Macrophage-Secreted Lipocalin-2 Promotes Regeneration of Injured Primary Murine Renal Tubular Epithelial Cells

 and
and
Abstract
:1. Introduction
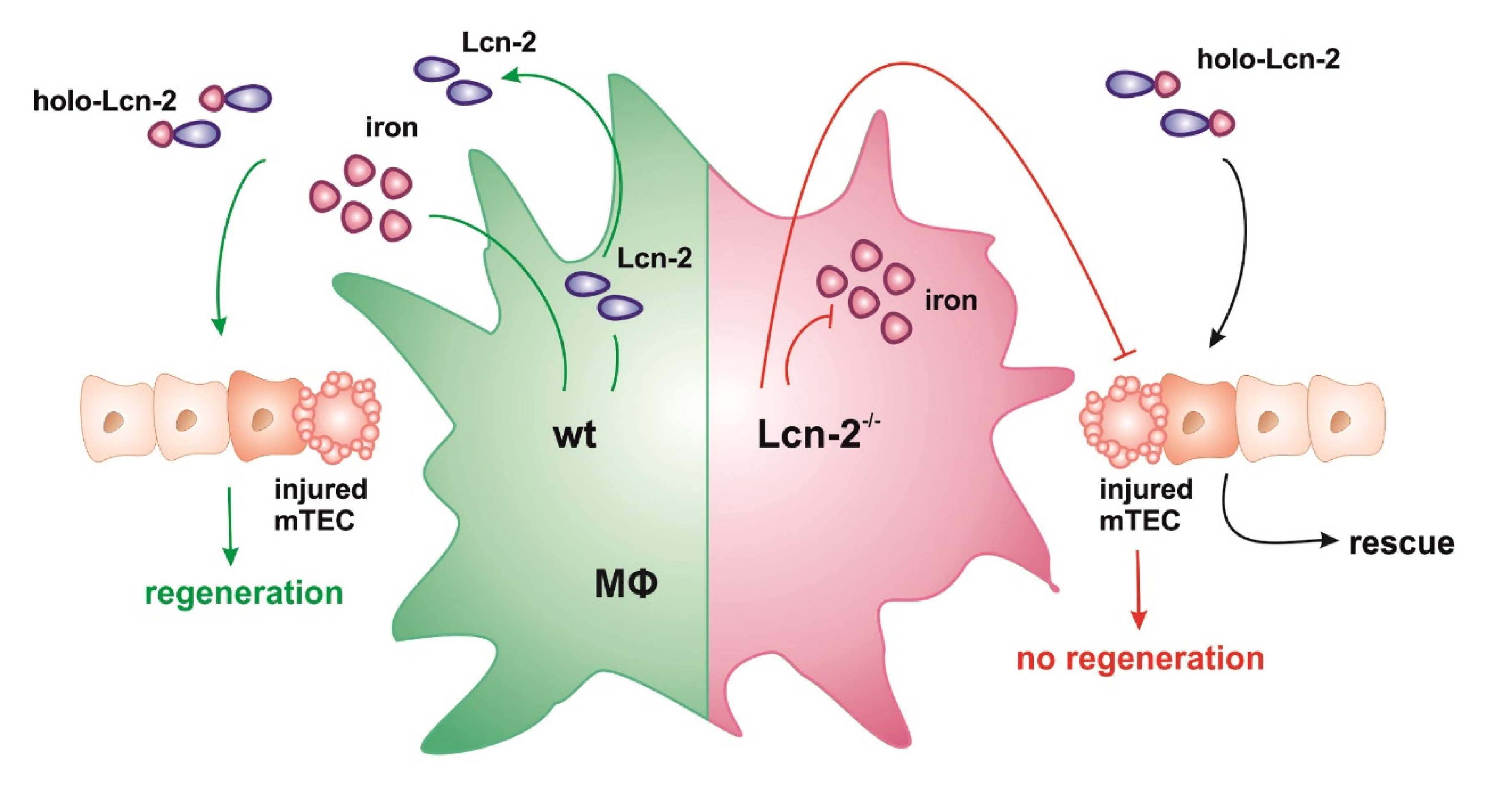
2. Results
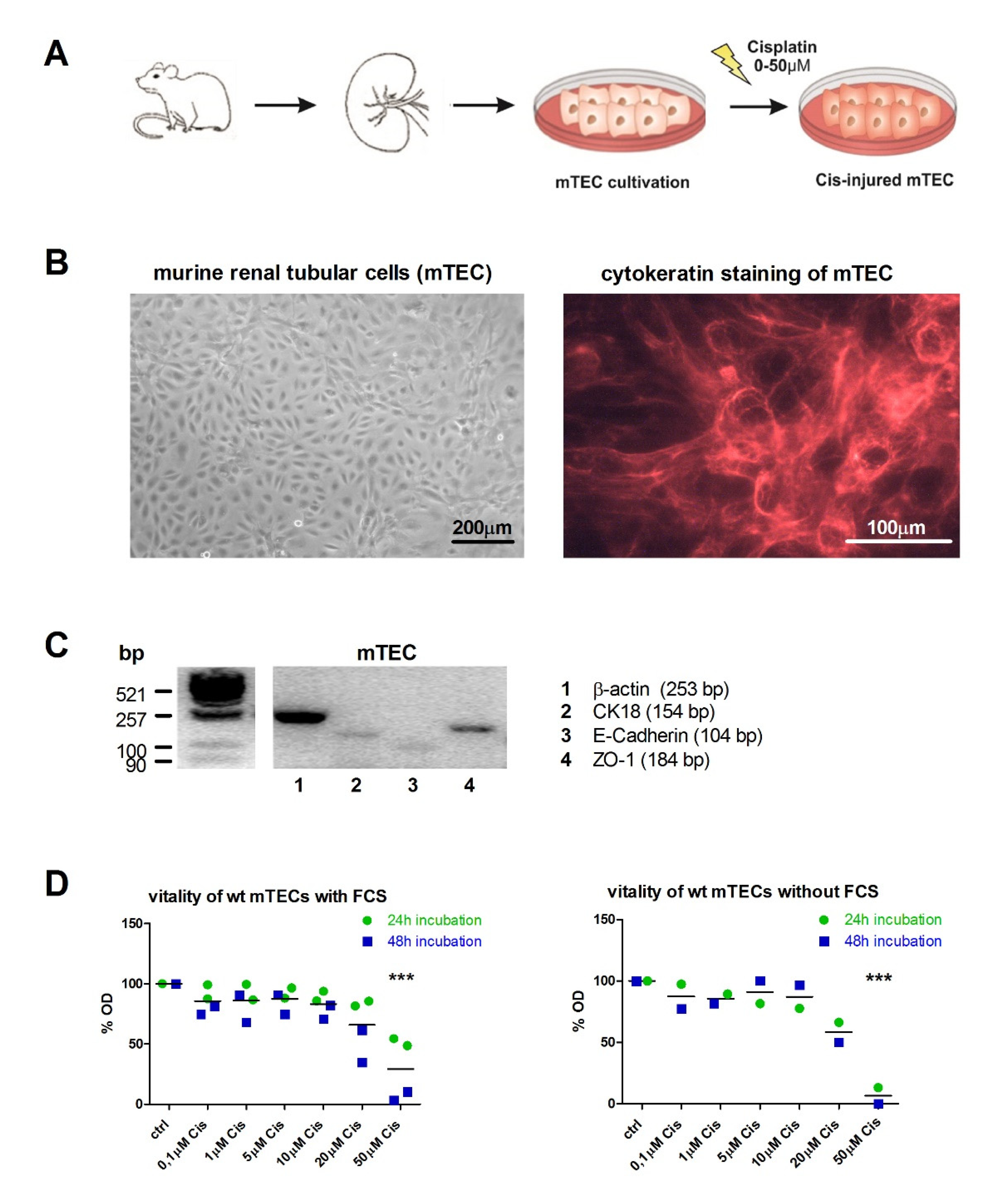
2.1. Dose-Dependent Injury of Primary Mouse Tubular Epithelial Cells (mTECs) upon Incubation with Cisplatin
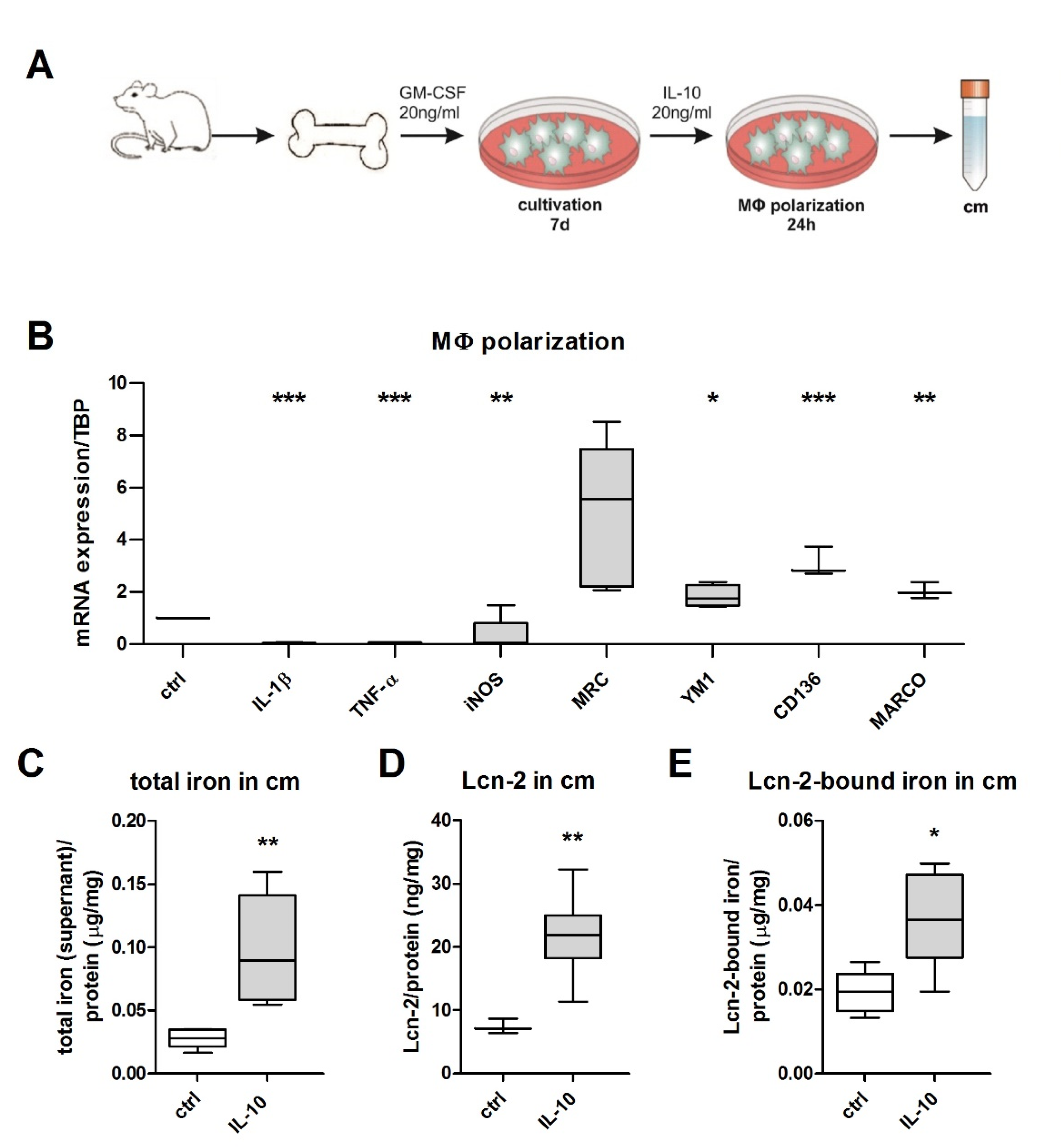
2.2. Establishment of Conditioned Media and Macrophage Polarization
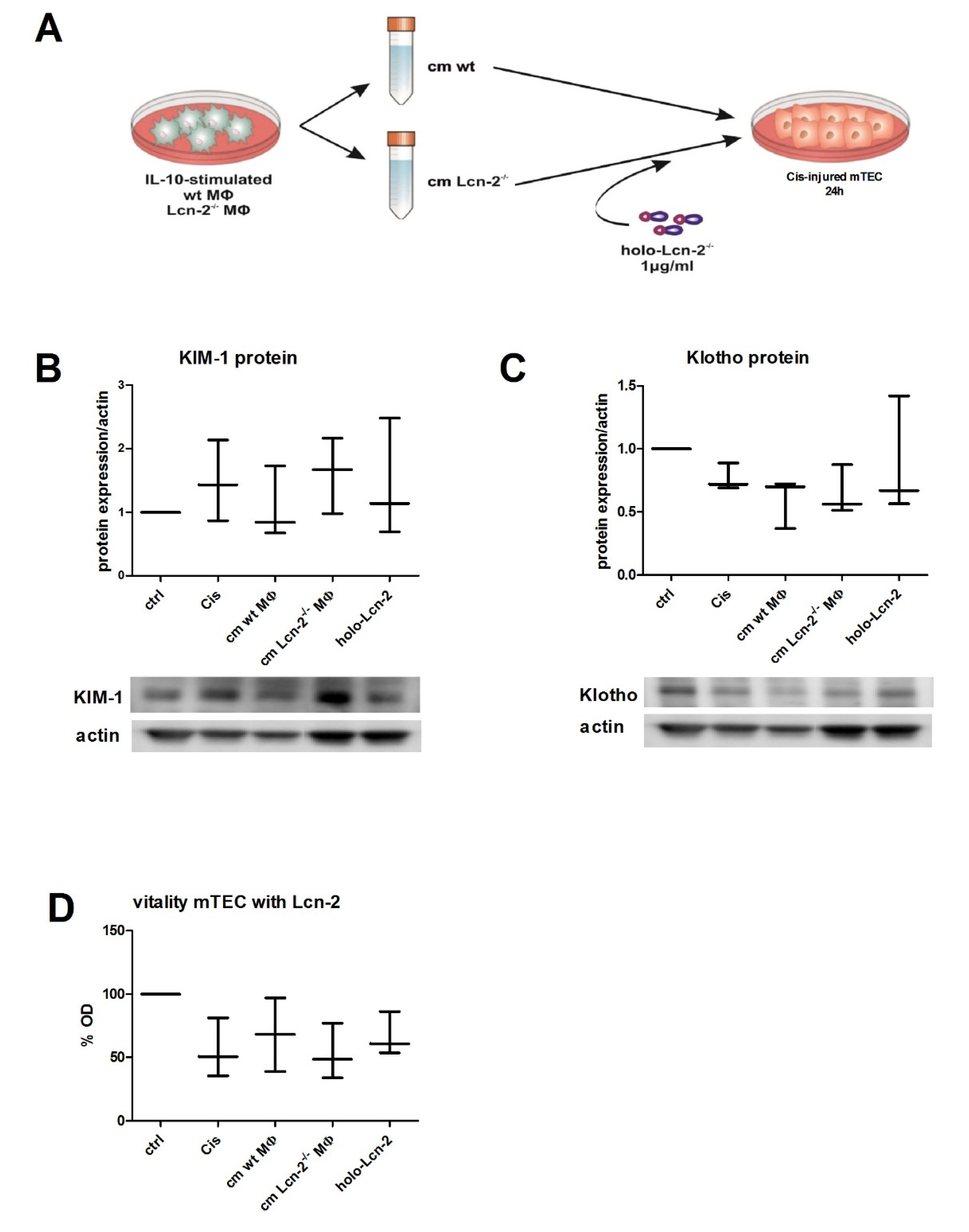
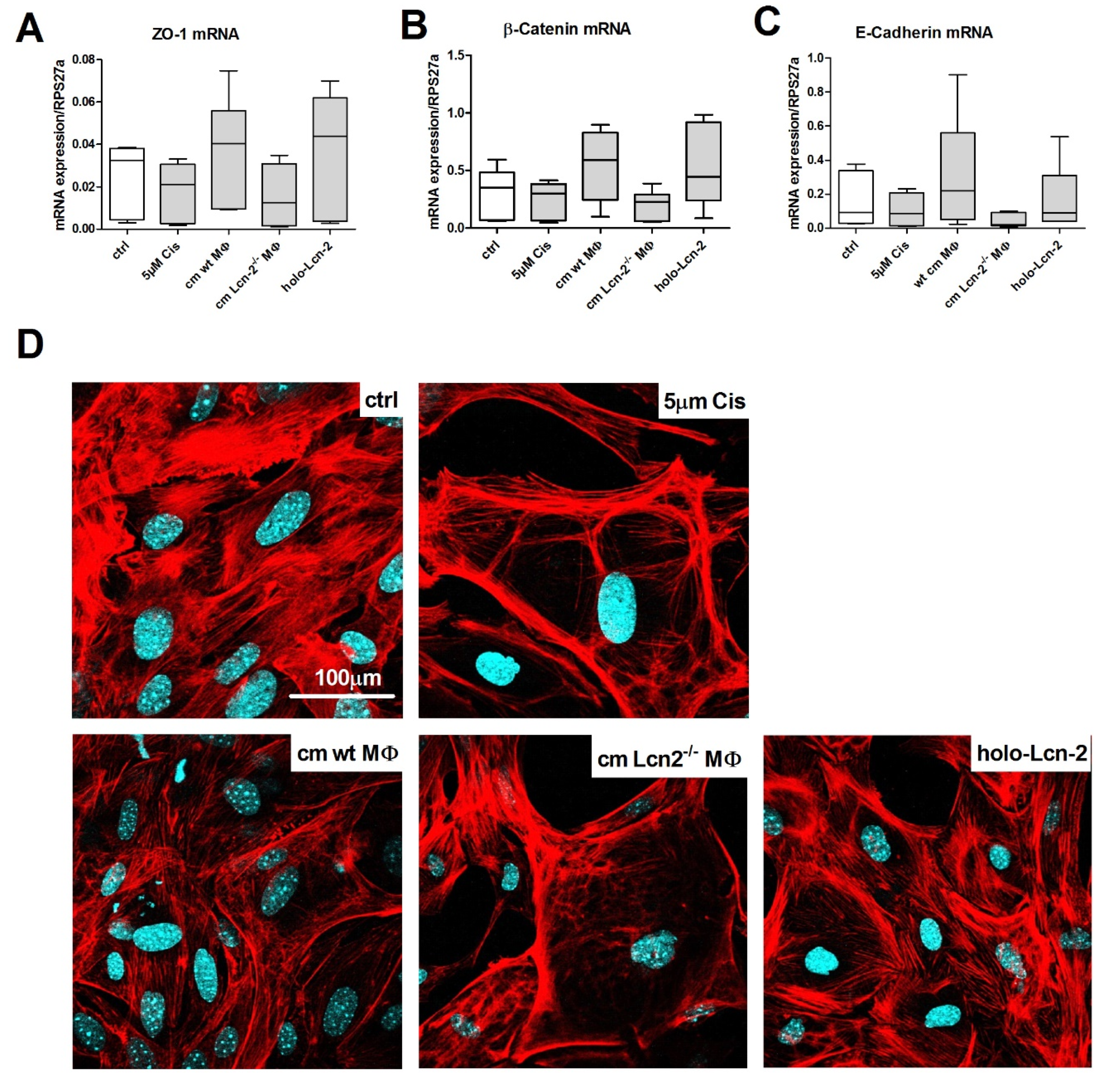
2.3. Conditioned Medium From wt MΦ or the Supply of Holo-Lcn-2 Tends to Promote Epithelial Viability upon Cisplatin Treatment
2.4. Lcn-2-Mediated Iron Uptake Promote Proliferation of Cisplatin-Injured mTECs
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Isolation and Culture of Murine Proximal Tubular Epithelial Cells
4.3. Establishment of Cisplatin Injury in mTECs Model
4.4. Generation of Murine BMDM and Generation of Conditioned Media
4.5. Generation of Recombinant Lcn-2
4.6. Lcn-2 Immunoprecipitation
4.7. Atomic Absorption Spectrometry
4.8. Establishment of Rescue Model Following Cisplatin Injury in mTECs
4.9. RNA Extraction and Quantitative Real-Time PCR (qPCR)
4.10. Western Blot
4.11. Lcn-2 ELISA
4.12. Phalloidin-Staining
4.13. Immunofluoresecence Cytokeratin Stain
4.14. xCELLigence Proliferation Assay
4.15. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AAS | Atomic absorption spectrometry |
| BMDM | Bone marrow-derived macrophages |
| Cis | Cisplatin |
| Cm | Conditioned medium |
| ctrl | Control |
| IL-10 | Interleukin 10 |
| IRI | Ischemia-reperfusion injury |
| KIM-1 | Kidney injury molecule 1 |
| Lcn-2 | Lipocalin-2 |
| mTEC | Murine tubular epithelial cells |
| MΦ | Macrophages |
| wt | Wildtype |
References
- Urbschat, A.; Obermuller, N.; Haferkamp, A. Biomarkers of kidney injury. Biomarkers 2011, 16, S22–S30. [Google Scholar] [CrossRef] [PubMed]
- Ronco, C.; Bellomo, R.; Kellum, J.A. Acute kidney injury. Lancet 2019, 394, 1949–1964. [Google Scholar] [CrossRef]
- Bonventre, J.V. Dedifferentiation and proliferation of surviving epithelial cells in acute renal failure. J. Am. Soc. Nephrol. 2003, 14, S55–S61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huen, S.C.; Cantley, L.G. Macrophages in Renal Injury and Repair. Annu. Rev. Physiol. 2017, 79, 449–469. [Google Scholar] [CrossRef] [PubMed]
- Ysebaert, D.K.; De Greef, K.E.; Vercauteren, S.R.; Ghielli, M.; Verpooten, G.A.; Eyskens, E.J.; De Broe, M.E. Identification and kinetics of leukocytes after severe ischaemia/reperfusion renal injury. Nephrol. Dial. Transpl. 2000, 15, 1562–1574. [Google Scholar] [CrossRef] [PubMed]
- Vinuesa, E.; Hotter, G.; Jung, M.; Herrero-Fresneda, I.; Torras, J.; Sola, A. Macrophage involvement in the kidney repair phase after ischaemia/reperfusion injury. J. Pathol. 2008, 214, 104–113. [Google Scholar] [CrossRef]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef]
- Labonte, A.C.; Tosello-Trampont, A.C.; Hahn, Y.S. The role of macrophage polarization in infectious and inflammatory diseases. Mol. Cells 2014, 37, 275–285. [Google Scholar] [CrossRef] [Green Version]
- Jung, M.; Brune, B.; Hotter, G.; Sola, A. Macrophage-derived Lipocalin-2 contributes to ischemic resistance mechanisms by protecting from renal injury. Sci. Rep. 2016, 6, 21950. [Google Scholar] [CrossRef]
- Jung, M.; Sola, A.; Hughes, J.; Kluth, D.C.; Vinuesa, E.; Vinas, J.L.; Perez-Ladaga, A.; Hotter, G. Infusion of IL-10-expressing cells protects against renal ischemia through induction of lipocalin-2. Kidney Int. 2012, 81, 969–982. [Google Scholar] [CrossRef]
- Sola, A.; Weigert, A.; Jung, M.; Vinuesa, E.; Brecht, K.; Weis, N.; Brune, B.; Borregaard, N.; Hotter, G. Sphingosine-1-phosphate signalling induces the production of Lcn-2 by macrophages to promote kidney regeneration. J. Pathol. 2011, 225, 597–608. [Google Scholar] [CrossRef] [PubMed]
- Recalcati, S.; Locati, M.; Cairo, G. Systemic and cellular consequences of macrophage control of iron metabolism. Semin. Immunol. 2012, 24, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Haase, M.; Devarajan, P.; Haase-Fielitz, A.; Bellomo, R.; Cruz, D.N.; Wagener, G.; Krawczeski, C.D.; Koyner, J.L.; Murray, P.; Zappitelli, M.; et al. The outcome of neutrophil gelatinase-associated lipocalin-positive subclinical acute kidney injury: A multicenter pooled analysis of prospective studies. J. Am. Coll. Cardiol. 2011, 57, 1752–1761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paragas, N.; Qiu, A.; Zhang, Q.; Samstein, B.; Deng, S.X.; Schmidt-Ott, K.M.; Viltard, M.; Yu, W.; Forster, C.S.; Gong, G.; et al. The Ngal reporter mouse detects the response of the kidney to injury in real time. Nat. Med. 2011, 17, 216–222. [Google Scholar] [CrossRef] [Green Version]
- Mishra, J.; Ma, Q.; Prada, A.; Mitsnefes, M.; Zahedi, K.; Yang, J.; Barasch, J.; Devarajan, P. Identification of neutrophil gelatinase-associated lipocalin as a novel early urinary biomarker for ischemic renal injury. J. Am. Soc. Nephrol. 2003, 14, 2534–2543. [Google Scholar] [CrossRef] [Green Version]
- Mishra, J.; Mori, K.; Ma, Q.; Kelly, C.; Barasch, J.; Devarajan, P. Neutrophil gelatinase-associated lipocalin: A novel early urinary biomarker for cisplatin nephrotoxicity. Am. J. Nephrol. 2004, 24, 307–315. [Google Scholar] [CrossRef]
- Mishra, J.; Mori, K.; Ma, Q.; Kelly, C.; Yang, J.; Mitsnefes, M.; Barasch, J.; Devarajan, P. Amelioration of ischemic acute renal injury by neutrophil gelatinase-associated lipocalin. J. Am. Soc. Nephrol. 2004, 15, 3073–3082. [Google Scholar] [CrossRef]
- Mori, K.; Lee, H.T.; Rapoport, D.; Drexler, I.R.; Foster, K.; Yang, J.; Schmidt-Ott, K.M.; Chen, X.; Li, J.Y.; Weiss, S.; et al. Endocytic delivery of lipocalin-siderophore-iron complex rescues the kidney from ischemia-reperfusion injury. J. Clin. Investig. 2005, 115, 610–621. [Google Scholar] [CrossRef]
- Gwira, J.A.; Wei, F.; Ishibe, S.; Ueland, J.M.; Barasch, J.; Cantley, L.G. Expression of neutrophil gelatinase-associated lipocalin regulates epithelial morphogenesis in vitro. J. Biol. Chem. 2005, 280, 7875–7882. [Google Scholar] [CrossRef] [Green Version]
- Jung, M.; Oren, B.; Mora, J.; Mertens, C.; Dziumbla, S.; Popp, R.; Weigert, A.; Grossmann, N.; Fleming, I.; Brune, B. Lipocalin 2 from macrophages stimulated by tumor cell-derived sphingosine 1-phosphate promotes lymphangiogenesis and tumor metastasis. Sci. Signal. 2016, 9, ra64. [Google Scholar] [CrossRef]
- Oren, B.; Urosevic, J.; Mertens, C.; Mora, J.; Guiu, M.; Gomis, R.R.; Weigert, A.; Schmid, T.; Grein, S.; Brune, B.; et al. Tumour stroma-derived lipocalin-2 promotes breast cancer metastasis. J. Pathol. 2016, 239, 274–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinuesa, E.; Sola, A.; Jung, M.; Alfaro, V.; Hotter, G. Lipocalin-2-induced renal regeneration depends on cytokines. Am. J. Physiol. Ren. Physiol. 2008, 295, F1554–F1562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devireddy, L.R.; Gazin, C.; Zhu, X.; Green, M.R. A cell-surface receptor for lipocalin 24p3 selectively mediates apoptosis and iron uptake. Cell 2005, 123, 1293–1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paller, M.S.; Hedlund, B.E. Role of iron in postischemic renal injury in the rat. Kidney Int. 1988, 34, 474–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paller, M.S.; Hedlund, B.E. Extracellular iron chelators protect kidney cells from hypoxia/reoxygenation. Free Radic. Biol. Med. 1994, 17, 597–603. [Google Scholar] [CrossRef]
- Lieberthal, W.; Triaca, V.; Levine, J. Mechanisms of death induced by cisplatin in proximal tubular epithelial cells: Apoptosis vs. necrosis. Am. J. Physiol. 1996, 270, F700–F708. [Google Scholar] [CrossRef]
- Borch, R.F.; Pleasants, M.E. Inhibition of cis-platinum nephrotoxicity by diethyldithiocarbamate rescue in a rat model. Proc. Natl. Acad. Sci. USA 1979, 76, 6611–6614. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Kato, A.; Yasuda, H.; Miyaji, T.; Fujigaki, Y.; Yamamoto, T.; Yonemura, K.; Hishida, A. The induction of cell cycle regulatory and DNA repair proteins in cisplatin-induced acute renal failure. Toxicol. Appl. Pharm. 2004, 200, 111–120. [Google Scholar] [CrossRef] [Green Version]
- Kashiwagi, E.; Tonomura, Y.; Kondo, C.; Masuno, K.; Fujisawa, K.; Tsuchiya, N.; Matsushima, S.; Torii, M.; Takasu, N.; Izawa, T.; et al. Involvement of neutrophil gelatinase-associated lipocalin and osteopontin in renal tubular regeneration and interstitial fibrosis after cisplatin-induced renal failure. Exp. Toxicol. Pathol. 2014, 66, 301–311. [Google Scholar] [CrossRef]
- Molitoris, B.A.; Marrs, J. The role of cell adhesion molecules in ischemic acute renal failure. Am. J. Med. 1999, 106, 583–592. [Google Scholar] [CrossRef]
- Bonventre, J.V.; Kelly, K.J. Adhesion molecules and acute renal failure. Adv. Nephrol. Necker Hosp. 1996, 25, 159–176. [Google Scholar] [PubMed]
- Kruidering, M.; van de Water, B.; Zhan, Y.; Baelde, J.J.; Heer, E.; Mulder, G.J.; Stevens, J.L.; Nagelkerke, J.F. Cisplatin effects on F-actin and matrix proteins precede renal tubular cell detachment and apoptosis in vitro. Cell Death Differ. 1998, 5, 601–614. [Google Scholar] [CrossRef] [PubMed]
- Jung, M.; Hotter, G.; Vinas, J.L.; Sola, A. Cisplatin upregulates mitochondrial nitric oxide synthase and peroxynitrite formation to promote renal injury. Toxicol. Appl. Pharm. 2009, 234, 236–246. [Google Scholar] [CrossRef] [PubMed]
- Curmi, P.A.; Gavet, O.; Charbaut, E.; Ozon, S.; Lachkar-Colmerauer, S.; Manceau, V.; Siavoshian, S.; Maucuer, A.; Sobel, A. Stathmin and its phosphoprotein family: General properties, biochemical and functional interaction with tubulin. Cell Struct. Funct. 1999, 24, 345–357. [Google Scholar] [CrossRef] [Green Version]
- Peschanski, M.; Hirsch, E.; Dusart, I.; Doye, V.; Marty, S.; Manceau, V.; Sobel, A. Stathmin: Cellular localization of a major phosphoprotein in the adult rat and human CNS. J. Comp. Neurol. 1993, 337, 655–668. [Google Scholar] [CrossRef]
- Mertens, C.; Akam, E.A.; Rehwald, C.; Brune, B.; Tomat, E.; Jung, M. Intracellular Iron Chelation Modulates the Macrophage Iron Phenotype with Consequences on Tumor Progression. PLoS ONE 2016, 11, e0166164. [Google Scholar] [CrossRef] [Green Version]
- Recalcati, S.; Locati, M.; Marini, A.; Santambrogio, P.; Zaninotto, F.; De Pizzol, M.; Zammataro, L.; Girelli, D.; Cairo, G. Differential regulation of iron homeostasis during human macrophage polarized activation. Eur. J. Immunol. 2010, 40, 824–835. [Google Scholar] [CrossRef]
- Rehwald, C.; Schnetz, M.; Urbschat, A.; Mertens, C.; Meier, J.K.; Bauer, R.; Baer, P.; Winslow, S.; Roos, F.C.; Zwicker, K.; et al. The iron load of lipocalin-2 (LCN-2) defines its pro-tumour function in clear-cell renal cell carcinoma. Br. J. Cancer 2019, 122, 421–433. [Google Scholar] [CrossRef]
- Mertens, C.; Mora, J.; Oren, B.; Grein, S.; Winslow, S.; Scholich, K.; Weigert, A.; Malmstrom, P.; Forsare, C.; Ferno, M.; et al. Macrophage-derived lipocalin-2 transports iron in the tumor microenvironment. Oncoimmunology 2018, 7, e1408751. [Google Scholar] [CrossRef] [Green Version]
- Baer, P.C.; Nockher, W.A.; Haase, W.; Scherberich, J.E. Isolation of proximal and distal tubule cells from human kidney by immunomagnetic separation. Technical note. Kidney Int. 1997, 52, 1321–1331. [Google Scholar] [CrossRef] [Green Version]
- Dekel, B.; Zangi, L.; Shezen, E.; Reich-Zeliger, S.; Eventov-Friedman, S.; Katchman, H.; Jacob-Hirsch, J.; Amariglio, N.; Rechavi, G.; Margalit, R.; et al. Isolation and characterization of nontubular sca-1+lin- multipotent stem/progenitor cells from adult mouse kidney. J. Am. Soc. Nephrol. 2006, 17, 3300–3314. [Google Scholar] [CrossRef] [Green Version]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharm. 2014, 740, 364–378. [Google Scholar] [CrossRef] [Green Version]
- Volarevic, V.; Djokovic, B.; Jankovic, M.G.; Harrell, C.R.; Fellabaum, C.; Djonov, V.; Arsenijevic, N. Molecular mechanisms of cisplatin-induced nephrotoxicity: A balance on the knife edge between renoprotection and tumor toxicity. J. Biomed. Sci. 2019, 26, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zahedi, K.; Wang, Z.; Barone, S.; Tehrani, K.; Yokota, N.; Petrovic, S.; Rabb, H.; Soleimani, M. Identification of stathmin as a novel marker of cell proliferation in the recovery phase of acute ischemic renal failure. Am. J. Physiol. Cell Physiol. 2004, 286, C1203–C1211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miura, K.; Goldstein, R.S.; Pasino, D.A.; Hook, J.B. Cisplatin nephrotoxicity: Role of filtration and tubular transport of cisplatin in isolated perfused kidneys. Toxicology 1987, 44, 147–158. [Google Scholar] [CrossRef]
- Witzgall, R.; Brown, D.; Schwarz, C.; Bonventre, J.V. Localization of proliferating cell nuclear antigen, vimentin, c-Fos, and clusterin in the postischemic kidney. Evidence for a heterogenous genetic response among nephron segments, and a large pool of mitotically active and dedifferentiated cells. J. Clin. Investig. 1994, 93, 2175–2188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caracausi, M.; Piovesan, A.; Antonaros, F.; Strippoli, P.; Vitale, L.; Pelleri, M.C. Systematic identification of human housekeeping genes possibly useful as references in gene expression studies. Mol. Med. Rep. 2017, 16, 2397–2410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, M.; Weigert, A.; Tausendschon, M.; Mora, J.; Oren, B.; Sola, A.; Hotter, G.; Muta, T.; Brune, B. Interleukin-10-induced neutrophil gelatinase-associated lipocalin production in macrophages with consequences for tumor growth. Mol. Cell Biol. 2012, 32, 3938–3948. [Google Scholar] [CrossRef] [Green Version]
- Tanase, D.M.; Gosav, E.M.; Radu, S.; Costea, C.F.; Ciocoiu, M.; Carauleanu, A.; Lacatusu, C.M.; Maranduca, M.A.; Floria, M.; Rezus, C. The Predictive Role of the Biomarker Kidney Molecule-1 (KIM-1) in Acute Kidney Injury (AKI) Cisplatin-Induced Nephrotoxicity. Int. J. Mol. Sci. 2019, 20, 5238. [Google Scholar] [CrossRef] [Green Version]
- Mitobe, M.; Yoshida, T.; Sugiura, H.; Shirota, S.; Tsuchiya, K.; Nihei, H. Oxidative stress decreases klotho expression in a mouse kidney cell line. Nephron Exp. Nephrol. 2005, 101, e67–e74. [Google Scholar] [CrossRef]
- Sugiura, H.; Yoshida, T.; Tsuchiya, K.; Mitobe, M.; Nishimura, S.; Shirota, S.; Akiba, T.; Nihei, H. Klotho reduces apoptosis in experimental ischaemic acute renal failure. Nephrol. Dial. Transpl. 2005, 20, 2636–2645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, M.C.; Shi, M.; Zhang, J.; Quinones, H.; Kuro-o, M.; Moe, O.W. Klotho deficiency is an early biomarker of renal ischemia-reperfusion injury and its replacement is protective. Kidney Int. 2010, 78, 1240–1251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duffield, J.S. Macrophages in kidney repair and regeneration. J. Am. Soc. Nephrol. 2011, 22, 199–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Sequence |
|---|---|
| β-actin | Forward: 5-CCACCATGTACCCAGGCATT-3 Reverse: 5-AGGGTGTAAAACGCAGCTCA-3 |
| β-Catenin | Forward: 5-TCTGAGGACAAGCCACAGGATTACA-3 Reverse: 5-GGGCACCAATGTCCAGTCCAA-3 |
| CK18 | Forward: 5-TTGTCACCACCAAGTCTGCC-3 Reverse: 5-TTGTATCGGGCCTCCACATC-3 |
| E-Cadherin (real-time) | Forward: 5-TGAAGAAGGAAGAAGA-3 Reverse: 5-TGGGAGCCACTTTCGA-3 |
| E-Cadherin (qRT) | Forward: CAACGATCCTGACCAGCAGT Reverse: TGTATTGCTGCTTGGCCTCA |
| IL-1β | Forward: 5-AGGCCACAGGTATTTTGTCG-3 Reverse: 5-GACCTTCCAGGATGAGGACA-3 |
| iNOS | Forward: 5-ACCCTAAGAGTCACAAAATGGC-3 Reverse: 5-TTGATCCTCACATACTGTGGACG-3 |
| Lcn-2 | qHsaCED0045408 |
| MRC | Forward: 5-GGAGTGATGGAACCCCAGTG-3 Reverse: 5-CTGTCCGCCCAGTATCCATC-3 |
| PCNA | Forward: 5-AATGGGGTGAAGTTTTCTGC-3 Reverse: 5-CAGTGGAGTGGCTTTTGTGA-3 |
| RPS27a | Forward: 5-GACCCTTACGGGGAAAACCAT-3 Reverse: 5-AGACAAAGTCCGGCCATCTTC-3 |
| Stathmin | Forward: 5-CTTGCGAGAGAAGGACAAGC-3 Reverse: 5-CGGTCCTACATCGGCTTCTA-3 |
| TBP | Forward: 5-GGGCCGCCGGTTAACT-3 Reverse: 5-AGCCCTGAGCGTGGCA-3 |
| TNFα | Forward: 5-CCATTCCTGAGTTCTGCAAAGG-3 Reverse: 5-AGGTAGGAAGGCCTGAGATCTTATC-3 |
| YM-1 | Forward: 5-GGGCATACCTTTATCCTGAG-3 Reverse: 5-CCACTGAAGTCATCCATGTC-3 |
| ZO-1 (real-time) | Forward: 5-GCCATTACACGGTCCTCTGA-3 Reverse: 5-GCGAAAGGTAAGGGACTGG-3 |
| ZO-1 (qPCR) | Forward: GCCATTACACGGTCCTCTGA Reverse: GCGAAAGGTAAGGGACTGGA |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Urbschat, A.; Thiemens, A.-K.; Mertens, C.; Rehwald, C.; Meier, J.K.; Baer, P.C.; Jung, M. Macrophage-Secreted Lipocalin-2 Promotes Regeneration of Injured Primary Murine Renal Tubular Epithelial Cells. Int. J. Mol. Sci. 2020, 21, 2038. https://doi.org/10.3390/ijms21062038
Urbschat A, Thiemens A-K, Mertens C, Rehwald C, Meier JK, Baer PC, Jung M. Macrophage-Secreted Lipocalin-2 Promotes Regeneration of Injured Primary Murine Renal Tubular Epithelial Cells. International Journal of Molecular Sciences. 2020; 21(6):2038. https://doi.org/10.3390/ijms21062038
Chicago/Turabian StyleUrbschat, Anja, Anne-Kathrin Thiemens, Christina Mertens, Claudia Rehwald, Julia K. Meier, Patrick C. Baer, and Michaela Jung. 2020. "Macrophage-Secreted Lipocalin-2 Promotes Regeneration of Injured Primary Murine Renal Tubular Epithelial Cells" International Journal of Molecular Sciences 21, no. 6: 2038. https://doi.org/10.3390/ijms21062038