Application of Thin-Layer Chromatography-Flame Ionization Detection (TLC-FID) to Total Lipid Quantitation in Mycolic-Acid Synthesizing Rhodococcus and Williamsia Species

 , ,
, ,
Abstract
: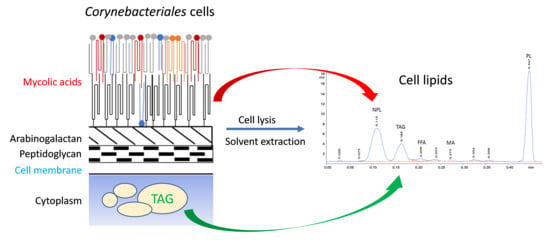
1. Introduction
2. Results
2.1. Separation of Lipid Standards and Bacterial Extracts Using Plate TLC
2.2. Separation of Standards and Lipids in Bacterial Extracts by TLC-FID
2.3. Identification of Lipids in the Non-Polar Lipid Peak
2.4. Comparison of the Efficiency of Bead Beating with Sonication and Quantification of Bacterial Lipid Classes
3. Discussion
4. Materials and Methods
4.1. Bacterial Strains and Media
4.2. Cell Lysis by Bead Beating and Sonication
4.3. Separation of Lipid Classes Using Plate TLC
4.4. Separation of Lipid Classes Using TLC-FID
4.5. GC-MS Analysis to Identify Non-polar Lipid Peaks in Bacterial Extracts
4.6. Identification of Non-Polar Lipids Using UPLC-MS/MS and NMR
4.7. Preparation of the TLC-FID Calibration Curve to Quantify Lipid Classes
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fahy, E.; Cotter, D.; Sud, M.; Subramaniam, S. Lipid classification, structures and tools. Biochim. Biophys. Acta 2011, 1811, 637–647. [Google Scholar] [CrossRef] [PubMed]
- Cronan, J.E., Jr.; Gelmann, E.P. Physical properties of membrane lipids: Biological relevance and regulation. Bacteriol. Rev. 1975, 39, 232–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.-M.; Rock, C.O. Membrane lipid homeostasis in bacteria. Nat. Rev. Microbiol. 2008, 6, 222–233. [Google Scholar] [CrossRef] [PubMed]
- Minnikin, D. Chemical principles in the organization of lipid components in the mycobacterial cell envelope. Res. Microbiol. 1991, 142, 423–427. [Google Scholar] [CrossRef]
- Minnikin, D. Complex lipids: Their chemistry, biosynthesis and roles. In The Biology of Mycobacteria; Ratledge, C., Stanford, J., Eds.; Academic: London, UK, 1982; pp. 94–184. [Google Scholar]
- Li, Q.; Du, W.; Liu, D. Perspectives of microbial oils for biodiesel production. Appl. Microbiol. Biotechnol. 2008, 80, 749–756. [Google Scholar] [CrossRef]
- Alvarez, H.; Steinbüchel, A. Triacylglycerols in prokaryotic microorganisms. Appl. Microbiol. Biotechnol. 2002, 60, 367–376. [Google Scholar] [CrossRef]
- Castro, A.R.; Rocha, I.; Alves, M.M.; Pereira, M.A. Rhodococcus opacus B4: A promising bacterium for production of biofuels and biobased chemicals. AMB Express 2016, 6, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Kurosawa, K.; Boccazzi, P.; de Almeida, N.M.; Sinskey, A.J. High-cell-density batch fermentation of Rhodococcus opacus PD630 using a high glucose concentration for triacylglycerol production. J. Biotechnol. 2010, 147, 212–218. [Google Scholar] [CrossRef]
- Kurosawa, K.; Radek, A.; Plassmeier, J.K.; Sinskey, A.J. Improved glycerol utilization by a triacylglycerol-producing Rhodococcus opacus strain for renewable fuels. Biotechnol. Biofuels 2015, 8, 31. [Google Scholar] [CrossRef] [Green Version]
- Wältermann, M.; Luftmann, H.; Baumeister, D.; Kalscheuer, R.; Steinbüchel, A. Rhodococcus opacus strain PD630 as a new source of high-value single-cell oil? Isolation and characterization of triacylglycerols and other storage lipids. Microbiology 2000, 146, 1143–1149. [Google Scholar] [CrossRef] [Green Version]
- Abel, K.; Peterson, J. Classification of microorganisms by analysis of chemical composition I. Feasibility of utilizing gas chromatography. J. Bacteriol. 1963, 85, 1039–1044. [Google Scholar] [CrossRef] [Green Version]
- Shaw, N. Lipid composition as a guide to the classification of bacteria. Adv. Appl. Microbiol. 1974, 17, 63–108. [Google Scholar] [PubMed]
- Watson, A. Lipidomics: A global approach to lipid analysis in biological systems. J. Lipid Res. 2006, 47, 2101–2111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Han, J.; Wang, Z.; Liu, J.A.; Wei, J.; Xiong, S.; Zhao, Z. Mass spectrometry methodology in lipid analysis. Int. J. Mol. Sci. 2014, 15, 10492–10507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gidden, J.; Denson, J.; Liyanage, R.; Ivey, D.M.; Lay, J.O. Lipid compositions in Escherichia coli and Bacillus subtilis during growth as determined by MALDI-TOF and TOF/TOF mass spectrometry. Int. J. Mass Spectrom. 2009, 283, 178–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, R.C.; Mathews, W.R.; Rokach, J.; Fenselau, C. Comparison of biological-derived and synthetic leukotriene C4 by fast atom bombardment mass spectrometry. Prostaglandins 1982, 23, 201–206. [Google Scholar] [CrossRef]
- Whitehouse, C.M.; Dreyer, R.; Yamashita, M.; Fenn, J. Electrospray ionization for mass-spectrometry of large biomolecules. Science 1989, 246, 64–71. [Google Scholar] [CrossRef]
- Karas, M.; Hillenkamp, F. Laser desorption ionization of proteins with molecular masses exceeding 10,000 daltons. Anal. Chem. 1988, 60, 2299–2301. [Google Scholar] [CrossRef]
- Jouhet, J.; Lupette, J.; Clerc, O.; Magneschi, L.; Bedhomme, M.; Collin, S.; Roy, S.; Maréchal, E.; Rébeillé, F. LC-MS/MS versus TLC plus GC methods: Consistency of glycerolipid and fatty acid profiles in microalgae and higher plant cells and effect of a nitrogen starvation. PLoS ONE 2017, 12, e0182423. [Google Scholar] [CrossRef]
- Goren, M.; Brennan, P. Mycobacterial lipids: Chemistry and biologic activities. In Tuberculosis; Youmans, G., Ed.; W.B. Saunders: Philadelphia, PA, USA, 1979; pp. 63–193. [Google Scholar]
- Goodfellow, M. The family Nocardiacae. In The Prokaryotes; Rosenberg, E., DeLong, E.F., Lory, S., Stackebrandt, E., Thompson, F., Eds.; Springer: Berlin, Germany, 2014; pp. 595–650. [Google Scholar] [CrossRef]
- Brennan, P.J.; Nikaido, H. The envelope of mycobacteria. Annu. Rev. Biochem. 1995, 64, 29–63. [Google Scholar] [CrossRef]
- Marrakchi, H.; Laneelle, M.A.; Daffe, M. Mycolic acids: Structures, biosynthesis, and beyond. Chem. Biol. 2014, 21, 67–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minnikin, D.; Alshamaony, L.; Goodfellow, M. Differentiation of Mycobacterium, Nocardia, and related taxa by thin-layer chromatographic analysis of whole-organism methanolysates. Microbiology 1975, 88, 200–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaneda, K.; Naito, S.; Imaizumi, S.; Yano, I.; Mizuno, S.; Tomiyasu, I.; Baba, T.; Kusunose, E.; Kusunose, M. Determination of molecular species composition of C80 or longer-chain alpha-mycolic acids in Mycobacterium spp. by gas chromatography-mass spectrometry and mass chromatography. J. Clin. Microbiol. 1986, 24, 1060–1070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butler, W.R.; Guthertz, L.S. Mycolic acid analysis by high-performance liquid chromatography for identification of Mycobacterium species. Clin. Microbiol. Rev. 2001, 14, 704–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, F.-F.; Soehl, K.; Turk, J.; Haas, A. Characterization of mycolic acids from the pathogen Rhodococcus equi by tandem mass spectrometry with electrospray ionization. Anal. Biochem. 2011, 409, 112–122. [Google Scholar] [CrossRef] [Green Version]
- Guerrant, G.O.; Lambert, M.; Moss, C.W. Gas-chromatographic analysis of mycolic acid cleavage products in mycobacteria. J. Clin. Microbiol. 1981, 13, 899–907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barry, C.E.; Lee, R.E.; Mdluli, K.; Sampson, A.E.; Schroeder, B.G.; Slayden, R.A.; Yuan, Y. Mycolic acids: Structure, biosynthesis and physiological functions. Prog. Lipid Res. 1998, 37, 143–179. [Google Scholar] [CrossRef] [Green Version]
- Bligh, E.G.; Dyer, W.J. A rapid method of total lipid extraction and purification. Can. J. Biochem. 1959, 37, 911–917. [Google Scholar]
- Butler, W.; Ahearn, D.; Kilburn, J. High-performance liquid chromatography of mycolic acids as a tool in the identification of Corynebacterium, Nocardia, Rhodococcus, and Mycobacterium species. J. Clin. Microbiol. 1986, 23, 182–185. [Google Scholar] [CrossRef] [Green Version]
- Nikaido, H.; Kim, S.H.; Rosenberg, E.Y. Physical organization of lipids in the cell wall of Mycobacterium chelonae. Mol. Microbiol. 1993, 8, 1025–1030. [Google Scholar] [CrossRef]
- McNeil, M.; Brennan, P. Structure, function and biogenesis of the cell envelope of mycobacteria in relation to bacterial physiology, pathogenesis and drug resistance; some thoughts and possibilities arising from recent structural information. Res. Microbiol. 1991, 142, 451–463. [Google Scholar] [CrossRef]
- Bansal-Mutalik, R.; Nikaido, H. Quantitative lipid composition of cell envelopes of Corynebacterium glutamicum elucidated through reverse micelle extraction. Proc. Natl. Acad. Sci. USA 2011, 108, 15360–15365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bansal-Mutalik, R.; Nikaido, H. Mycobacterial outer membrane is a lipid bilayer and the inner membrane is unusually rich in diacyl phosphatidylinositol dimannosides. Proc. Natl. Acad. Sci. USA 2014, 111, 4958–4963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sokolovská, I.; Rozenberg, R.; Riez, C.; Rouxhet, P.G.; Agathos, S.N.; Wattiau, P. Carbon source-induced modifications in the mycolic acid content and cell wall permeability of Rhodococcus erythropolis E1. Appl. Environ. Microbiol. 2003, 69, 7019–7027. [Google Scholar] [CrossRef] [Green Version]
- Volkman, J.K.; Nichols, P.D. Applications of thin layer chromatography-flame ionization detection to the analysis of lipids and pollutants in marine and environmental samples. J. Planar Chromatogr. 1991, 4, 19–26. [Google Scholar]
- Nichols, D.S.; Nichols, P.D.; McMeekin, T.A. A new n-C-31:9 polyene hydrocarbon from Antarctic bacteria. FEMS Microbiol. Lett. 1995, 125, 281–285. [Google Scholar] [CrossRef]
- Parrish, C.; Ackman, R. Chromarod separations for the analysis of marine lipid classes by Iatroscan chromatography-flame ionization detection. J. Chromatogr. 1983, 262, 103–112. [Google Scholar] [CrossRef]
- Striby, L.; Lafont, R.; Goutx, M. Improvement in the Iatroscan thin-layer chromatographic–flame ionisation detection analysis of marine lipids. Separation and quantitation of monoacylglycerols and diacylglycerols in standards and natural samples. J. Chromatogr. 1999, 849, 371–380. [Google Scholar] [CrossRef]
- Sinanoglou, V.J.; Strati, I.F.; Bratakos, S.M.; Proestos, C.; Zoumpoulakis, P.; Miniadis-Meimaroglou, S. On the combined application of Iatroscan TLC-FID and GC-FID to identify total, neutral, and polar lipids and their fatty acids extracted from foods. ISRN Chromatography 2013, 2013, 859024. [Google Scholar] [CrossRef]
- Parrish, C.C. Separation of aquatic lipid classes by chromarod thin-layer chromatography with measurement by latroscan flame ionization detection. Can. J. Fish. Aquat. Sci. 1987, 44, 722–731. [Google Scholar] [CrossRef]
- Indrasena, W.M.; Henneberry, K.; Barrow, C.J.; Kralovec, J.A. Qualitative and quantitative analysis of lipid classes in fish oils by thin-layer chromatography with an Iatroscan flame ionization detector (TLC-FID) and liquid chromatography with an evaporative light scattering detector (LC-ELSD). J. Liq. Chromatogr. Relat. Technol. 2003, 28, 2581–2595. [Google Scholar] [CrossRef] [Green Version]
- Llorens-Fons, M.; Julián, E.; Luquin, M.; Pérez-Trujillo, M. Molecule confirmation and structure characterization of pentatriacontatrienyl mycolate in Mycobacterium smegmatis. Chem. Phys. Lipids 2018, 212, 138–143. [Google Scholar] [CrossRef] [PubMed]
- Bhamidi, S.; Scherman, M.S.; Jones, V.; Crick, D.C.; Belisle, J.T.; Brennan, P.J.; McNeil, M.R. Detailed structural and quantitative analysis reveals the spatial organization of the cell walls of in vivo grown Mycobacterium leprae and in vitro grown Mycobacterium tuberculosis. J. Biol. Chem. 2011, 286, 23168–23177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khanuja, S.P.S.; Srivastava, S.; Kumar, T.R.S.; Shasany, A.K. Quick and Sensitive Method of Quantifying Mycolic Acid to Develop Anti-Microbial Agents and a Diagnostic Kit Thereof. U.S. Patent No. 6833249, 21 December 2004. [Google Scholar]
- Lee, R.E.; Li, W.; Chatterjee, D.; Lee, R.E. Rapid structural characterization of the arabinogalactan and lipoarabinomannan in live mycobacterial cells using 2D and 3D HR-MAS NMR: Structural changes in the arabinan due to ethambutol treatment and gene mutation are observed. Glycobiology 2004, 15, 139–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parcher, J.F.; Wang, M.; Chittiboyina, A.G.; Khan, I.A. In-source collision-induced dissociation (IS-CID): Applications, issues and structure elucidation with single-stage mass analyzers. Drug Test Anal. 2018, 10, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Byreddy, A.R.; Gupta, A.; Barrow, C.J.; Puri, M. Comparison of cell disruption methods for improving lipid extraction from Thraustochytrid strains. Mar. Drugs 2015, 13, 5111–5127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldev, E.; MubarakAli, D.; Dhivya, M.; Kanimozhi, M.; Shakena-Fathima, T.; Alharbi, N.S.; Arunachalam, C.; Alharbi, S.A.; Thajuddin, N. Facile and novel strategy for methods of extraction of biofuel grade lipids from microalgae-an experimental report. Int. J. Biotechnol. Wellness Ind. 2015, 3, 121–127. [Google Scholar]
- Kramer, J.; Fouchard, R.; Farnworth, E. Effect of solvents on the resolution of neutral lipids on chromarods. J. Chromatogr. 1980, 198, 279–285. [Google Scholar] [CrossRef]
- Herrero, O.M.; Moncalián, G.; Alvarez, H.M. Physiological and genetic differences amongst Rhodococcus species for using glycerol as a source for growth and triacylglycerol production. Microbiology 2016, 162, 384–397. [Google Scholar] [CrossRef]
- Alvarez, H.M.; Mayer, F.; Fabritius, D.; Steinbüchel, A. Formation of intracytoplasmic lipid inclusions by Rhodococcus opacus strain PD630. Arch. Microbiol. 1996, 165, 377–386. [Google Scholar] [CrossRef]
- Shields-Menard, S.A.; Amirsadeghi, M.; Sukhbaatar, B.; Revellame, E.; Hernandez, R.; Donaldson, J.R.; French, W.T. Lipid accumulation by Rhodococcus rhodochrous grown on glucose. J. Ind. Microbiol. Biotechnol. 2015, 42, 693–699. [Google Scholar] [CrossRef] [PubMed]
- Round, J.; Roccor, R.; Li, S.-N.; Eltis, L.D. A fatty acyl coenzyme A reductase promotes wax ester accumulation in Rhodococcus jostii RHA1. Appl. Environ. Microbiol. 2017, 83, e00902-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nahar, A.; Baker, A.L.; Charleston, M.A.; Bowman, J.P.; Britz, M.L. Draft genome sequences of three sub-Antarctic Rhodococcus spp., including two novel psychrophilic genomospecies. Microbiol. Res. Announc. 2017, 5, e00898-17. [Google Scholar] [CrossRef] [PubMed]
- Nahar, A.; Baker, A.L.; Charleston, M.A.; Bowman, J.P.; Britz, M.L. Draft genome sequences of two novel Sub-Antarctic Williamsia species. Microbiol. Res. Announc. 2017, 5, e01047-17. [Google Scholar] [CrossRef] [Green Version]
- Nahar, A.; Baker, A.L.; Charleston, M.A.; Britz, M.L. Draft genome sequence of subantarctic Rhodococcus sp. strain 1139. Microbiol. Res. Announc. 2017, 5, e00090-17. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lipid Extraction Methods | Total Peak Area (µV/s) | Detectable Lipid Classes (mg/500 µL) by TLC-FID | Lipid % of Cell Dry Weight | |||||
|---|---|---|---|---|---|---|---|---|
| NPL | TAG | FFA | MA | PL/TDM | Total | |||
| Bead beating (unbound) | 24410 | 2.20 | 0.37 | 0.69 | - | 2.45 | 5.71 | 28.6 |
| Bead beating (bound) a | 284 | - | - | - | - | - | - | 1.2 |
| Sonication (unbound) | 11789 | 1.24 | 0.26 | 0.59 | - | 1.50 | 3.59 | 18.0 |
| Sonication (bound) a | 5098 | 0.49 | - | 0.97 | - | 0.37 | 1.83 | 9.2 |
| Strains | Total Peak Area (µV/s) | Detectable Lipid Classes (mg/500 µL) by TLC-FID | Lipid % of Cell Dry Weight | |||||
|---|---|---|---|---|---|---|---|---|
| NPL | TAG | FFA | MA | PL/TDM | Total | |||
| Williamsia sp. 1135 | 23042 | 1.91 | 0.66 | 0.67 | 0.84 | 2.2 | 6.28 | 31.4 |
| Williamsia sp. 1138 | 21250 | 2.01 | 0.60 | 0.66 | - | 1.60 | 4.87 | 24.4 |
| Rhodococcus sp. 1159 | 22228 | 1.89 | 0.44 | - | - | 2.61 | 4.94 | 24.7 |
| Rhodococcus sp. 1163 | 16343 | 1.34 | 0.44 | 0.65 | - | 2.09 | 4.52 | 22.6 |
| Rhodococcus sp. 1168 | 16151 | 1.57 | 0.78 | 0.64 | - | 1.60 | 4.59 | 23.0 |
| C. glutamicum | 14697 | 1.71 | - | - | - | 1.71 | 3.42 | 17.1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nahar, A.; Baker, A.L.; Nichols, D.S.; Bowman, J.P.; Britz, M.L. Application of Thin-Layer Chromatography-Flame Ionization Detection (TLC-FID) to Total Lipid Quantitation in Mycolic-Acid Synthesizing Rhodococcus and Williamsia Species. Int. J. Mol. Sci. 2020, 21, 1670. https://doi.org/10.3390/ijms21051670
Nahar A, Baker AL, Nichols DS, Bowman JP, Britz ML. Application of Thin-Layer Chromatography-Flame Ionization Detection (TLC-FID) to Total Lipid Quantitation in Mycolic-Acid Synthesizing Rhodococcus and Williamsia Species. International Journal of Molecular Sciences. 2020; 21(5):1670. https://doi.org/10.3390/ijms21051670
Chicago/Turabian StyleNahar, Akhikun, Anthony L. Baker, David S. Nichols, John P. Bowman, and Margaret L. Britz. 2020. "Application of Thin-Layer Chromatography-Flame Ionization Detection (TLC-FID) to Total Lipid Quantitation in Mycolic-Acid Synthesizing Rhodococcus and Williamsia Species" International Journal of Molecular Sciences 21, no. 5: 1670. https://doi.org/10.3390/ijms21051670