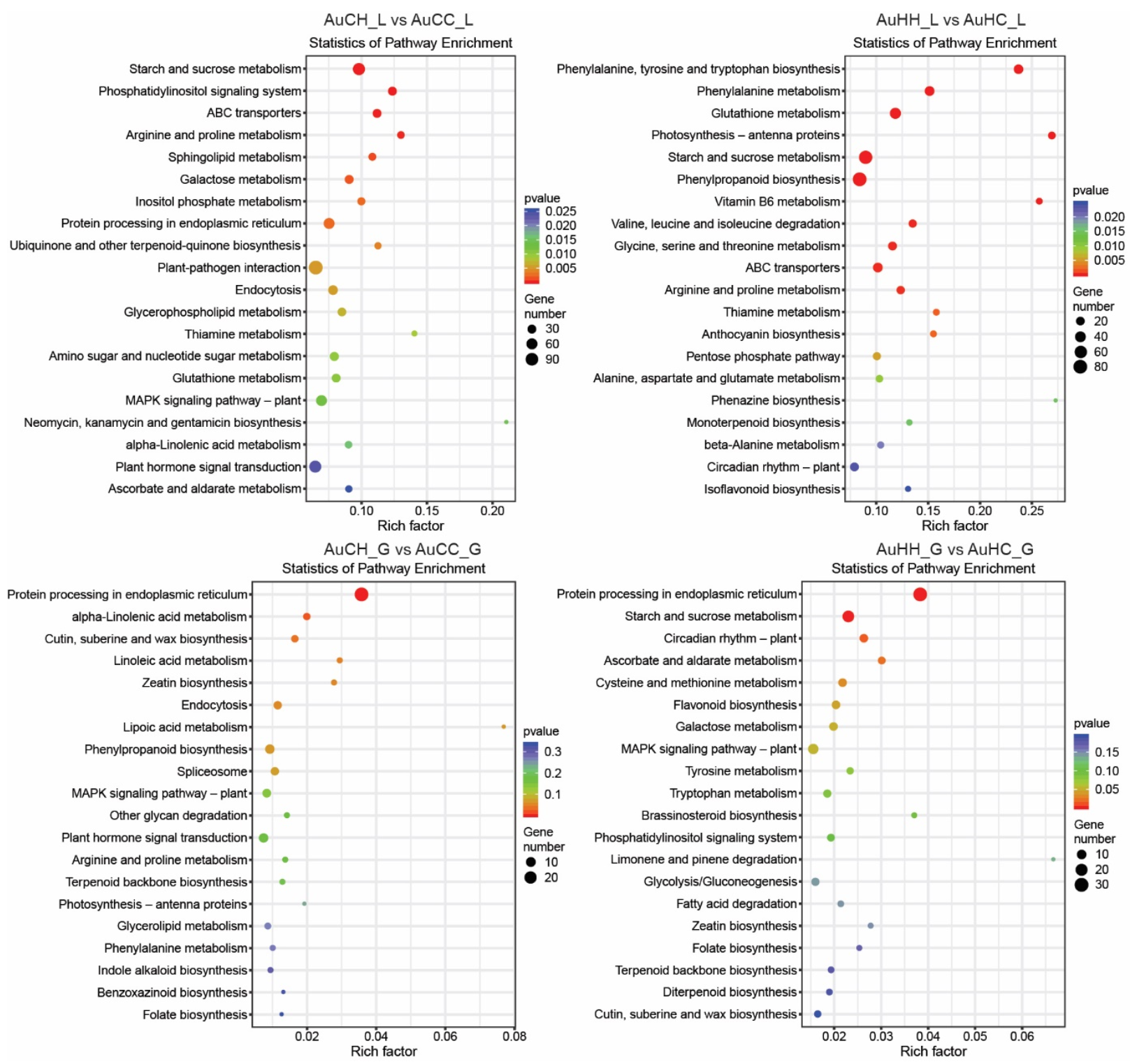
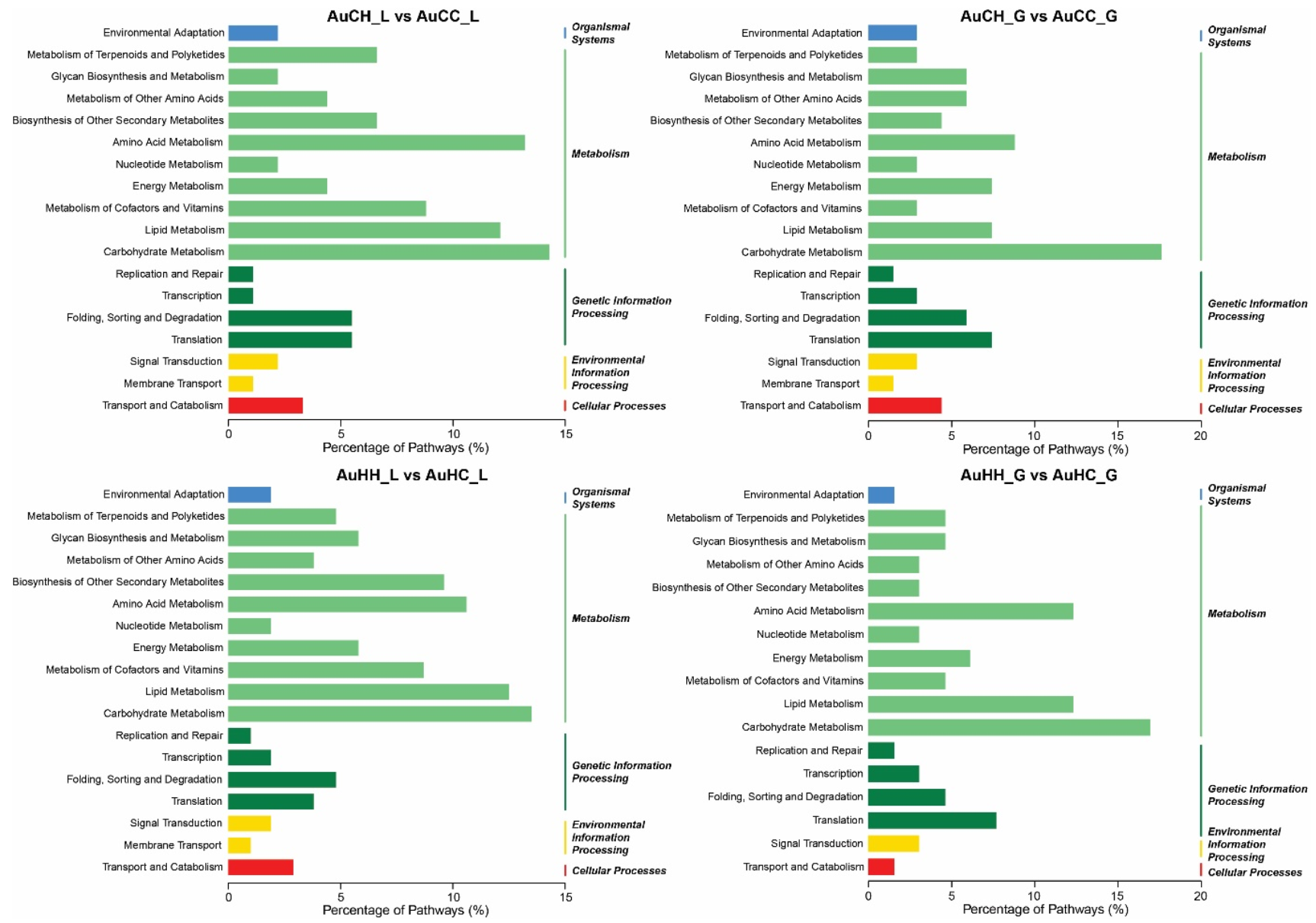
2.3. Analysis of the mRNA Transcriptome, Degradome and Differentially Expressed Genes (DEGs)
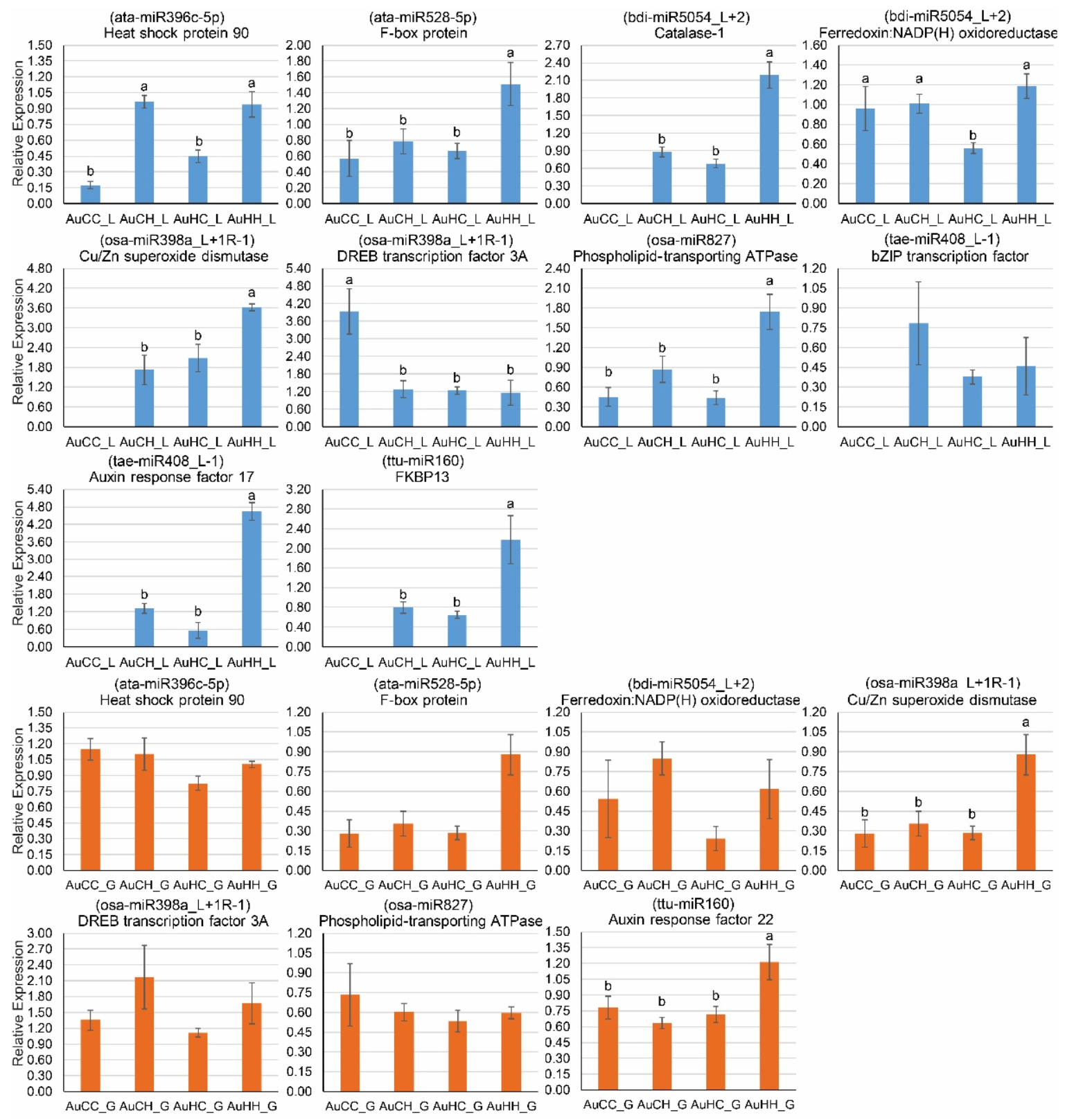
2.4. Identification of Key miRNA-mRNA Modules with Integrated Omics Analysis
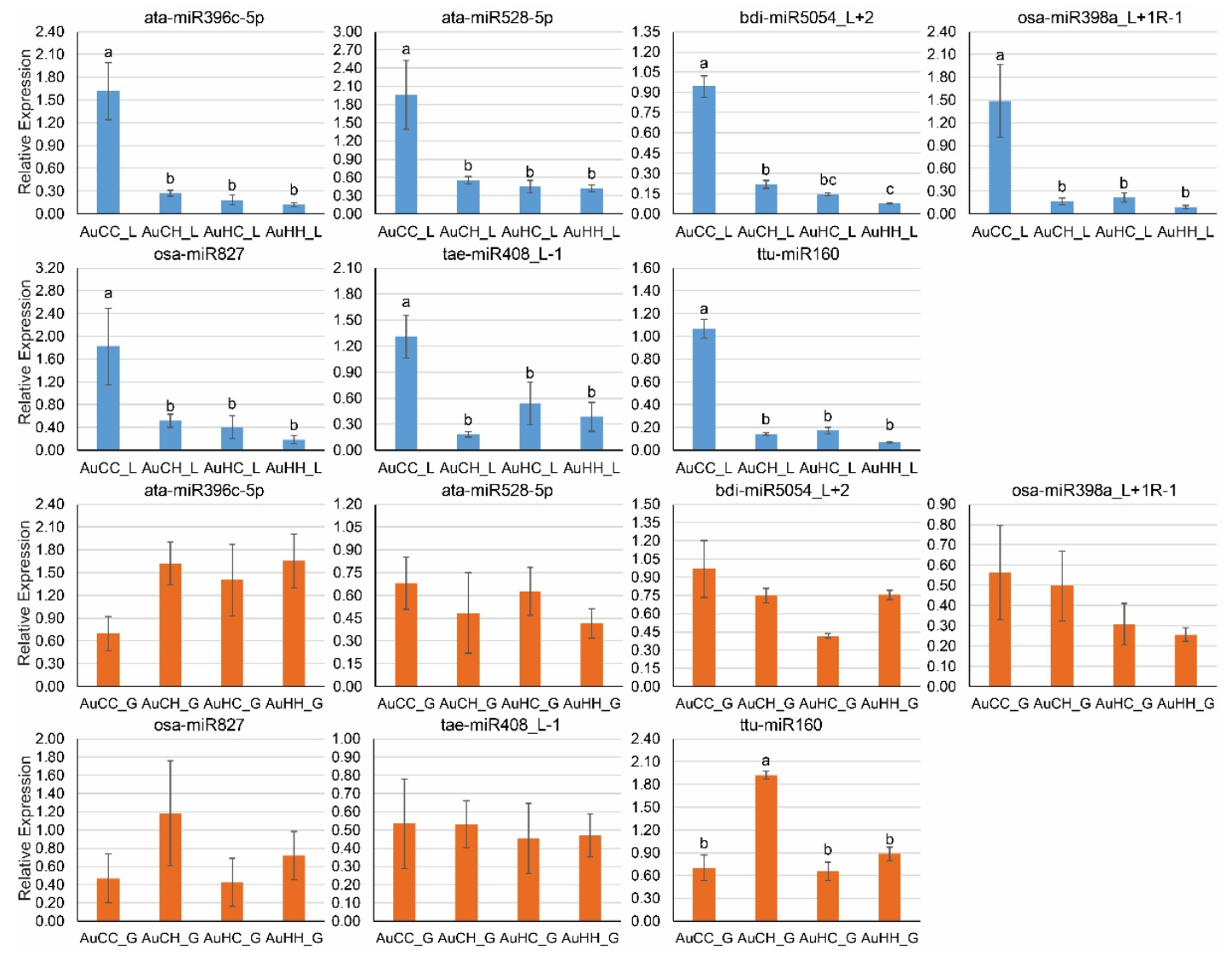
2.5. qPCR Profiling of Stress-Responsive DEMs and DEGs
3. Discussion
4. Materials and Methods
4.1. Glasshouse Growing Conditions and Stress Treatments
4.2. Evaluation of Crop Performance and Statistical Analysis
4.3. High-Throughput Small RNA Sequencing, Transcriptome Sequencing and Degradome Sequencing
4.4. Intergrated Omics Analysis and Functional Enrichment Analyses of Genes
4.5. qPCR Profiling of DEMs and DEGs
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Guzmán, C.; Autrique, J.E.; Mondal, S.; Singh, R.P.; Govindan, V.; Morales-Dorantes, A.; Posadas-Romano, G.; Crossa, J.; Ammar, K.; Peña, R.J. Response to drought and heat stress on wheat quality, with special emphasis on bread-making quality, in durum wheat. Field Crops Res. 2016, 186, 157–165. [Google Scholar] [CrossRef]
- Sissons, M. Role of durum wheat composition on the quality of pasta and bread. Food 2008, 2, 75–90. [Google Scholar]
- Troccoli, A.; Borrelli, G.; De Vita, P.; Fares, C.; Di Fonzo, N. Mini review: Durum wheat quality: A multidisciplinary concept. J. Cereal Sci. 2000, 32, 99–113. [Google Scholar] [CrossRef]
- Longin, C.F.H.; Sieber, A.N.; Reif, J.C. Combining frost tolerance, high grain yield and good pasta quality in durum wheat. Plant Breed. 2013, 132, 353–358. [Google Scholar] [CrossRef]
- Li, Y.-F.; Wu, Y.; Hernandez-Espinosa, N.; Peña, R.J. Heat and drought stress on durum wheat: Responses of genotypes, yield, and quality parameters. J. Cereal Sci. 2013, 57, 398–404. [Google Scholar] [CrossRef]
- Liu, H.; Searle, I.R.; Mather, D.E.; Able, A.J.; Able, J.A. Morphological, physiological and yield responses of durum wheat to pre-anthesis water-deficit stress are genotype-dependent. Crop Pasture Sci. 2015, 66, 1024–1038. [Google Scholar] [CrossRef]
- Liu, H.; Bruce, D.R.; Sissons, M.; Able, A.J.; Able, J.A. Genotype-dependent changes in the phenolic content of durum under water-deficit stress. Cereal Chem. 2018, 95, 59–78. [Google Scholar] [CrossRef]
- Liu, H.; Able, A.J.; Able, J.A. Genotypic performance of Australian durum under single and combined water-deficit and heat stress during reproduction. Sci. Rep. 2019, 9, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Stone, P.; Nicolas, M. Wheat cultivars vary widely in their responses of grain yield and quality to short periods of post-anthesis heat stress. Funct. Plant Biol. 1994, 21, 887–900. [Google Scholar] [CrossRef]
- Barnabás, B.; Jäger, K.; Fehér, A. The effect of drought and heat stress on reproductive processes in cereals. Plant Cell Environ. 2008, 31, 11–38. [Google Scholar] [CrossRef]
- Farooq, M.; Bramley, H.; Palta, J.A.; Siddique, K.H. Heat stress in wheat during reproductive and grain-filling phases. Crit. Rev. Plant Sci. 2011, 30, 491–507. [Google Scholar] [CrossRef]
- Talukder, A.; McDonald, G.K.; Gill, G.S. Effect of short-term heat stress prior to flowering and early grain set on the grain yield of wheat. Field Crops Res. 2014, 160, 54–63. [Google Scholar] [CrossRef]
- Aiqing, S.; Somayanda, I.; Sebastian, S.V.; Singh, K.; Gill, K.; Prasad, P.; Jagadish, S.K. Heat stress during flowering affects time of day of flowering, seed set, and grain quality in spring wheat. Crop Sci. 2018, 58, 380–392. [Google Scholar]
- Porter, J.R.; Gawith, M. Temperatures and the growth and development of wheat: A review. Eur. J. Agron. 1999, 10, 23–36. [Google Scholar] [CrossRef]
- Akter, N.; Islam, M.R. Heat stress effects and management in wheat. A review. Agron. Sustain. Dev. 2017, 37, 1–17. [Google Scholar] [CrossRef]
- Almeselmani, M.; Deshmukh, P.; Sairam, R. High temperature stress tolerance in wheat genotypes: Role of antioxidant defence enzymes. Acta Agron. Hung. 2009, 57, 1–14. [Google Scholar] [CrossRef]
- Kumar, R.R.; Goswami, S.; Dubey, K.; Singh, K.; Singh, J.P.; Kumar, A.; Rai, G.K.; Singh, S.D.; Bakshi, S.; Singh, B. RuBisCo activase—A catalytic chaperone involved in modulating the RuBisCo activity and heat stress-tolerance in wheat. J. Plant Biochem. Biotechnol. 2019, 28, 63–75. [Google Scholar] [CrossRef]
- Wardlaw, I.F.; Blumenthal, C.; Larroque, O.; Wrigley, C.W. Contrasting effects of chronic heat stress and heat shock on kernel weight and flour quality in wheat. Funct. Plant Biol. 2002, 29, 25–34. [Google Scholar] [CrossRef]
- Corbellini, M.; Canevar, M.; Mazza, L.; Ciaffi, M.; Lafiandra, D.; Borghi, B. Effect of the duration and intensity of heat shock during grain filling on dry matter and protein accumulation, technological quality and protein composition in bread and durum wheat. Funct. Plant Biol. 1997, 24, 245–260. [Google Scholar] [CrossRef]
- Kruszka, K.; Pacak, A.; Swida-Barteczka, A.; Nuc, P.; Alaba, S.; Wroblewska, Z.; Karlowski, W.; Jarmolowski, A.; Szweykowska-Kulinska, Z. Transcriptionally and post-transcriptionally regulated microRNAs in heat stress response in barley. J. Exp. Bot. 2014, 65, 6123–6135. [Google Scholar] [CrossRef] [Green Version]
- Rangan, P.; Furtado, A.; Henry, R. Transcriptome profiling of wheat genotypes under heat stress during grain-filling. J. Cereal Sci. 2020, 91, 102895. [Google Scholar] [CrossRef]
- Racette, K.; Zurweller, B.; Tillman, B.; Rowland, D. Transgenerational stress memory of water deficit in peanut production. Field Crops Res. 2020, 248, 107712. [Google Scholar] [CrossRef]
- Lämke, J.; Bäurle, I. Epigenetic and chromatin-based mechanisms in environmental stress adaptation and stress memory in plants. Genome Biol. 2017, 18, 124. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Able, A.J.; Able, J.A. Nitrogen starvation-responsive microRNAs are affected by transgenerational stress in durum wheat seedlings. Plants 2021, 10, 826. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Able, A.J.; Able, J.A. Small RNAs and their targets are associated with the transgenerational effects of water-deficit stress in durum wheat. Sci. Rep. 2021, 11, 1–17. [Google Scholar]
- Liu, H.; Able, A.J.; Able, J.A. Transgenerational effects of water-deficit and heat stress on germination and seedling vigour—new insights from durum wheat microRNAs. Plants 2020, 9, 189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wijewardana, C.; Reddy, K.R.; Krutz, L.J.; Gao, W.; Bellaloui, N. Drought stress has transgenerational effects on soybean seed germination and seedling vigor. PLoS ONE 2019, 14, e0214977. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Xin, C.; Cai, J.; Zhou, Q.; Dai, T.; Cao, W.; Jiang, D. Heat priming induces trans-generational tolerance to high temperature stress in wheat. Front. Plant Sci. 2016, 7, 501. [Google Scholar] [CrossRef] [Green Version]
- Crisp, P.A.; Ganguly, D.; Eichten, S.R.; Borevitz, J.O.; Pogson, B.J. Reconsidering plant memory: Intersections between stress recovery, RNA turnover, and epigenetics. Sci. Adv. 2016, 2, e1501340. [Google Scholar] [CrossRef] [Green Version]
- Molinier, J.; Ries, G.; Zipfel, C.; Hohn, B. Transgeneration memory of stress in plants. Nature 2006, 442, 1046–1049. [Google Scholar] [CrossRef]
- Fleta-Soriano, E.; Munné-Bosch, S. Stress memory and the inevitable effects of drought: A physiological perspective. Front. Plant Sci. 2016, 7, 143. [Google Scholar] [CrossRef] [Green Version]
- Bäurle, I.; Trindade, I. Chromatin regulation of somatic abiotic stress memory. J. Exp. Bot. 2020, 71, 5269–5279. [Google Scholar] [CrossRef] [Green Version]
- Hatzig, S.V.; Nuppenau, J.-N.; Snowdon, R.J.; Schießl, S.V. Drought stress has transgenerational effects on seeds and seedlings in winter oilseed rape (Brassica napus L.). BMC Plant Biol. 2018, 18, 297. [Google Scholar] [CrossRef]
- Nosalewicz, A.; Siecińska, J.; Śmiech, M.; Nosalewicz, M.; Wiącek, D.; Pecio, A.; Wach, D. Transgenerational effects of temporal drought stress on spring barley morphology and functioning. Environ. Exp. Bot. 2016, 131, 120–127. [Google Scholar] [CrossRef]
- Liu, H.; Able, A.J.; Able, J.A. SMARTER de-stressed cereal breeding. Trends Plant Sci. 2016, 21, 909–925. [Google Scholar] [CrossRef]
- Zhang, B.; Pan, X.; Cannon, C.H.; Cobb, G.P.; Anderson, T.A. Conservation and divergence of plant microRNA genes. Plant J. 2006, 46, 243–259. [Google Scholar] [CrossRef]
- Zhang, B.; Pan, X.; Cobb, G.P.; Anderson, T.A. Plant microRNA: A small regulatory molecule with big impact. Dev. Biol. 2006, 289, 3–16. [Google Scholar] [CrossRef]
- Alptekin, B.; Akpinar, B.A.; Budak, H. A comprehensive prescription for plant miRNA identification. Front. Plant Sci. 2017, 7, 2058. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Li, K.; Chen, L.; Zou, Y.; Liu, H.; Tian, Y.; Li, D.; Wang, R.; Zhao, F.; Ferguson, B.J.; et al. microRNA167-directed regulation of the auxin response factors, GmARF8a and GmARF8b, is required for soybean nodulation and lateral root development. Plant Physiol. 2015, 168, 984–999. [Google Scholar] [CrossRef] [Green Version]
- Budak, H.; Zhang, B. MicroRNAs in Model and Complex Organisms; Springer: Berlin/Heidelberg, Germany, 2017. [Google Scholar]
- Alptekin, B.; Langridge, P.; Budak, H. Abiotic stress miRNomes in the Triticeae. Funct. Integr. Genom. 2016, 17, 1–26. [Google Scholar] [CrossRef] [Green Version]
- Budak, H.; Akpinar, B.A. Plant miRNAs: Biogenesis, organization and origins. Funct. Integr. Genom. 2015, 15, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Brant, E.J.; Budak, H. Plant small non-coding RNAs and their roles in biotic stresses. Front. Plant Sci. 2018, 9, 1038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Budak, H.; Kantar, M.; Bulut, R.; Akpinar, B.A. Stress responsive miRNAs and isomiRs in cereals. Plant Sci. 2015, 235, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Akpinar, B.A.; Kantar, M.; Budak, H. Root precursors of microRNAs in wild emmer and modern wheats show major differences in response to drought stress. Funct. Integr. Genom. 2015, 15, 587–598. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Able, A.J.; Able, J.A. Water-deficit stress responsive microRNAs and their targets in four durum wheat genotypes. Funct. Integr. Genom. 2017, 17, 237–251. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Able, A.J.; Able, J.A. Genotypic water-deficit stress responses in durum wheat: Association between physiological traits, microRNA regulatory modules and yield components. Funct. Plant Biol. 2017, 44, 538–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Able, A.J.; Able, J.A. Multi-omics analysis of small RNA, transcriptome, and degradome in T. turgidum—regulatory networks of grain development and abiotic stress response. Int. J. Mol. 2020, 21, 7772. [Google Scholar] [CrossRef]
- Liu, H.; Able, A.J.; Able, J.A. Integrated analysis of small RNA, transcriptome, and degradome sequencing reveals the water-deficit and heat stress response network in durum wheat. Int. J. Mol. 2020, 21, 6017. [Google Scholar] [CrossRef]
- Liu, H.; Searle, I.R.; Watson-Haigh, N.S.; Baumann, U.; Mather, D.E.; Able, A.J.; Able, J.A. Genome-wide identification of microRNAs in leaves and the developing head of four durum genotypes during water deficit stress. PLoS ONE 2015, 10, e0142799. [Google Scholar]
- Zuluaga, D.L.; Sonnante, G. The use of nitrogen and its regulation in cereals: Structural genes, transcription factors, and the role of miRNAs. Plants 2019, 8, 294. [Google Scholar] [CrossRef] [Green Version]
- Zuluaga, D.L.; Liuzzi, V.; Curci, P.L.; Sonnante, G. MicroRNAs in durum wheat seedlings under chronic and short-term nitrogen stress. Funct. Integr. Genom. 2018, 18, 645–657. [Google Scholar] [CrossRef]
- Zuluaga, D.L.; De Paola, D.; Janni, M.; Curci, P.L.; Sonnante, G. Durum wheat miRNAs in response to nitrogen starvation at the grain filling stage. PLoS ONE 2017, 12, e0183253. [Google Scholar]
- De Paola, D.; Zuluaga, D.L.; Sonnante, G. The miRNAome of durum wheat: Isolation and characterisation of conserved and novel microRNAs and their target genes. BMC Genom. 2016, 17, 505. [Google Scholar] [CrossRef] [Green Version]
- Ravichandran, S.; Ragupathy, R.; Edwards, T.; Domaratzki, M.; Cloutier, S. MicroRNA-guided regulation of heat stress response in wheat. BMC Genom. 2019, 20, 488. [Google Scholar] [CrossRef] [Green Version]
- Budak, H.; Khan, Z.; Kantar, M. History and current status of wheat miRNAs using next-generation sequencing and their roles in development and stress. Brief. Funct. Genom. 2015, 14, 189–198. [Google Scholar] [CrossRef] [Green Version]
- Ebrahimi Khaksefidi, R.; Mirlohi, S.; Khalaji, F.; Fakhari, Z.; Shiran, B.; Fallahi, H.; Rafiei, F.; Budak, H.; Ebrahimie, E. Differential expression of seven conserved microRNAs in response to abiotic stress and their regulatory network in Helianthus annuus. Front. Plant Sci. 2015, 6, 741. [Google Scholar] [CrossRef] [Green Version]
- Bilichak, A.; Ilnytskyy, Y.; Wóycicki, R.; Kepeshchuk, N.; Fogen, D.; Kovalchuk, I. The elucidation of stress memory inheritance in Brassica rapa plants. Front. Plant Sci. 2015, 6, 5. [Google Scholar] [CrossRef] [Green Version]
- Stief, A.; Altmann, S.; Hoffmann, K.; Pant, B.D.; Scheible, W.-R.; Bäurle, I. Arabidopsis miR156 regulates tolerance to recurring environmental stress through SPL transcription factors. Plant Cell 2014, 26, 1792–1807. [Google Scholar] [CrossRef] [Green Version]
- Byeon, B.; Bilichak, A.; Kovalchuk, I. Transgenerational response to heat stress in the form of differential expression of noncoding RNA fragments in Brassica rapa plants. Plant Genome 2019, 12, 180022. [Google Scholar] [CrossRef] [Green Version]
- Tabassum, T.; Farooq, M.; Ahmad, R.; Zohaib, A.; Wahid, A. Seed priming and transgenerational drought memory improves tolerance against salt stress in bread wheat. Plant Physiol. Biochem. 2017, 118, 362–369. [Google Scholar] [CrossRef]
- Hossain, M.A.; Li, Z.-G.; Hoque, T.S.; Burritt, D.J.; Fujita, M.; Munné-Bosch, S. Heat or cold priming-induced cross-tolerance to abiotic stresses in plants: Key regulators and possible mechanisms. Protoplasma 2018, 255, 399–412. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Vignjevic, M.; Liu, F.; Jacobsen, S.; Jiang, D.; Wollenweber, B. Drought priming at vegetative growth stages improves tolerance to drought and heat stresses occurring during grain filling in spring wheat. Plant Growth Regul. 2015, 75, 677–687. [Google Scholar] [CrossRef]
- Chen, Y.; Fanourakis, D.; Tsaniklidis, G.; Aliniaeifard, S.; Yang, Q.; Li, T. Low UVA intensity during cultivation improves the lettuce shelf-life, an effect that is not sustained at higher intensity. Postharvest Biol. Technol. 2021, 172, 111376. [Google Scholar] [CrossRef]
- Wang, Q.-L.; Chen, J.-H.; He, N.-Y.; Guo, F.-Q. Metabolic reprogramming in chloroplasts under heat stress in plants. Int. J. Mol. 2018, 19, 849. [Google Scholar] [CrossRef] [Green Version]
- Feng, B.; Liu, P.; Li, G.; Dong, S.; Wang, F.; Kong, L.; Zhang, J. Effect of heat stress on the photosynthetic characteristics in flag leaves at the grain-filling stage of different heat-resistant winter wheat varieties. J. Agron. Crop Sci. 2014, 200, 143–155. [Google Scholar] [CrossRef]
- Ou, X.; Zhang, Y.; Xu, C.; Lin, X.; Zang, Q.; Zhuang, T.; Jiang, L.; Von Wettstein, D.; Liu, B. Transgenerational inheritance of modified DNA methylation patterns and enhanced tolerance induced by heavy metal stress in rice (Oryza sativa L.). PLoS ONE 2012, 7, e41143. [Google Scholar]
- Murgia, I.; Giacometti, S.; Balestrazzi, A.; Paparella, S.; Pagliano, C.; Morandini, P.A. Analysis of the transgenerational iron deficiency stress memory in Arabidopsis thaliana plants. Front. Plant Sci. 2015, 6, 745. [Google Scholar] [CrossRef] [Green Version]
- Hernández-Espinosa, N.; Mondal, S.; Autrique, E.; Gonzalez-Santoyo, H.; Crossa, J.; Huerta-Espino, J.; Singh, R.P.; Guzmán, C. Milling, processing and end-use quality traits of CIMMYT spring bread wheat germplasm under drought and heat stress. Field Crops Res. 2018, 215, 104–112. [Google Scholar] [CrossRef]
- Viswanathan, C.; Khanna-Chopra, R. Effect of heat stress on grain growth, starch synthesis and protein synthesis in grains of wheat (Triticum aestivum L.) varieties differing in grain weight stability. J. Agron. Crop Sci. 2001, 186, 1–7. [Google Scholar] [CrossRef]
- Wang, X.; Cai, J.; Liu, F.; Jin, M.; Yu, H.; Jiang, D.; Wollenweber, B.; Dai, T.; Cao, W. Pre-anthesis high temperature acclimation alleviates the negative effects of post-anthesis heat stress on stem stored carbohydrates remobilization and grain starch accumulation in wheat. J. Cereal Sci. 2012, 55, 331–336. [Google Scholar] [CrossRef]
- Lechner, E.; Achard, P.; Vansiri, A.; Potuschak, T.; Genschik, P. F-box proteins everywhere. Curr. Opin. Plant Biol. 2006, 9, 631–638. [Google Scholar] [CrossRef]
- Zhou, S.-M.; Kong, X.-Z.; Kang, H.-H.; Sun, X.-D.; Wang, W. The involvement of wheat F-box protein gene TaFBA1 in the oxidative stress tolerance of plants. PLoS ONE 2015, 10, e0122117. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Wang, W.; Wang, W.; Zhang, G.; Liu, Y.; Wang, Y.; Wang, W. Wheat F-box protein gene TaFBA1 is involved in plant tolerance to heat stress. Front. Plant Sci. 2018, 9, 521. [Google Scholar] [CrossRef] [Green Version]
- Cui, H.-R.; Zhang, Z.-R.; Xu, J.-N.; Wang, X.-Y. Genome-wide characterization and analysis of F-box protein-encoding genes in the Malus domestica genome. Mol. Genet. Genom. 2015, 290, 1435–1446. [Google Scholar] [CrossRef]
- Gill, S.S.; Tuteja, N. Reactive oxygen species and antioxidant machinery in abiotic stress tolerance in crop plants. Plant Physiol. Biochem. 2010, 48, 909–930. [Google Scholar] [CrossRef]
- Sun, A.-Z.; Guo, F.-Q. Chloroplast retrograde regulation of heat stress responses in plants. Front. Plant Sci. 2016, 7, 398. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.; Li, J.; Zhang, X.; Wei, H.; Cui, L. Effects of heat acclimation pretreatment on changes of membrane lipid peroxidation, antioxidant metabolites, and ultrastructure of chloroplasts in two cool-season turfgrass species under heat stress. Environ. Exp. Bot. 2006, 56, 274–285. [Google Scholar] [CrossRef]
- Sharma, D.K.; Andersen, S.B.; Ottosen, C.O.; Rosenqvist, E. Wheat cultivars selected for high Fv/Fm under heat stress maintain high photosynthesis, total chlorophyll, stomatal conductance, transpiration and dry matter. Physiol. Plant 2015, 153, 284–298. [Google Scholar] [CrossRef]
- RNAfold Web Server. Available online: http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi (accessed on 2 February 2021).








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Grain Weight (g) | Grain Number | Biomass (g) | Harvest Index | 1000-Grain Weight (g) | |
|---|---|---|---|---|---|
| AtCC | 12.82 ± 0.31 a | 330.38 ± 6.72 a | 25.92 ± 0.36 a | 0.496 ± 0.017 a | 38.98 ± 1.48 |
| AtCH | 10.36 ± 0.72 b | 273.43 ± 13.29 bc | 23.56 ± 0.52 b | 0.437 ± 0.021 b | 37.99 ± 2.00 |
| AtHC | 12.18 ± 0.27 a | 306.13 ± 5.54 ab | 25.73 ± 0.37 a | 0.473 ± 0.005 ab | 39.84 ± 0.85 |
| AtHH | 10.50 ± 0.49 b | 271.50 ± 18.50 b | 23.52 ± 0.71 b | 0.446 ± 0.013 b | 39.40 ± 2.01 |
| F pr. | 0.001 | 0.005 | 0.002 | 0.037 | 0.879 |
| LSD | 1.35 | 35.27 | 1.48 | 0.043 | n.a. |
| AuCC | 13.67 ± 0.23 b | 283.00 ± 6.34 b | 24.65 ± 0.23 b | 0.555 ± 0.008 ab | 48.35 ± 0.54 |
| AuCH | 11.23 ± 0.43 c | 250.50 ± 12.45 c | 21.16 ± 0.47 c | 0.530 ± 0.013 bc | 45.12 ± 1.11 |
| AuHC | 14.80 ± 0.18 a | 325.25 ± 8.03 a | 26.38 ± 0.30 a | 0.561 ± 0.002 a | 45.66 ± 1.03 |
| AuHH | 12.94 ± 0.43 b | 286.88 ± 15.19 b | 24.69 ± 0.54 b | 0.523 ± 0.008 c | 45.52 ± 1.30 |
| F pr. | <0.001 | <0.001 | <0.001 | 0.013 | 0.132 |
| LSD | 0.97 | 32.07 | 1.18 | 0.026 | n.a. |
| Grain Protein Content (GPC%) | Total Starch Content (TSC%) | Flour Yellowness (*b) | Plant Height (cm) | Tiller Number | Main Spike Length (cm) | |
|---|---|---|---|---|---|---|
| AtCC | 11.66 ± 0.31 b | 52.03 ± 6.72 c | 22.89 ± 0.23 | 70.40 ± 1.00 | 5.50 ± 0.27 | 8.79 ± 0.07 b |
| AtCH | 14.21 ± 1.06 a | 55.10 ± 13.29 bc | 22.99 ± 0.26 | 71.46 ± 1.32 | 5.14 ± 0.14 | 8.83 ± 0.14 b |
| AtHC | 11.72 ± 0.26 b | 61.18 ± 1.61 a | 23.09 ± 0.31 | 70.39 ± 1.23 | 5.50 ± 0.19 | 9.09 ± 0.10 ab |
| AtHH | 13.39 ± 0.33 ab | 58.65 ± 1.31 ab | 23.35 ± 0.24 | 73.30 ± 1.28 | 5.38 ± 0.32 | 9.35 ± 0.15 a |
| F pr. | 0.024 | 0.011 | 0.644 | 0.286 | 0.726 | 0.007 |
| LSD | 1.826 | 5.125 | n.a. | n.a. | n.a. | 0.34 |
| AuCC | 11.90 ± 0.23 b | 54.03 ± 1.32 ab | 21.24 ± 0.36 | 71.04 ± 1.56 | 5.50 ± 0.19 | 8.80 ± 0.13 |
| AuCH | 13.18 ± 0.58 a | 51.82 ± 1.16 b | 21.27 ± 0.30 | 74.21 ± 0.75 | 5.25 ± 0.37 | 8.93 ± 0.13 |
| AuHC | 11.82 ± 0.22 b | 56.13 ± 0.91 a | 21.93 ± 0.28 | 75.78 ± 1.02 | 5.63 ± 0.26 | 9.01 ± 0.17 |
| AuHH | 12.85 ± 0.28 ab | 56.41 ± 1.06 a | 21.99 ± 0.29 | 73.43 ± 1.33 | 6.00 ± 0.38 | 9.01 ± 0.10 |
| F pr. | 0.045 | 0.045 | 0.210 | 0.066 | 0.397 | 0.654 |
| LSD | 1.102 | 3.456 | n.a. | n.a. | n.a. | n.a. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, H.; Able, A.J.; Able, J.A. Small RNA, Transcriptome and Degradome Analysis of the Transgenerational Heat Stress Response Network in Durum Wheat. Int. J. Mol. Sci. 2021, 22, 5532. https://doi.org/10.3390/ijms22115532
Liu H, Able AJ, Able JA. Small RNA, Transcriptome and Degradome Analysis of the Transgenerational Heat Stress Response Network in Durum Wheat. International Journal of Molecular Sciences. 2021; 22(11):5532. https://doi.org/10.3390/ijms22115532
Chicago/Turabian StyleLiu, Haipei, Amanda J. Able, and Jason A. Able. 2021. "Small RNA, Transcriptome and Degradome Analysis of the Transgenerational Heat Stress Response Network in Durum Wheat" International Journal of Molecular Sciences 22, no. 11: 5532. https://doi.org/10.3390/ijms22115532