Alternatively Constructed Estrogen Receptor Alpha-Driven Super-Enhancers Result in Similar Gene Expression in Breast and Endometrial Cell Lines



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. General Comparison of the Putative ERα Binding Sites in MCF-7 and Ishikawa Cells
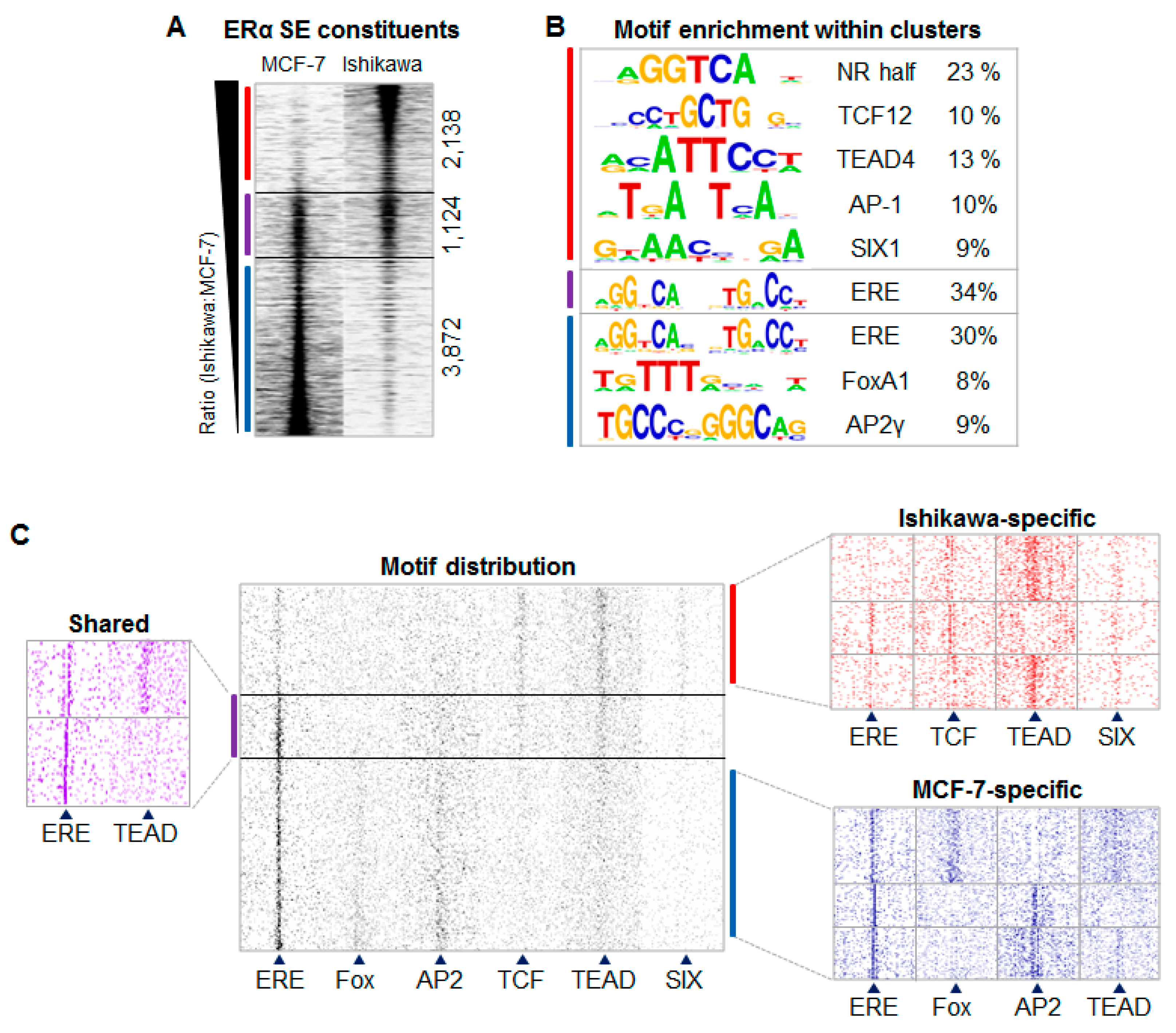
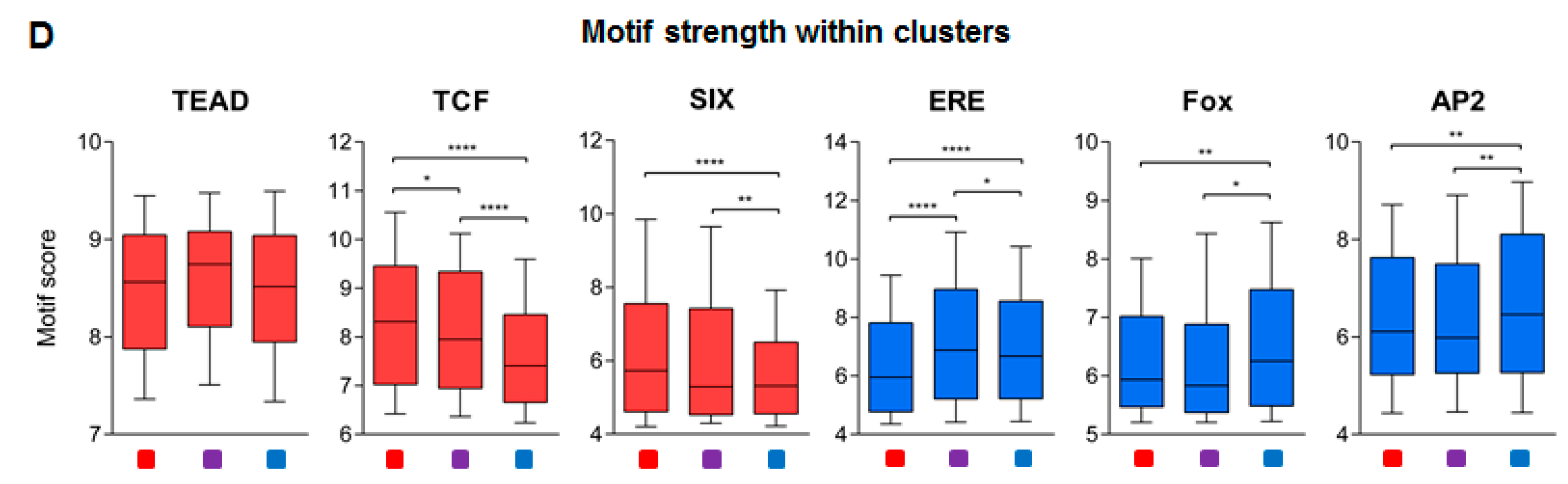
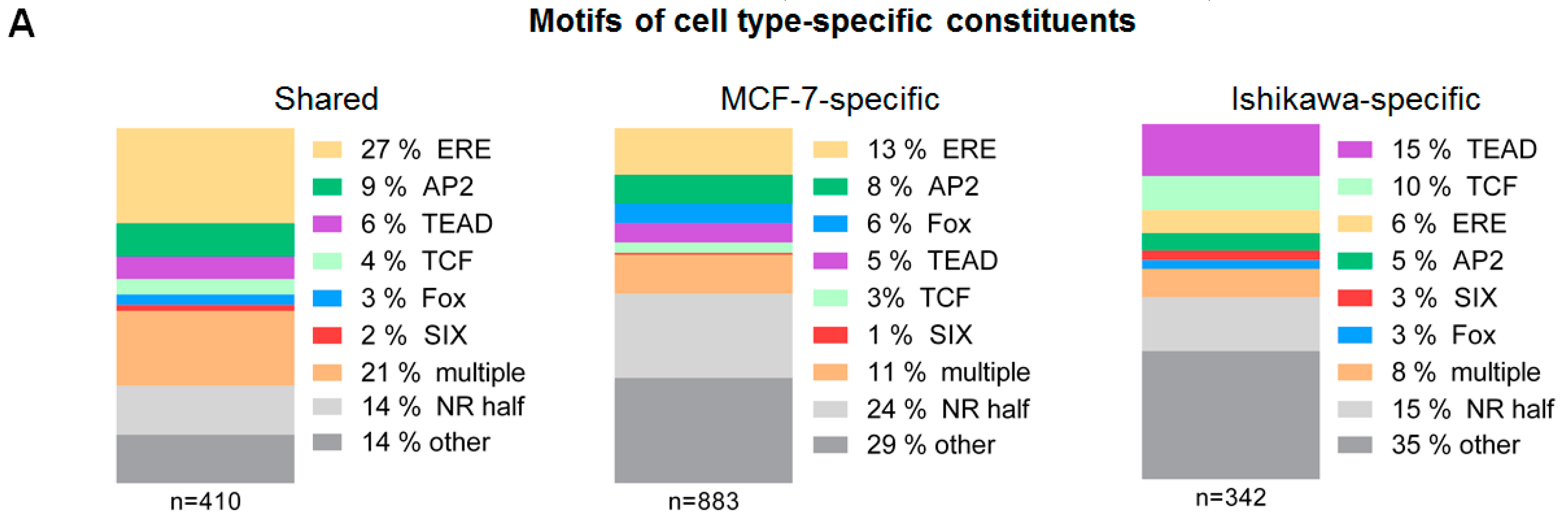
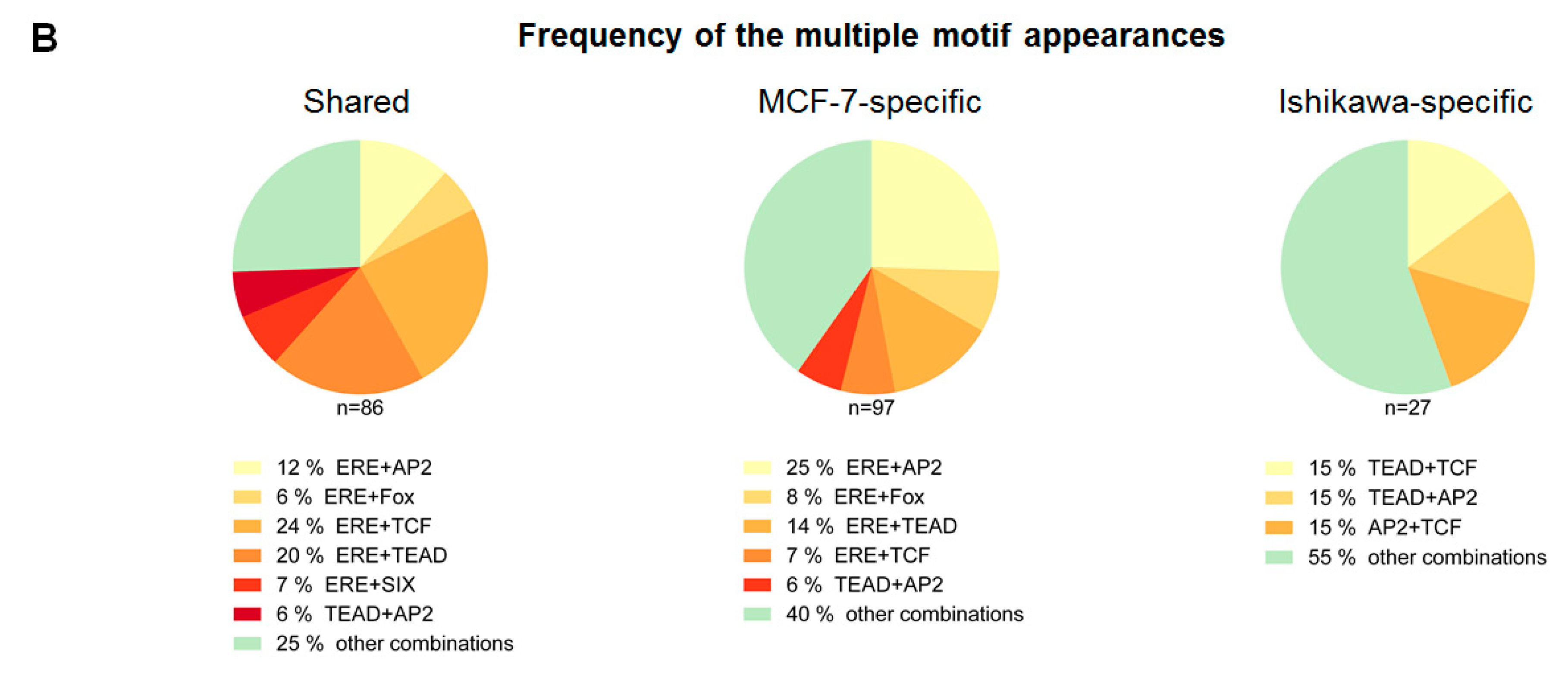
2.2. ERα-driven SE Constituents have Different Motif Preferences in MCF-7 and Ishikawa Cells
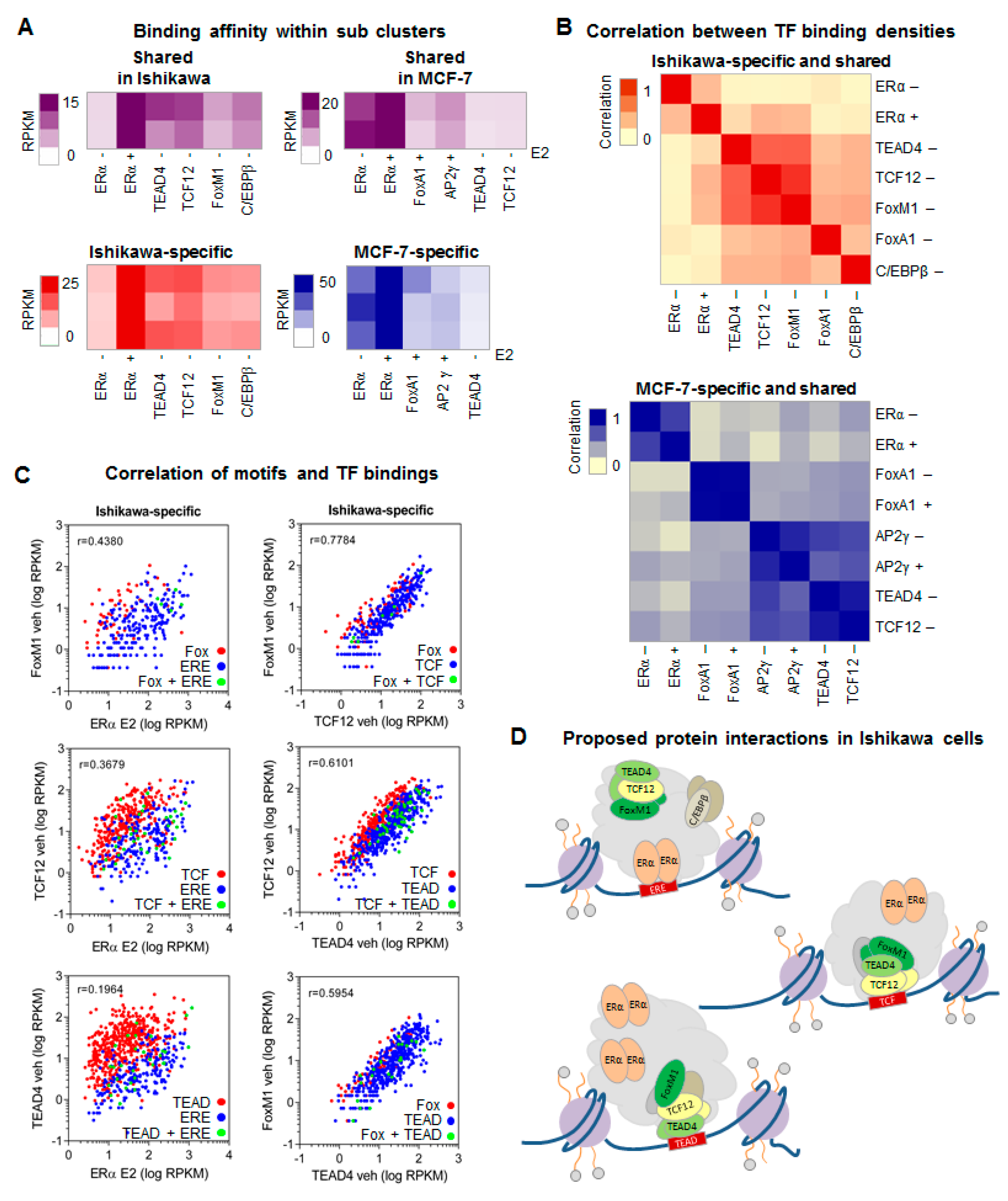
2.3. Response Elements Determine the Hierarchy between FoxM1/TCF12/TEAD4 Proteins in Ishikawa Cells
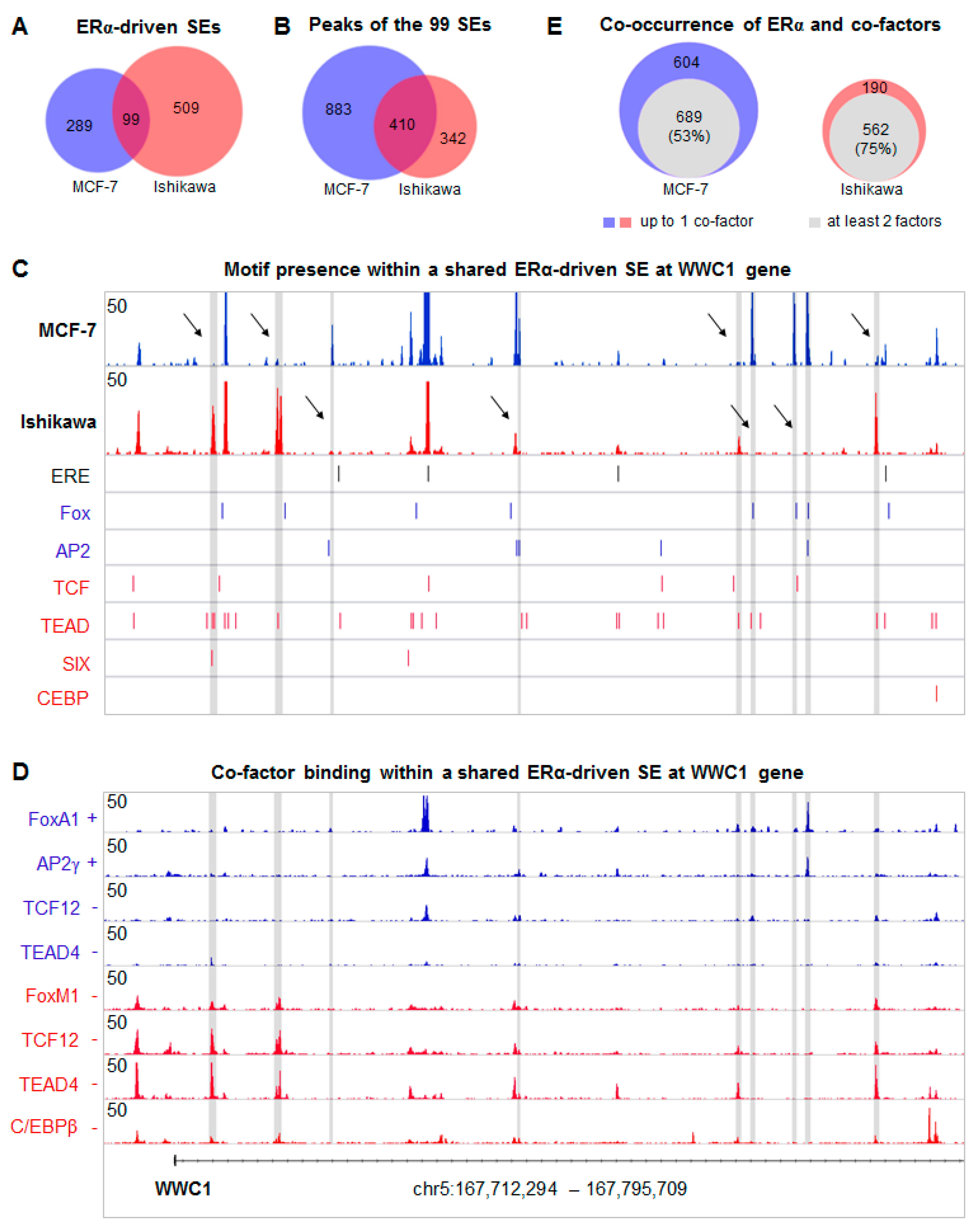
2.4. Overlapping SE Regions Are Composed of Different ERα Binding Sites
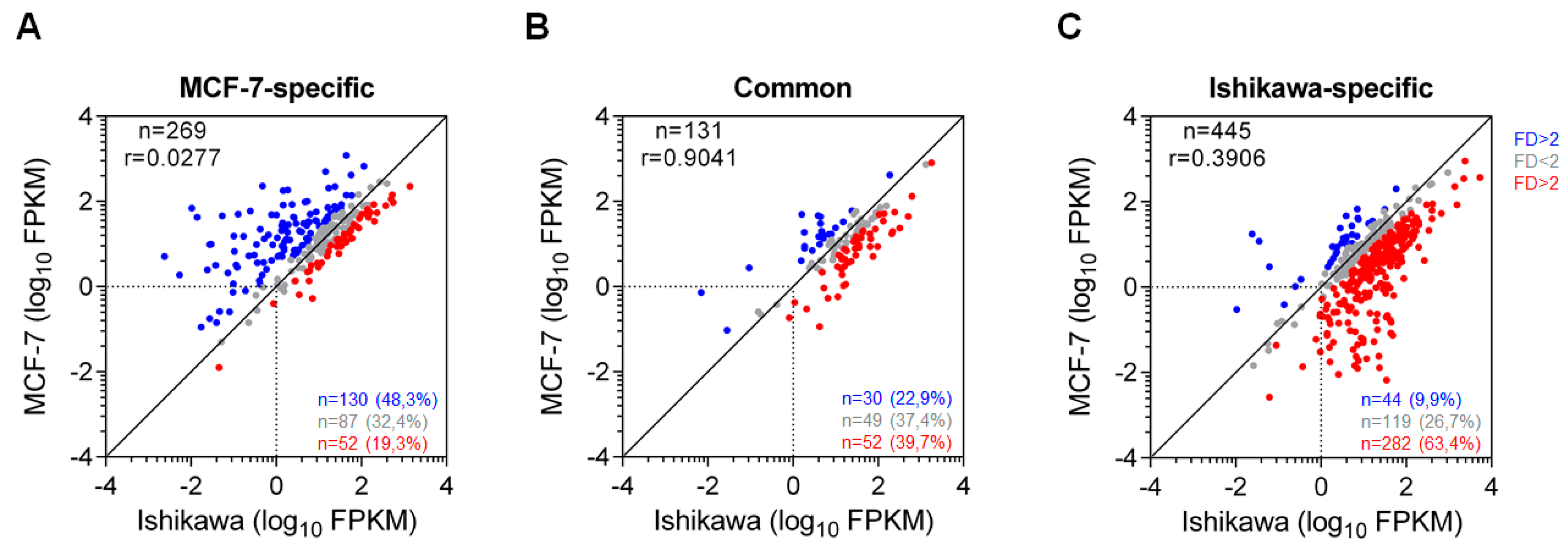
2.5. Common ERα-driven SEs Generate Similar Transcriptional Output in MCF-7 and Ishikawa Cells
3. Discussion
4. Materials and Methods
4.1. Data Selection
4.2. ChIP-seq Analysis
4.3. Super-Enhancer Prediction
4.4. Motif Analysis
4.5. DNase-seq Analysis
4.6. RNA-seq Analysis
4.7. Gene Annotation
4.8. Visualization
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ERα | estrogen receptor alpha |
| ERE | estrogen response element |
| E2 | estradiol (17β-estradiol) |
| H3K27ac | acetylation at the 27th lysine residue of the histone H3 protein |
| NR | nuclear receptor |
| SE | super-enhancer |
| TF | transcription factor |
| TFBS | transcription factor binding site |
References
- Germain, P.; Staels, B.; Dacquet, C.; Spedding, M.; Laudet, V. Overview of nomenclature of nuclear receptors. Pharmacol. Rev. 2006, 58, 685–704. [Google Scholar] [CrossRef] [PubMed]
- Ross-Innes, C.S.; Stark, R.; Teschendorff, A.E.; Holmes, K.A.; Ali, H.R.; Dunning, M.J.; Brown, G.D.; Gojis, O.; Ellis, I.O.; Green, A.R.; et al. Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature 2012, 481, 389–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Zhao, D.; Gong, C.; Zhang, F.; He, J.; Zhang, W.; Zhao, Y.; Sun, J. Prognostic role of hormone receptors in endometrial cancer: A systematic review and meta-analysis. World J. Surg. Oncol. 2015, 13, 208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reed, B.G.; Carr, B.R. The Normal Menstrual Cycle and the Control of Ovulation; MDText.com Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Väänänen, H.K.; Härkönen, P.L. Estrogen and bone metabolism. Maturitas 1996, 23, S65–S69. [Google Scholar] [CrossRef]
- Groothuis, P.G.; Dassen, H.H.N.M.; Romano, A.; Punyadeera, C. Estrogen and the endometrium: Lessons learned from gene expression profiling in rodents and human. Hum. Reprod. Update 2007, 13, 405–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein-Hitpass, L.; Schorpp, M.; Wagner, U.; Ryffel, G.U. An estrogen-responsive element derived from the 5’ flanking region of the Xenopus vitellogenin A2 gene functions in transfected human cells. Cell 1986, 46, 1053–1061. [Google Scholar] [CrossRef]
- Kumar, V.; Chambon, P. The estrogen receptor binds tightly to its responsive element as a ligand-induced homodimer. Cell 1988, 55, 145–156. [Google Scholar] [CrossRef]
- Klinge, C.M. Estrogen receptor interaction with co-activators and co-repressors. Steroids 2000, 65, 227–251. [Google Scholar] [CrossRef]
- Gaub, M.P.; Bellard, M.; Scheuer, I.; Chambon, P.; Sassone-Corsi, P. Activation of the ovalbumin gene by the estrogen receptor involves the fos-jun complex. Cell 1990, 63, 1267–1276. [Google Scholar] [CrossRef]
- Klinge, C.M. Estrogen receptor interaction with estrogen response elements. Nucleic Acids Res. 2001, 29, 2905–2919. [Google Scholar] [CrossRef] [Green Version]
- Vega, V.B.; Lin, C.-Y.; Lai, K.S.; Kong, S.L.; Xie, M.; Su, X.; Teh, H.F.; Thomsen, J.S.; Yeo, A.L.; Sung, W.K.; et al. Multiplatform genome-wide identification and modeling of functional human estrogen receptor binding sites. Genome Biol. 2006, 7, R82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joseph, R.; Orlov, Y.L.; Huss, M.; Sun, W.; Kong, S.L.; Ukil, L.; Pan, Y.F.; Li, G.; Lim, M.; Thomsen, J.S.; et al. Integrative model of genomic factors for determining binding site selection by estrogen receptor-α. Mol. Syst. Biol. 2010, 6, 456. [Google Scholar] [CrossRef] [PubMed]
- Bourdeau, V.; Deschênes, J.; Métivier, R.; Nagai, Y.; Nguyen, D.; Bretschneider, N.; Gannon, F.; White, J.H.; Mader, S. Genome-wide identification of high-affinity estrogen response elements in human and mouse. Mol. Endocrinol. 2004, 18, 1411–1427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bojcsuk, D.; Nagy, G.; Balint, B.L. Inducible super-enhancers are organized based on canonical signal-specific transcription factor binding elements. Nucleic Acids Res. 2017, 45, 3699–3706. [Google Scholar] [CrossRef] [Green Version]
- Bojcsuk, D.; Bálint, B.L. Classification of different types of estrogen receptor alpha binding sites in MCF-7 cells. J. Biotechnol. 2019, 299, 13–20. [Google Scholar] [CrossRef]
- Lovén, J.; Hoke, H.A.; Lin, C.Y.; Lau, A.; Orlando, D.A.; Vakoc, C.R.; Bradner, J.E.; Lee, T.I.; Young, R.A. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 2013, 153, 320–334. [Google Scholar] [CrossRef] [Green Version]
- Pott, S.; Lieb, J.D. What are super-enhancers? Nat. Genet. 2015, 47, 8–12. [Google Scholar] [CrossRef]
- Kagey, M.H.; Newman, J.J.; Bilodeau, S.; Zhan, Y.; Orlando, D.A.; van Berkum, N.L.; Ebmeier, C.C.; Goossens, J.; Rahl, P.B.; Levine, S.S.; et al. Mediator and cohesin connect gene expression and chromatin architecture. Nature 2010, 467, 430–435. [Google Scholar] [CrossRef] [Green Version]
- Di Micco, R.; Fontanals-Cirera, B.; Low, V.; Ntziachristos, P.; Yuen, S.K.; Lovell, C.D.; Dolgalev, I.; Yonekubo, Y.; Zhang, G.; Rusinova, E.; et al. Control of embryonic stem cell identity by BRD4-dependent transcriptional elongation of super-enhancer-associated pluripotency genes. Cell Rep. 2014, 9, 234–247. [Google Scholar] [CrossRef] [Green Version]
- Tsai, C.-J.; Nussinov, R. Gene-specific transcription activation via long-range allosteric shape-shifting. Biochem. J. 2011, 439, 15–25. [Google Scholar] [CrossRef]
- Sabari, B.R.; Dall’Agnese, A.; Boija, A.; Klein, I.A.; Coffey, E.L.; Shrinivas, K.; Abraham, B.J.; Hannett, N.M.; Zamudio, A.V.; Manteiga, J.C.; et al. Coactivator condensation at super-enhancers links phase separation and gene control. Science 2018, 361, eaar3958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hnisz, D.; Shrinivas, K.; Young, R.A.; Chakraborty, A.K.; Sharp, P.A. A phase separation model for transcriptional control. Cell 2017, 169, 13–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hnisz, D.; Abraham, B.J.; Lee, T.I.; Lau, A.; Saint-André, V.; Sigova, A.A.; Hoke, H.A.; Young, R.A. Super-enhancers in the control of cell identity and disease. Cell 2013, 155, 934–947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siersbæk, R.; Baek, S.; Rabiee, A.; Nielsen, R.; Traynor, S.; Clark, N.; Sandelin, A.; Jensen, O.N.; Sung, M.-H.; Hager, G.L.; et al. Molecular architecture of transcription factor hotspots in early adipogenesis. Cell Rep. 2014, 7, 1434–1442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willi, M.; Yoo, K.H.; Reinisch, F.; Kuhns, T.M.; Lee, H.K.; Wang, C.; Hennighausen, L. Facultative CTCF sites moderate mammary super-enhancer activity and regulate juxtaposed gene in non-mammary cells. Nat. Commun. 2017, 8, 16069. [Google Scholar] [CrossRef] [Green Version]
- Dowen, J.M.; Fan, Z.P.; Hnisz, D.; Ren, G.; Abraham, B.J.; Zhang, L.N.; Weintraub, A.S.; Schuijers, J.; Lee, T.I.; Zhao, K.; et al. Control of cell identity genes occurs in insulated neighborhoods in mammalian chromosomes. Cell 2014, 159, 374–387. [Google Scholar] [CrossRef] [Green Version]
- Shin, H.Y. Targeting super-enhancers for disease treatment and diagnosis. Mol. Cells 2018, 41, 506–514. [Google Scholar]
- Sengupta, S.; George, R.E. Super-enhancer-driven transcriptional dependencies in cancer. Trends Cancer 2017, 3, 269–281. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Li, K.; Cai, W.; Liu, X.; Zhang, Y.; Orkin, S.H.; Xu, J.; Yuan, G.-C. Dissecting super-enhancer hierarchy based on chromatin interactions. Nat. Commun. 2018, 9, 943. [Google Scholar] [CrossRef] [Green Version]
- Nord, A.S.; Blow, M.J.; Attanasio, C.; Akiyama, J.A.; Holt, A.; Hosseini, R.; Phouanenavong, S.; Plajzer-Frick, I.; Shoukry, M.; Afzal, V.; et al. Rapid and pervasive changes in genome-wide enhancer usage during mammalian development. Cell 2013, 155, 1521–1531. [Google Scholar] [CrossRef] [Green Version]
- Carroll, J.S.; Liu, X.S.; Brodsky, A.S.; Li, W.; Meyer, C.A.; Szary, A.J.; Eeckhoute, J.; Shao, W.; Hestermann, E.V.; Geistlinger, T.R.; et al. Chromosome-wide mapping of estrogen receptor binding reveals long-range regulation requiring the forkhead protein FoxA1. Cell 2005, 122, 33–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurtado, A.; Holmes, K.A.; Ross-Innes, C.S.; Schmidt, D.; Carroll, J.S. FoxA1 is a key determinant of estrogen receptor function and endocrine response. Nat. Genet. 2011, 43, 27–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, S.K.; Lin, Z.H.; Chang, C.W.; Varang, V.; Chng, K.R.; Pan, Y.F.; Yong, E.L.; Sung, W.K.; Sung, W.K.; Cheung, E. AP-2γ regulates oestrogen receptor-mediated long-range chromatin interaction and gene transcription. EMBO J. 2011, 30, 2569–2581. [Google Scholar] [CrossRef] [PubMed]
- Ross-Innes, C.S.; Stark, R.; Holmes, K.A.; Schmidt, D.; Spyrou, C.; Russell, R.; Massie, C.E.; Vowler, S.L.; Eldridge, M.; Carroll, J.S. Cooperative interaction between retinoic acid receptor- and estrogen receptor in breast cancer. Genes Dev. 2010, 24, 171–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theodorou, V.; Stark, R.; Menon, S.; Carroll, J.S. GATA3 acts upstream of FOXA1 in mediating ESR1 binding by shaping enhancer accessibility. Genome Res. 2013, 23, 12–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Merkurjev, D.; Yang, F.; Li, W.; Oh, S.; Friedman, M.J.; Song, X.; Zhang, F.; Ma, Q.; Ohgi, K.A.; et al. Enhancer activation requires trans-recruitment of a mega transcription factor complex. Cell 2014, 159, 358–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Droog, M.; Nevedomskaya, E.; Kim, Y.; Severson, T.; Flach, K.D.; Opdam, M.; Schuurman, K.; Gradowska, P.; Hauptmann, M.; Dackus, G.; et al. Comparative cistromics reveals genomic cross-talk between FoxA1 and ER in tamoxifen-associated endometrial carcinomas. Cancer Res. 2016, 76, 3773–3784. [Google Scholar] [CrossRef] [Green Version]
- Khushi, M.; Clarke, C.L.; Graham, J.D. Bioinformatic analysis of cis-regulatory interactions between progesterone and estrogen receptors in breast cancer. Peer J. 2014, 2, e654. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Li, H.; Rajurkar, M.; Li, Q.; Cotton, J.L.; Ou, J.; Zhu, L.J.; Goel, H.L.; Mercurio, A.M.; Park, J.-S.; et al. Tead and AP1 coordinate transcription and motility. Cell Rep. 2016, 14, 1169–1180. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Liang, Y.; Zhang, Y.; Jiang, M.; Han, Z.; Liang, Y.; Wan, Y.; Yin, J.; He, H.-C.; Zhong, W. Decreased expression of TCF12 contributes to progression and predicts biochemical recurrence in patients with prostate cancer. Tumor Biol. 2017, 39, 101042831770392. [Google Scholar] [CrossRef] [Green Version]
- Kompass, K.S.; Witte, J.S. Co-regulatory expression quantitative trait loci mapping: Method and application to endometrial cancer. BMC Med. Genom. 2011, 4, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suen, A.A.; Jefferson, W.N.; Wood, C.E.; Padilla-Banks, E.; Bae-Jump, V.L.; Williams, C.J. SIX1 oncoprotein as a biomarker in a model of hormonal carcinogenesis and in human endometrial cancer. Mol. Cancer Res. 2016, 14, 849–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stender, J.D.; Kim, K.; Charn, T.H.; Komm, B.; Chang, K.C.N.; Kraus, W.L.; Benner, C.; Glass, C.K.; Katzenellenbogen, B.S. Genome-wide analysis of estrogen receptor alpha DNA binding and tethering mechanisms identifies Runx1 as a novel tethering factor in receptor-mediated transcriptional activation. Mol. Cell. Biol. 2010, 30, 3943–3955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lupien, M.; Eeckhoute, J.; Meyer, C.A.; Wang, Q.; Zhang, Y.; Li, W.; Carroll, J.S.; Liu, X.S.; Brown, M. FoxA1 translates epigenetic Signatures into enhancer-driven lineage-specific transcription. Cell 2008, 132, 958–970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tewari, A.K.; Yardimci, G.; Shibata, Y.; Sheffield, N.C.; Song, L.; Taylor, B.S.; Georgiev, S.G.; Coetzee, G.A.; Ohler, U.; Furey, T.S.; et al. Chromatin accessibility reveals insights into androgen receptor activation and transcriptional specificity. Genome Biol. 2012, 13, R88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahu, B.; Laakso, M.; Pihlajamaa, P.; Ovaska, K.; Sinielnikov, I.; Hautaniemi, S.; Janne, O.A. FoxA1 Specifies unique androgen and glucocorticoid receptor binding events in prostate cancer cells. Cancer Res. 2013, 73, 1570–1580. [Google Scholar] [CrossRef] [Green Version]
- Bärlund, M.; Monni, O.; Weaver, J.D.; Kauraniemi, P.; Sauter, G.; Heiskanen, M.; Kallioniemi, O.-P.; Kallioniemi, A. Cloning of BCAS3 (17q23) and BCAS4 (20q13) genes that undergo amplification, overexpression, and fusion in breast cancer†. Genes Chromosom. Cancer 2002, 35, 311–317. [Google Scholar] [CrossRef]
- Vendrell, J.A.; Thollet, A.; Nguyen, N.T.; Ghayad, S.E.; Vinot, S.; Bieche, I.; Grisard, E.; Josserand, V.; Coll, J.-L.; Roux, P.; et al. ZNF217 is a marker of poor prognosis in breast cancer that drives epithelial-mesenchymal transition and invasion. Cancer Res. 2012, 72, 3593–3606. [Google Scholar] [CrossRef] [Green Version]
- Alonso-Alconada, L.; Santacana, M.; Garcia-Sanz, P.; Muinelo-Romay, L.; Colas, E.; Mirantes, C.; Monge, M.; Cueva, J.; Oliva, E.; Soslow, R.A.; et al. Annexin-A2 as predictor biomarker of recurrent disease in endometrial cancer. Int. J. Cancer 2015, 136, 1863–1873. [Google Scholar] [CrossRef]
- Liu, B.; Chen, P.; Xi, D.; Zhu, H.; Gao, Y. ATF4 regulates CCL2 expression to promote endometrial cancer growth by controlling macrophage infiltration. Exp. Cell Res. 2017, 360, 105–112. [Google Scholar] [CrossRef]
- Silva, T.M.; Cirenajwis, H.; Wallace, H.M.; Oredsson, S.; Persson, L. A role for antizyme inhibitor in cell proliferation. Amino. Acids 2015, 47, 1341–1352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, L.; Wang, Y.; Gao, Y.; Chen, X. Overexpression of CXXC5 is a strong poor prognostic factor in ER+ breast cancer. Oncol. Lett. 2018, 16, 395–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomaskovic-Crook, E.; Thompson, E.W.; Thiery, J.P. Epithelial to mesenchymal transition and breast cancer. Breast Cancer Res. 2009, 11, 213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korch, C.; Spillman, M.A.; Jackson, T.A.; Jacobsen, B.M.; Murphy, S.K.; Lessey, B.A.; Jordan, V.C.; Bradford, A.P. DNA profiling analysis of endometrial and ovarian cell lines reveals misidentification, redundancy and contamination. Gynecol. Oncol. 2012, 127, 241–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barta, E. Command line analysis of ChIP-seq results. EMB Net. J. 2011, 17, 13–17. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 genome project data processing subgroup the sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Heinz, S.; Benner, C.; Spann, N.; Bertolino, E.; Lin, Y.C.; Laslo, P.; Cheng, J.X.; Murre, C.; Singh, H.; Glass, C.K. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 2010, 38, 576–589. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nusbaum, C.; Myers, R.M.; Brown, M.; Li, W.; et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, R137. [Google Scholar] [CrossRef] [Green Version]
- Dunham, I.; Kundaje, A.; Aldred, S.F.; Collins, P.J.; Davis, C.A.; Doyle, F.; Epstein, C.B.; Frietze, S.; Harrow, J.; Kaul, R.; et al. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stark, R.; Brown, G. DiffBind: Differential Binding Analysis of ChIP- Seq Peak Data. Available online: https://bioconductor.statistik.tu-dortmund.de/packages/2.13/bioc/vignettes/DiffBind/inst/doc/DiffBind.pdf (accessed on 22 February 2020).
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salmon-Divon, M.; Dvinge, H.; Tammoja, K.; Bertone, P. PeakAnalyzer: Genome-wide annotation of chromatin binding and modification loci. BMC Bioinform. 2010, 11, 415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saldanha, A.J. Java Treeview—Extensible visualization of microarray data. Bioinformatics 2004, 20, 3246–3248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hulsen, T.; de Vlieg, J.; Alkema, W. BioVenn—A web application for the comparison and visualization of biological lists using area-proportional Venn diagrams. BMC Genom. 2008, 9, 488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorvaldsdottir, H.; Robinson, J.T.; Mesirov, J.P. Integrative genomics viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bojcsuk, D.; Nagy, G.; Bálint, B.L. Alternatively Constructed Estrogen Receptor Alpha-Driven Super-Enhancers Result in Similar Gene Expression in Breast and Endometrial Cell Lines. Int. J. Mol. Sci. 2020, 21, 1630. https://doi.org/10.3390/ijms21051630
Bojcsuk D, Nagy G, Bálint BL. Alternatively Constructed Estrogen Receptor Alpha-Driven Super-Enhancers Result in Similar Gene Expression in Breast and Endometrial Cell Lines. International Journal of Molecular Sciences. 2020; 21(5):1630. https://doi.org/10.3390/ijms21051630
Chicago/Turabian StyleBojcsuk, Dóra, Gergely Nagy, and Bálint László Bálint. 2020. "Alternatively Constructed Estrogen Receptor Alpha-Driven Super-Enhancers Result in Similar Gene Expression in Breast and Endometrial Cell Lines" International Journal of Molecular Sciences 21, no. 5: 1630. https://doi.org/10.3390/ijms21051630