Podocyte Lysosome Dysfunction in Chronic Glomerular Diseases

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
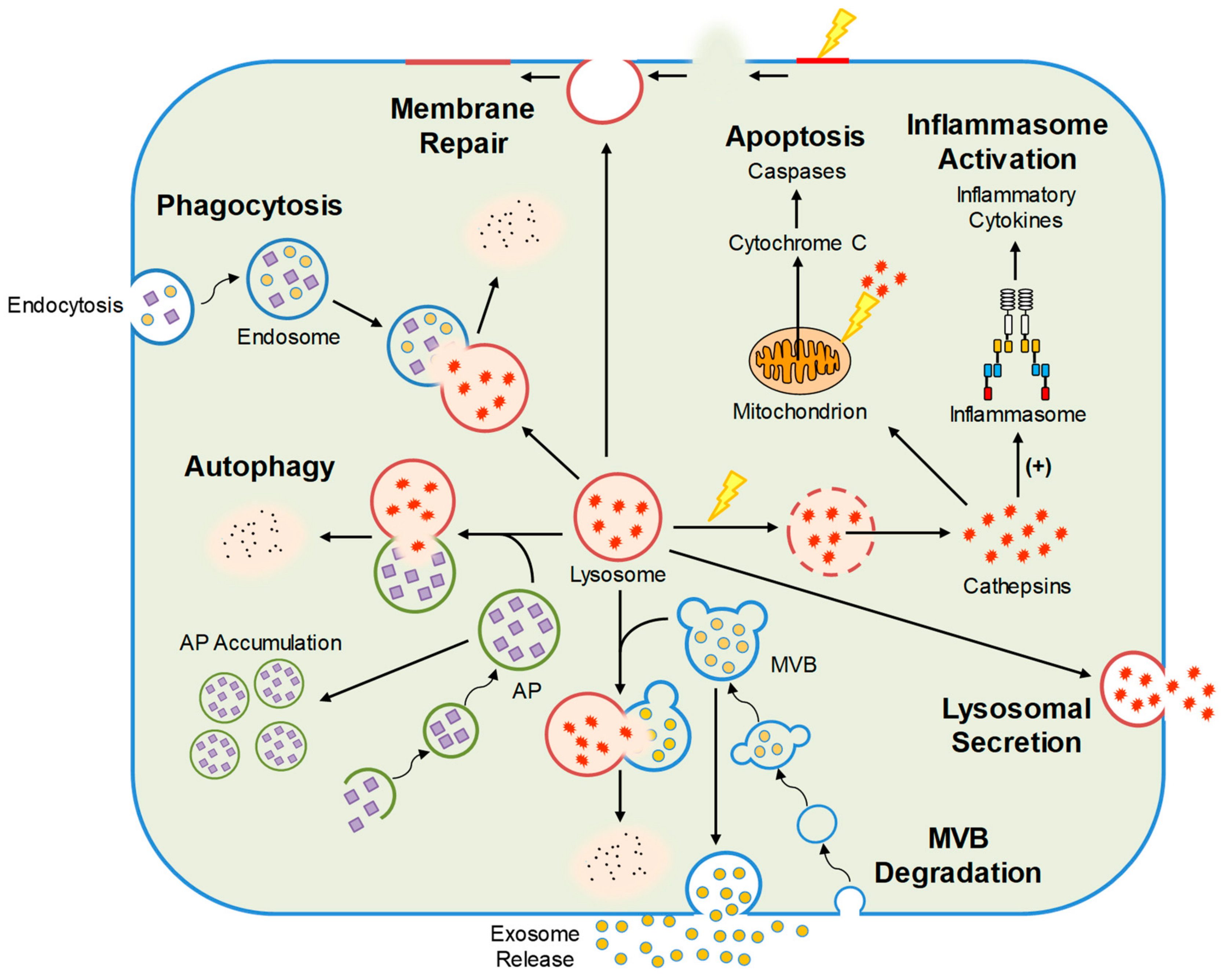
2. Different Types of Lysosomes
3. Cellular Functions Regulated by Lysosomes
3.1. Autophagic Flux
3.2. Lipid Metabolism
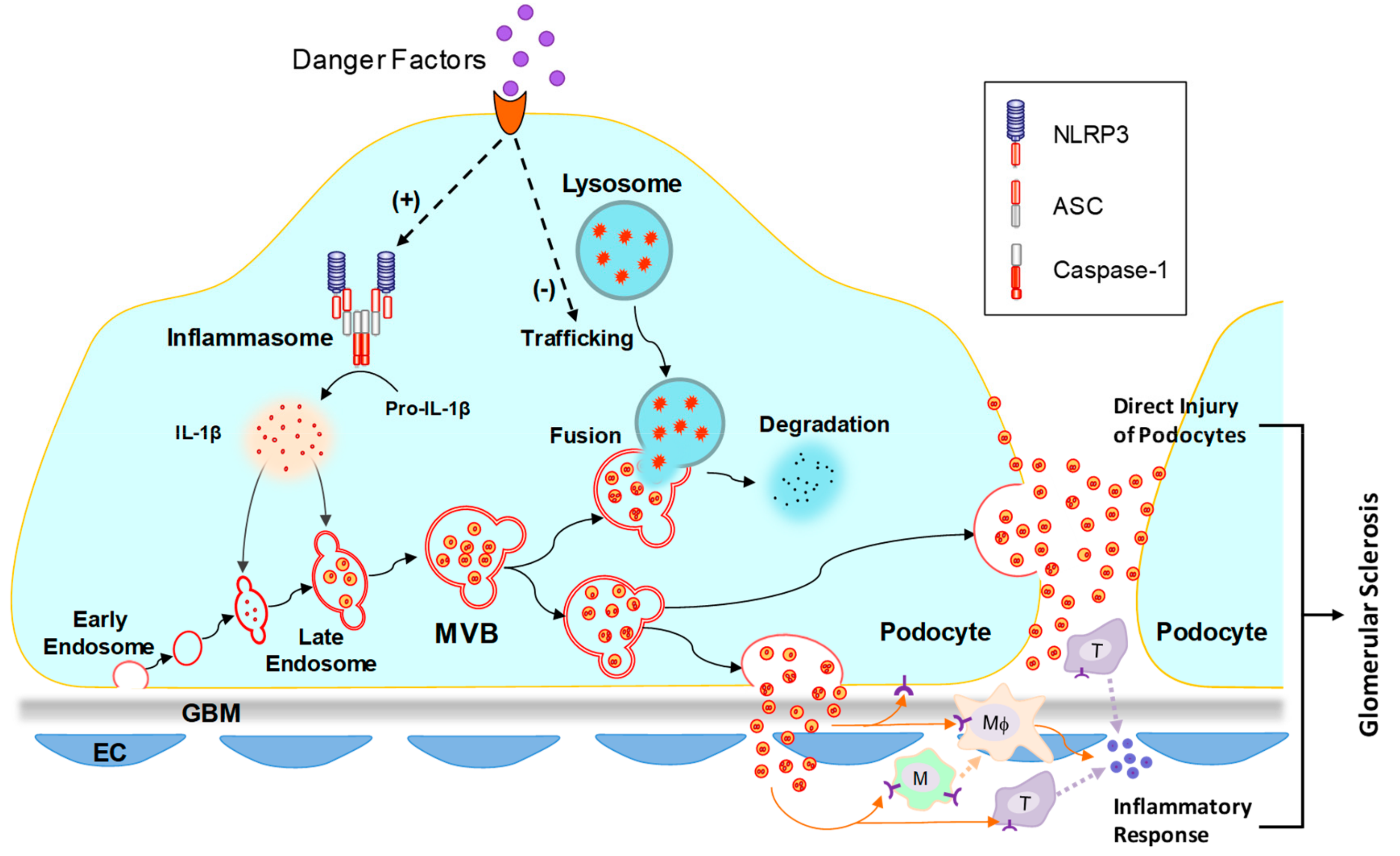
3.3. Inflammasome Activation
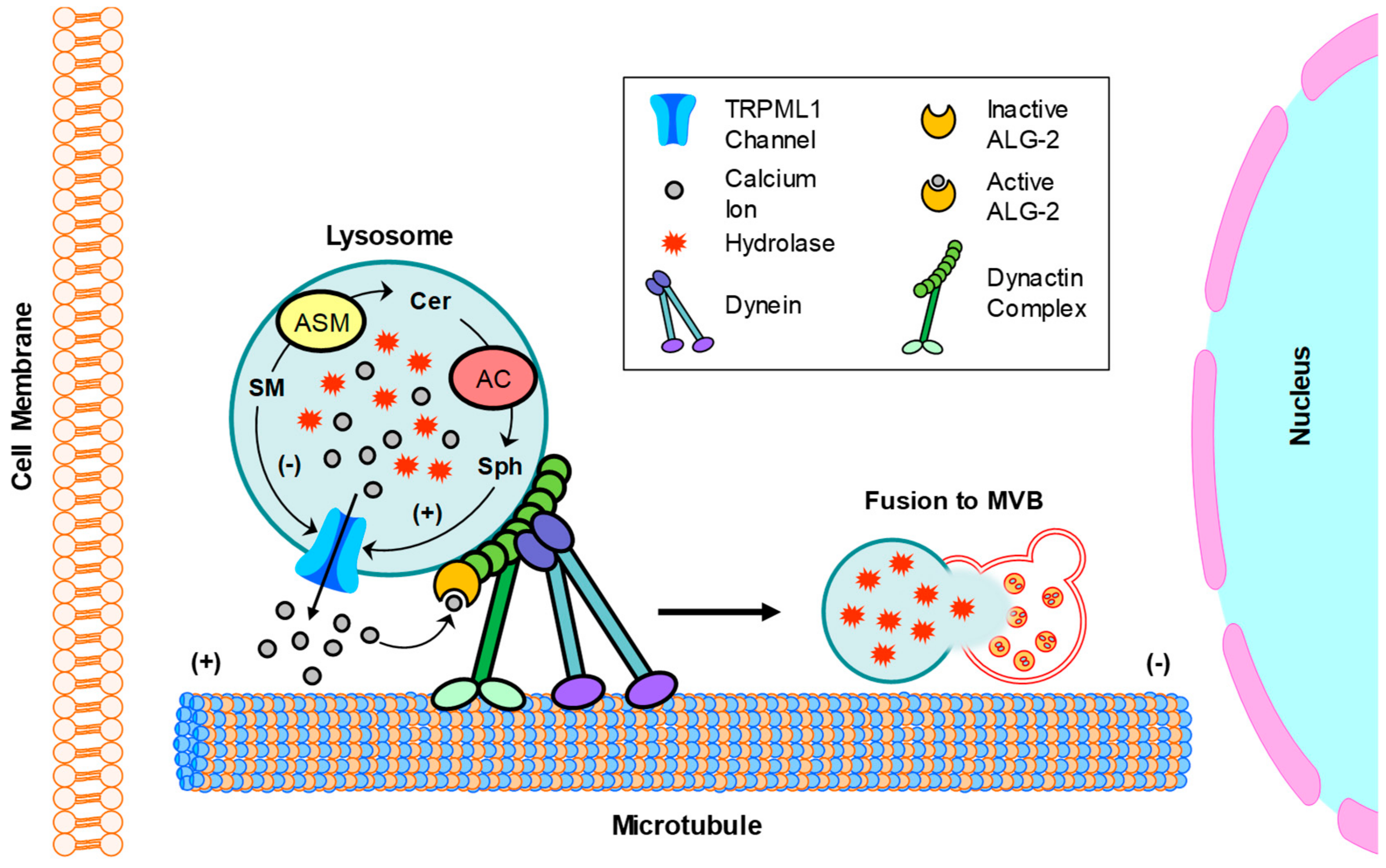
3.4. Exosome Release
4. Podocyte Lysosome Dysfunction in Chronic Glomerular Diseases
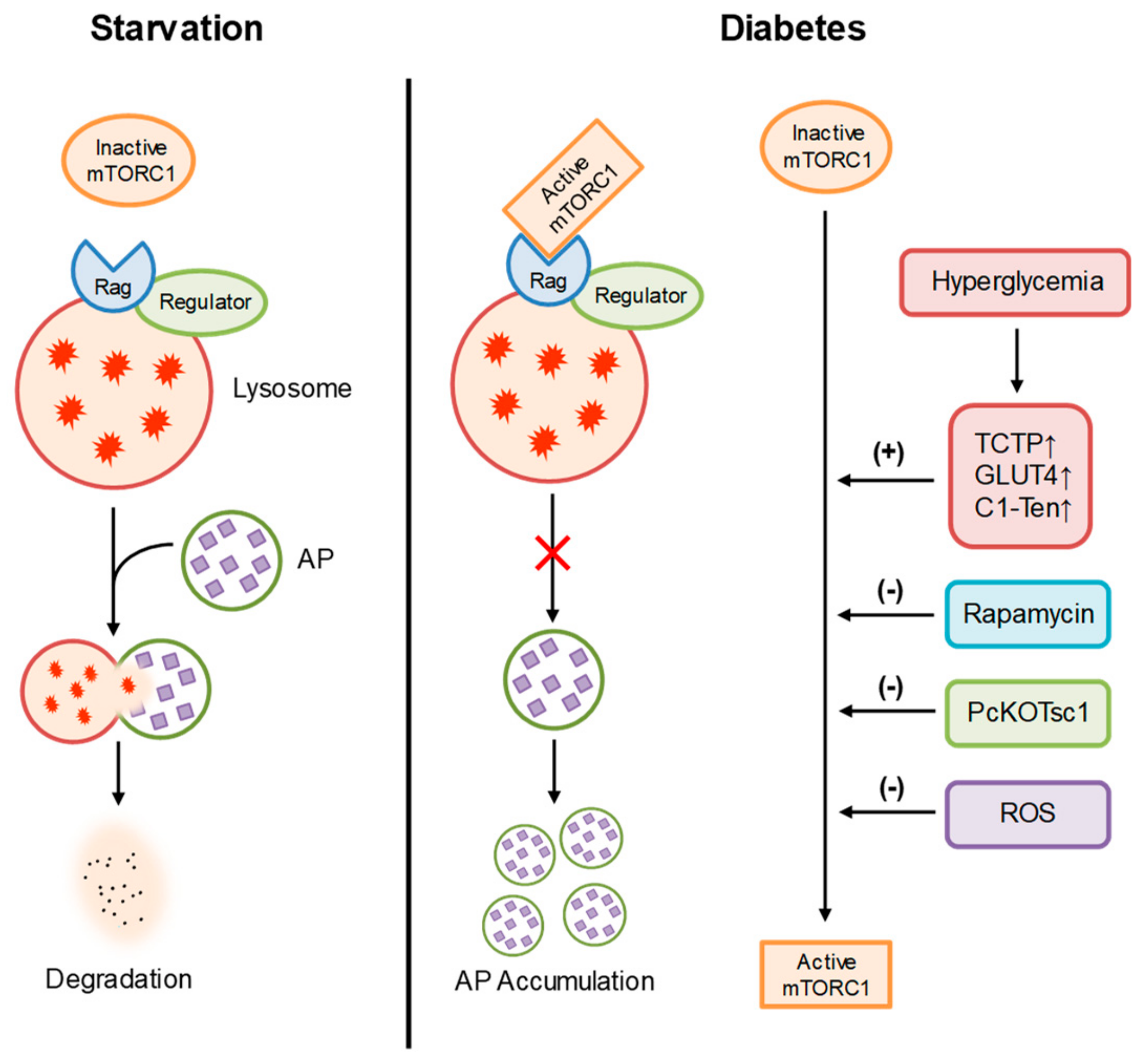
4.1. Diabetic Nephropathy
4.2. Glomerular Injury during Fabry Disease
4.3. Hyperhomocysteinemic Nephropathy
4.4. Obesity-Induced Podocyte Injury and Glomerular Disease
4.5. APOL1-Associated Nephropathy
5. Therapeutic Potential of Chronic Glomerular Diseases by Restoring Lysosome Function
6. Concluding Remarks
Funding
Conflicts of Interest
References
- Bainton, D.F. The discovery of lysosomes. J. Cell Biol. 1981, 91, 66s–76s. [Google Scholar] [CrossRef] [Green Version]
- De Duve, C. The lysosome turns fifty. Nat. Cell Biol. 2005, 7, 847–849. [Google Scholar] [CrossRef]
- Essner, E.; Novikoff, A.B. Localization of acid phosphatase activity in hepatic lysosomes by means of electron microscopy. J. Biophys Biochem. Cytol. 1961, 9, 773–784. [Google Scholar] [CrossRef]
- Straus, W. Isolation and biochemical properties of droplets from the cells of rat kidney. J. Biol. Chem. 1954, 207, 745–755. [Google Scholar]
- Mundel, P.; Shankland, S.J. Podocyte biology and response to injury. J. Am. Soc. Nephrol. 2002, 13, 3005–3015. [Google Scholar] [CrossRef] [Green Version]
- Pavenstadt, H.; Kriz, W.; Kretzler, M. Cell biology of the glomerular podocyte. Physiol. Rev. 2003, 83, 253–307. [Google Scholar] [CrossRef] [Green Version]
- Merscher, S.; Fornoni, A. Podocyte pathology and nephropathy - sphingolipids in glomerular diseases. Front. Endocrinol. (Lausanne) 2014, 5, 127. [Google Scholar] [CrossRef] [Green Version]
- Klionsky, D.J.; Emr, S.D. Autophagy as a regulated pathway of cellular degradation. Science 2000, 290, 1717–1721. [Google Scholar] [CrossRef]
- Stuart, L.M.; Ezekowitz, R.A. Phagocytosis: elegant complexity. Immunity 2005, 22, 539–550. [Google Scholar] [CrossRef] [Green Version]
- Huynh, C.; Roth, D.; Ward, D.M.; Kaplan, J.; Andrews, N.W. Defective lysosomal exocytosis and plasma membrane repair in Chediak-Higashi/beige cells. Proc. Natl. Acad. Sci. USA 2004, 101, 16795–16800. [Google Scholar] [CrossRef] [Green Version]
- Jaiswal, J.K.; Andrews, N.W.; Simon, S.M. Membrane proximal lysosomes are the major vesicles responsible for calcium-dependent exocytosis in nonsecretory cells. J. Cell Biol. 2002, 159, 625–635. [Google Scholar] [CrossRef] [Green Version]
- McNeil, P.L.; Kirchhausen, T. An emergency response team for membrane repair. Nat. Rev. Mol. Cell Biol. 2005, 6, 499–505. [Google Scholar] [CrossRef]
- Reddy, A.; Caler, E.V.; Andrews, N.W. Plasma membrane repair is mediated by Ca(2+)-regulated exocytosis of lysosomes. Cell 2001, 106, 157–169. [Google Scholar] [CrossRef] [Green Version]
- Bennett, E.M.; Lin, S.X.; Towler, M.C.; Maxfield, F.R.; Brodsky, F.M. Clathrin hub expression affects early endosome distribution with minimal impact on receptor sorting and recycling. Mol. Biol. Cell 2001, 12, 2790–2799. [Google Scholar] [CrossRef] [Green Version]
- Dunn, W.A., Jr. Autophagy and related mechanisms of lysosome-mediated protein degradation. Trends Cell Biol. 1994, 4, 139–143. [Google Scholar] [CrossRef]
- Luzio, J.P.; Pryor, P.R.; Bright, N.A. Lysosomes: Fusion and function. Nat. Rev. Mol. Cell Biol. 2007, 8, 622–632. [Google Scholar] [CrossRef]
- Mullins, C.; Bonifacino, J.S. The molecular machinery for lysosome biogenesis. Bioessays 2001, 23, 333–343. [Google Scholar] [CrossRef]
- Bao, J.X.; Jin, S.; Zhang, F.; Wang, Z.C.; Li, N.; Li, P.L. Activation of membrane NADPH oxidase associated with lysosome-targeted acid sphingomyelinase in coronary endothelial cells. Antioxid Redox Signal. 2010, 12, 703–712. [Google Scholar] [CrossRef] [Green Version]
- Dong, X.P.; Shen, D.; Wang, X.; Dawson, T.; Li, X.; Zhang, Q.; Cheng, X.; Zhang, Y.; Weisman, L.S.; Delling, M.; et al. PI(3,5)P(2) controls membrane trafficking by direct activation of mucolipin Ca(2+) release channels in the endolysosome. Nat. Commun. 2010, 1, 38. [Google Scholar] [CrossRef] [Green Version]
- Shen, D.; Wang, X.; Xu, H. Pairing phosphoinositides with calcium ions in endolysosomal dynamics: phosphoinositides control the direction and specificity of membrane trafficking by regulating the activity of calcium channels in the endolysosomes. Bioessays 2011, 33, 448–457. [Google Scholar] [CrossRef] [Green Version]
- Ward, D.M.; Pevsner, J.; Scullion, M.A.; Vaughn, M.; Kaplan, J. Syntaxin 7 and VAMP-7 are soluble N-ethylmaleimide-sensitive factor attachment protein receptors required for late endosome-lysosome and homotypic lysosome fusion in alveolar macrophages. Mol. Biol. Cell 2000, 11, 2327–2333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, W.B.; Deeter, C.; Gauthier, K.M.; Ingraham, R.H.; Falck, J.R.; Li, P.L. 14,15-Dihydroxyeicosatrienoic acid relaxes bovine coronary arteries by activation of K(Ca) channels. Am. J. Physiol. Heart Circ. Physiol. 2002, 282, H1656–H1664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Churchill, G.C.; Okada, Y.; Thomas, J.M.; Genazzani, A.A.; Patel, S.; Galione, A. NAADP mobilizes Ca(2+) from reserve granules, lysosome-related organelles, in sea urchin eggs. Cell 2002, 111, 703–708. [Google Scholar] [CrossRef] [Green Version]
- Dai, J.; Kuo, K.H.; Leo, J.M.; van Breemen, C.; Lee, C.H. Rearrangement of the close contact between the mitochondria and the sarcoplasmic reticulum in airway smooth muscle. Cell Calcium. 2005, 37, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Shen, D.; Wang, X.; Li, X.; Zhang, X.; Yao, Z.; Dibble, S.; Dong, X.P.; Yu, T.; Lieberman, A.P.; Showalter, H.D.; et al. Lipid storage disorders block lysosomal trafficking by inhibiting a TRP channel and lysosomal calcium release. Nat. Commun. 2012, 3, 731. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Li, C.X.; Xia, M.; Ritter, J.K.; Gehr, T.W.; Boini, K.; Li, P.L. Enhanced epithelial-to-mesenchymal transition associated with lysosome dysfunction in podocytes: role of p62/Sequestosome 1 as a signaling hub. Cell Physiol. Biochem. 2015, 35, 1773–1786. [Google Scholar] [CrossRef]
- Liu, W.J.; Li, Z.H.; Chen, X.C.; Zhao, X.L.; Zhong, Z.; Yang, C.; Wu, H.L.; An, N.; Li, W.Y.; Liu, H.F. Blockage of the lysosome-dependent autophagic pathway contributes to complement membrane attack complex-induced podocyte injury in idiopathic membranous nephropathy. Sci. Rep. 2017, 7, 8643. [Google Scholar] [CrossRef] [Green Version]
- Xiong, J.; Xia, M.; Xu, M.; Zhang, Y.; Abais, J.M.; Li, G.; Riebling, C.R.; Ritter, J.K.; Boini, K.M.; Li, P.L. Autophagy maturation associated with CD38-mediated regulation of lysosome function in mouse glomerular podocytes. J. Cell Mol. Med. 2013, 17, 1598–1607. [Google Scholar] [CrossRef]
- Fang, L.; Zhou, Y.; Cao, H.; Wen, P.; Jiang, L.; He, W.; Dai, C.; Yang, J. Autophagy attenuates diabetic glomerular damage through protection of hyperglycemia-induced podocyte injury. PLoS ONE 2013, 8, e60546. [Google Scholar] [CrossRef]
- Kang, Y.L.; Saleem, M.A.; Chan, K.W.; Yung, B.Y.; Law, H.K. The cytoprotective role of autophagy in puromycin aminonucleoside treated human podocytes. Biochem. Biophys Res. Commun. 2014, 443, 628–634. [Google Scholar] [CrossRef] [Green Version]
- Yi, M.; Zhang, L.; Liu, Y.; Livingston, M.J.; Chen, J.K.; Nahman, N.S., Jr.; Liu, F.; Dong, Z. Autophagy is activated to protect against podocyte injury in adriamycin-induced nephropathy. Am. J. Physiol. Renal. Physiol. 2017, 313, F74–F84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akchurin, O.; Reidy, K.J. Genetic causes of proteinuria and nephrotic syndrome: impact on podocyte pathobiology. Pediatr. Nephrol. 2015, 30, 221–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartleben, B.; Godel, M.; Meyer-Schwesinger, C.; Liu, S.; Ulrich, T.; Kobler, S.; Wiech, T.; Grahammer, F.; Arnold, S.J.; Lindenmeyer, M.T.; et al. Autophagy influences glomerular disease susceptibility and maintains podocyte homeostasis in aging mice. J. Clin. Invest. 2010, 120, 1084–1096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Li, Q.X.; Wang, X.J.; Zhang, C.; Duan, Y.Q.; Wang, Z.Y.; Zhang, Y.; Yu, X.; Li, N.J.; Sun, J.P.; et al. Beta-Arrestins promote podocyte injury by inhibition of autophagy in diabetic nephropathy. Cell Death Dis. 2016, 7, e2183. [Google Scholar] [CrossRef] [Green Version]
- Tagawa, A.; Yasuda, M.; Kume, S.; Yamahara, K.; Nakazawa, J.; Chin-Kanasaki, M.; Araki, H.; Araki, S.; Koya, D.; Asanuma, K.; et al. Impaired Podocyte Autophagy Exacerbates Proteinuria in Diabetic Nephropathy. Diabetes 2016, 65, 755–767. [Google Scholar] [CrossRef] [Green Version]
- Christensen, K.A.; Myers, J.T.; Swanson, J.A. pH-dependent regulation of lysosomal calcium in macrophages. J. Cell Sci. 2002, 115, 599–607. [Google Scholar]
- Ravikumar, B.; Sarkar, S.; Davies, J.E.; Futter, M.; Garcia-Arencibia, M.; Green-Thompson, Z.W.; Jimenez-Sanchez, M.; Korolchuk, V.I.; Lichtenberg, M.; Luo, S.; et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol. Rev. 2010, 90, 1383–1435. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Li, W.; Wen, J.; Yang, Z. Autophagy is involved in mouse kidney development and podocyte differentiation regulated by Notch signalling. J. Cell Mol. Med. 2017, 21, 1315–1328. [Google Scholar] [CrossRef]
- Wang, X.; Gao, Y.; Tian, N.; Wang, T.; Shi, Y.; Xu, J.; Wu, B. Astragaloside IV inhibits glucose-induced epithelial-mesenchymal transition of podocytes through autophagy enhancement via the SIRT-NF-kappaB p65 axis. Sci. Rep. 2019, 9, 323. [Google Scholar] [CrossRef] [Green Version]
- Kinnear, N.P.; Boittin, F.X.; Thomas, J.M.; Galione, A.; Evans, A.M. Lysosome-sarcoplasmic reticulum junctions. A trigger zone for calcium signaling by nicotinic acid adenine dinucleotide phosphate and endothelin-1. J. Biol. Chem. 2004, 279, 54319–54326. [Google Scholar] [CrossRef] [Green Version]
- Yamasaki, M.; Masgrau, R.; Morgan, A.J.; Churchill, G.C.; Patel, S.; Ashcroft, S.J.; Galione, A. Organelle selection determines agonist-specific Ca2+ signals in pancreatic acinar and beta cells. J. Biol. Chem. 2004, 279, 7234–7240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korolchuk, V.I.; Rubinsztein, D.C. Regulation of autophagy by lysosomal positioning. Autophagy 2011, 7, 927–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hollenbeck, P.J.; Swanson, J.A. Radial extension of macrophage tubular lysosomes supported by kinesin. Nature 1990, 346, 864–866. [Google Scholar] [CrossRef] [PubMed]
- Harada, A.; Takei, Y.; Kanai, Y.; Tanaka, Y.; Nonaka, S.; Hirokawa, N. Golgi vesiculation and lysosome dispersion in cells lacking cytoplasmic dynein. J. Cell Biol. 1998, 141, 51–59. [Google Scholar] [CrossRef] [Green Version]
- Heuser, J. Changes in lysosome shape and distribution correlated with changes in cytoplasmic pH. J. Cell Biol. 1989, 108, 855–864. [Google Scholar] [CrossRef] [Green Version]
- Pu, J.; Keren-Kaplan, T.; Bonifacino, J.S. A Ragulator-BORC interaction controls lysosome positioning in response to amino acid availability. J. Cell Biol. 2017, 216, 4183–4197. [Google Scholar] [CrossRef]
- Pu, J.; Guardia, C.M.; Keren-Kaplan, T.; Bonifacino, J.S. Mechanisms and functions of lysosome positioning. J. Cell Sci. 2016, 129, 4329–4339. [Google Scholar] [CrossRef] [Green Version]
- Futai, M.; Sun-Wada, G.H.; Wada, Y.; Matsumoto, N.; Nakanishi-Matsui, M. Vacuolar-type ATPase: A proton pump to lysosomal trafficking. Proc. Jpn Acad. Ser. B Phys. Biol. Sci. 2019, 95, 261–277. [Google Scholar] [CrossRef] [Green Version]
- Korolchuk, V.I.; Saiki, S.; Lichtenberg, M.; Siddiqi, F.H.; Roberts, E.A.; Imarisio, S.; Jahreiss, L.; Sarkar, S.; Futter, M.; Menzies, F.M.; et al. Lysosomal positioning coordinates cellular nutrient responses. Nat. Cell Biol. 2011, 13, 453–460. [Google Scholar] [CrossRef]
- Hong, Z.; Pedersen, N.M.; Wang, L.; Torgersen, M.L.; Stenmark, H.; Raiborg, C. PtdIns3P controls mTORC1 signaling through lysosomal positioning. J. Cell Biol. 2017, 216, 4217–4233. [Google Scholar] [CrossRef] [Green Version]
- Cinti, A.; Le Sage, V.; Milev, M.P.; Valiente-Echeverria, F.; Crossie, C.; Miron, M.J.; Pante, N.; Olivier, M.; Mouland, A.J. HIV-1 enhances mTORC1 activity and repositions lysosomes to the periphery by co-opting Rag GTPases. Sci. Rep. 2017, 7, 5515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, K.P.; Ho, M.Y.; Cho, H.C.; Yu, J.; Hung, J.T.; Yu, A.L. Fucosylation of LAMP-1 and LAMP-2 by FUT1 correlates with lysosomal positioning and autophagic flux of breast cancer cells. Cell Death Dis. 2016, 7, e2347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasuda, M.; Tanaka, Y.; Kume, S.; Morita, Y.; Chin-Kanasaki, M.; Araki, H.; Isshiki, K.; Araki, S.; Koya, D.; Haneda, M.; et al. Fatty acids are novel nutrient factors to regulate mTORC1 lysosomal localization and apoptosis in podocytes. Biochim. Biophys Acta 2014, 1842, 1097–1108. [Google Scholar] [CrossRef] [PubMed]
- Godel, M.; Hartleben, B.; Herbach, N.; Liu, S.; Zschiedrich, S.; Lu, S.; Debreczeni-Mor, A.; Lindenmeyer, M.T.; Rastaldi, M.P.; Hartleben, G.; et al. Role of mTOR in podocyte function and diabetic nephropathy in humans and mice. J. Clin. Invest. 2011, 121, 2197–2209. [Google Scholar] [CrossRef] [Green Version]
- Cina, D.P.; Onay, T.; Paltoo, A.; Li, C.; Maezawa, Y.; De Arteaga, J.; Jurisicova, A.; Quaggin, S.E. Inhibition of MTOR disrupts autophagic flux in podocytes. J. Am. Soc. Nephrol. 2012, 23, 412–420. [Google Scholar] [CrossRef] [Green Version]
- Bumbea, V.; Kamar, N.; Ribes, D.; Esposito, L.; Modesto, A.; Guitard, J.; Nasou, G.; Durand, D.; Rostaing, L. Long-term results in renal transplant patients with allograft dysfunction after switching from calcineurin inhibitors to sirolimus. Nephrol. Dial. Transplant. 2005, 20, 2517–2523. [Google Scholar] [CrossRef]
- Straathof-Galema, L.; Wetzels, J.F.; Dijkman, H.B.; Steenbergen, E.J.; Hilbrands, L.B. Sirolimus-associated heavy proteinuria in a renal transplant recipient: evidence for a tubular mechanism. Am. J. Transplant. 2006, 6, 429–433. [Google Scholar] [CrossRef]
- Fervenza, F.C.; Fitzpatrick, P.M.; Mertz, J.; Erickson, S.B.; Liggett, S.; Popham, S.; Wochos, D.N.; Synhavsky, A.; Hippler, S.; Larson, T.S.; et al. Acute rapamycin nephrotoxicity in native kidneys of patients with chronic glomerulopathies. Nephrol. Dial. Transplant. 2004, 19, 1288–1292. [Google Scholar] [CrossRef]
- Canaud, G.; Bienaime, F.; Viau, A.; Treins, C.; Baron, W.; Nguyen, C.; Burtin, M.; Berissi, S.; Giannakakis, K.; Muda, A.O.; et al. AKT2 is essential to maintain podocyte viability and function during chronic kidney disease. Nat. Med. 2013, 19, 1288–1296. [Google Scholar] [CrossRef]
- Reiser, J. Akt2 relaxes podocytes in chronic kidney disease. Nat. Med. 2013, 19, 1212–1213. [Google Scholar] [CrossRef]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy regulates lipid metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Evans, T.D.; Jeong, S.J.; Razani, B. Classical and alternative roles for autophagy in lipid metabolism. Curr. Opin. Lipidol. 2018, 29, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Kolter, T.; Sandhoff, K. Principles of lysosomal membrane digestion: stimulation of sphingolipid degradation by sphingolipid activator proteins and anionic lysosomal lipids. Annu. Rev. Cell Dev. Biol. 2005, 21, 81–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolter, T.; Sandhoff, K. Lysosomal degradation of membrane lipids. FEBS Lett. 2010, 584, 1700–1712. [Google Scholar] [CrossRef] [Green Version]
- Sonderfeld, S.; Conzelmann, E.; Schwarzmann, G.; Burg, J.; Hinrichs, U.; Sandhoff, K. Incorporation and metabolism of ganglioside GM2 in skin fibroblasts from normal and GM2 gangliosidosis subjects. Eur. J. Biochem. 1985, 149, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Riboni, L.; Bassi, R.; Prinetti, A.; Tettamanti, G. Salvage of catabolic products in ganglioside metabolism: a study on rat cerebellar granule cells in culture. FEBS Lett. 1996, 391, 336–340. [Google Scholar] [CrossRef] [Green Version]
- Tettamanti, G. Ganglioside/glycosphingolipid turnover: new concepts. Glycoconj. J. 2004, 20, 301–317. [Google Scholar] [CrossRef]
- Olivera, A.; Spiegel, S. Sphingosine-1-phosphate as second messenger in cell proliferation induced by PDGF and FCS mitogens. Nature 1993, 365, 557–560. [Google Scholar] [CrossRef]
- Kaipia, A.; Chun, S.Y.; Eisenhauer, K.; Hsueh, A.J. Tumor necrosis factor-alpha and its second messenger, ceramide, stimulate apoptosis in cultured ovarian follicles. Endocrinology 1996, 137, 4864–4870. [Google Scholar] [CrossRef]
- Merrill, A.H. Ceramide: a new lipid “second messenger”? Nutr. Rev. 1992, 50, 78–80. [Google Scholar] [CrossRef]
- Vanaja, S.K.; Rathinam, V.A.; Fitzgerald, K.A. Mechanisms of inflammasome activation: recent advances and novel insights. Trends Cell Biol. 2015, 25, 308–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saitoh, T.; Fujita, N.; Jang, M.H.; Uematsu, S.; Yang, B.G.; Satoh, T.; Omori, H.; Noda, T.; Yamamoto, N.; Komatsu, M.; et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature 2008, 456, 264–268. [Google Scholar] [CrossRef] [PubMed]
- Shibutani, S.T.; Saitoh, T.; Nowag, H.; Munz, C.; Yoshimori, T. Autophagy and autophagy-related proteins in the immune system. Nat. Immunol. 2015, 16, 1014–1024. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 2012, 36, 401–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, H.; Gris, D.; Lei, Y.; Jha, S.; Zhang, L.; Huang, M.T.; Brickey, W.J.; Ting, J.P. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat. Immunol. 2011, 12, 408–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lupfer, C.; Thomas, P.G.; Anand, P.K.; Vogel, P.; Milasta, S.; Martinez, J.; Huang, G.; Green, M.; Kundu, M.; Chi, H.; et al. Receptor interacting protein kinase 2-mediated mitophagy regulates inflammasome activation during virus infection. Nat. Immunol. 2013, 14, 480–488. [Google Scholar] [CrossRef] [Green Version]
- Shi, C.S.; Shenderov, K.; Huang, N.N.; Kabat, J.; Abu-Asab, M.; Fitzgerald, K.A.; Sher, A.; Kehrl, J.H. Activation of autophagy by inflammatory signals limits IL-1beta production by targeting ubiquitinated inflammasomes for destruction. Nat. Immunol. 2012, 13, 255–263. [Google Scholar] [CrossRef]
- Harris, J.; Hartman, M.; Roche, C.; Zeng, S.G.; O’Shea, A.; Sharp, F.A.; Lambe, E.M.; Creagh, E.M.; Golenbock, D.T.; Tschopp, J.; et al. Autophagy controls IL-1beta secretion by targeting pro-IL-1beta for degradation. J. Biol. Chem. 2011, 286, 9587–9597. [Google Scholar] [CrossRef] [Green Version]
- Hessvik, N.P.; Llorente, A. Current knowledge on exosome biogenesis and release. Cell Mol. Life Sci. 2018, 75, 193–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruno, S.; Porta, S.; Bussolati, B. Extracellular vesicles in renal tissue damage and regeneration. Eur. J. Pharmacol. 2016, 790, 83–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erdbrugger, U.; Le, T.H. Extracellular Vesicles in Renal Diseases: More than Novel Biomarkers? J. Am. Soc. Nephrol. 2016, 27, 12–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pomatto, M.A.C.; Gai, C.; Bussolati, B.; Camussi, G. Extracellular Vesicles in Renal Pathophysiology. Front. Mol. Biosci. 2017, 4, 37. [Google Scholar] [CrossRef]
- Alvarez-Erviti, L.; Seow, Y.; Schapira, A.H.; Gardiner, C.; Sargent, I.L.; Wood, M.J.; Cooper, J.M. Lysosomal dysfunction increases exosome-mediated alpha-synuclein release and transmission. Neurobiol. Dis. 2011, 42, 360–367. [Google Scholar] [CrossRef] [Green Version]
- Vingtdeux, V.; Hamdane, M.; Loyens, A.; Gele, P.; Drobeck, H.; Begard, S.; Galas, M.C.; Delacourte, A.; Beauvillain, J.C.; Buee, L.; et al. Alkalizing drugs induce accumulation of amyloid precursor protein by-products in luminal vesicles of multivesicular bodies. J. Biol. Chem. 2007, 282, 18197–18205. [Google Scholar] [CrossRef] [Green Version]
- Wei, Y.M.; Li, X.; Xu, M.; Abais, J.M.; Chen, Y.; Riebling, C.R.; Boini, K.M.; Li, P.L.; Zhang, Y. Enhancement of autophagy by simvastatin through inhibition of Rac1-mTOR signaling pathway in coronary arterial myocytes. Cell Physiol. Biochem. 2013, 31, 925–937. [Google Scholar] [CrossRef]
- Baixauli, F.; Lopez-Otin, C.; Mittelbrunn, M. Exosomes and autophagy: coordinated mechanisms for the maintenance of cellular fitness. Front. Immunol. 2014, 5, 403. [Google Scholar] [CrossRef] [Green Version]
- Fader, C.M.; Sanchez, D.; Furlan, M.; Colombo, M.I. Induction of autophagy promotes fusion of multivesicular bodies with autophagic vacuoles in k562 cells. Traffic 2008, 9, 230–250. [Google Scholar] [CrossRef]
- Murrow, L.; Debnath, J. ATG12-ATG3 connects basal autophagy and late endosome function. Autophagy 2015, 11, 961–962. [Google Scholar] [CrossRef] [Green Version]
- Hara, M.; Yamagata, K.; Tomino, Y.; Saito, A.; Hirayama, Y.; Ogasawara, S.; Kurosawa, H.; Sekine, S.; Yan, K. Urinary podocalyxin is an early marker for podocyte injury in patients with diabetes: establishment of a highly sensitive ELISA to detect urinary podocalyxin. Diabetologia 2012, 55, 2913–2919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stahl, A.L.; Johansson, K.; Mossberg, M.; Kahn, R.; Karpman, D. Exosomes and microvesicles in normal physiology, pathophysiology, and renal diseases. Pediatr. Nephrol. 2019, 34, 11–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hara, M.; Yanagihara, T.; Kihara, I.; Higashi, K.; Fujimoto, K.; Kajita, T. Apical cell membranes are shed into urine from injured podocytes: a novel phenomenon of podocyte injury. J. Am. Soc. Nephrol. 2005, 16, 408–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.; Han, K.H.; Lee, S.E.; Kim, S.H.; Kang, H.G.; Cheong, H.I. Urinary exosomal WT1 in childhood nephrotic syndrome. Pediatr. Nephrol. 2012, 27, 317–320. [Google Scholar] [CrossRef] [PubMed]
- Lytvyn, Y.; Xiao, F.; Kennedy, C.R.; Perkins, B.A.; Reich, H.N.; Scholey, J.W.; Cherney, D.Z.; Burger, D. Assessment of urinary microparticles in normotensive patients with type 1 diabetes. Diabetologia 2017, 60, 581–584. [Google Scholar] [CrossRef] [Green Version]
- Tkaczyk, M.; Baj, Z. Surface markers of platelet function in idiopathic nephrotic syndrome in children. Pediatr. Nephrol. 2002, 17, 673–677. [Google Scholar] [CrossRef]
- Wu, X.; Gao, Y.; Xu, L.; Dang, W.; Yan, H.; Zou, D.; Zhu, Z.; Luo, L.; Tian, N.; Wang, X.; et al. Exosomes from high glucose-treated glomerular endothelial cells trigger the epithelial-mesenchymal transition and dysfunction of podocytes. Sci. Rep. 2017, 7, 9371. [Google Scholar] [CrossRef] [Green Version]
- Castrop, H.; Schiessl, I.M. Novel routes of albumin passage across the glomerular filtration barrier. Acta Physiol. (Oxf) 2017, 219, 544–553. [Google Scholar] [CrossRef]
- Bao, J.X.; Zhang, Q.F.; Wang, M.; Xia, M.; Boini, K.M.; Gulbins, E.; Zhang, Y.; Li, P.L. Implication of CD38 gene in autophagic degradation of collagen I in mouse coronary arterial myocytes. Front. Biosci. (Landmark Ed.) 2017, 22, 558–569. [Google Scholar]
- Xu, M.; Li, X.; Walsh, S.W.; Zhang, Y.; Abais, J.M.; Boini, K.M.; Li, P.L. Intracellular two-phase Ca2+ release and apoptosis controlled by TRP-ML1 channel activity in coronary arterial myocytes. Am. J. Physiol. Cell Physiol. 2013, 304, C458–C466. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Xu, M.; Han, W.Q.; Li, P.L. Reconstitution of lysosomal NAADP-TRP-ML1 signaling pathway and its function in TRP-ML1(-/-) cells. Am. J. Physiol. Cell Physiol. 2011, 301, C421–C430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, M.; Goldin, E.; Stahl, S.; Falardeau, J.L.; Kennedy, J.C.; Acierno, J.S., Jr.; Bove, C.; Kaneski, C.R.; Nagle, J.; Bromley, M.C.; et al. Mucolipidosis type IV is caused by mutations in a gene encoding a novel transient receptor potential channel. Hum. Mol. Genet. 2000, 9, 2471–2478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galione, A. NAADP, a new intracellular messenger that mobilizes Ca2+ from acidic stores. Biochem. Soc. Trans. 2006, 34, 922–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, F.; Xia, M.; Li, P.L. Lysosome-dependent Ca(2+) release response to Fas activation in coronary arterial myocytes through NAADP: evidence from CD38 gene knockouts. Am. J. Physiol. Cell Physiol. 2010, 298, C1209–C1216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, F.; Zhang, G.; Zhang, A.Y.; Koeberl, M.J.; Wallander, E.; Li, P.L. Production of NAADP and its role in Ca2+ mobilization associated with lysosomes in coronary arterial myocytes. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H274–H282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, X.P.; Cheng, X.; Mills, E.; Delling, M.; Wang, F.; Kurz, T.; Xu, H. The type IV mucolipidosis-associated protein TRPML1 is an endolysosomal iron release channel. Nature 2008, 455, 992–996. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Rydzewski, N.; Hider, A.; Zhang, X.; Yang, J.; Wang, W.; Gao, Q.; Cheng, X.; Xu, H. A molecular mechanism to regulate lysosome motility for lysosome positioning and tubulation. Nat. Cell Biol. 2016, 18, 404–417. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Ouyang, J.; Li, S.; Wang, H.; Lian, B.; Liu, Z.; Xie, L. The analysis of risk factors for diabetic nephropathy progression and the construction of a prognostic database for chronic kidney diseases. J. Transl. Med. 2019, 17, 264. [Google Scholar] [CrossRef]
- Kim, W.Y.; Nam, S.A.; Song, H.C.; Ko, J.S.; Park, S.H.; Kim, H.L.; Choi, E.J.; Kim, Y.S.; Kim, J.; Kim, Y.K. The role of autophagy in unilateral ureteral obstruction rat model. Nephrology (Carlton) 2012, 17, 148–159. [Google Scholar] [CrossRef]
- Wang, Z.; Choi, M.E. Autophagy in kidney health and disease. Antioxid Redox Signal. 2014, 20, 519–537. [Google Scholar] [CrossRef] [Green Version]
- Wullschleger, S.; Loewith, R.; Hall, M.N. TOR signaling in growth and metabolism. Cell 2006, 124, 471–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sancak, Y.; Peterson, T.R.; Shaul, Y.D.; Lindquist, R.A.; Thoreen, C.C.; Bar-Peled, L.; Sabatini, D.M. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 2008, 320, 1496–1501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.J.; Feliers, D.; Mariappan, M.M.; Sataranatarajan, K.; Mahimainathan, L.; Musi, N.; Foretz, M.; Viollet, B.; Weinberg, J.M.; Choudhury, G.G.; et al. A role for AMP-activated protein kinase in diabetes-induced renal hypertrophy. Am. J. Physiol. Renal Physiol. 2007, 292, F617–F627. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K. Role of TSC-mTOR pathway in diabetic nephropathy. Diabetes Res. Clin. Pract 2008, 82 Suppl. 1, S59–S62. [Google Scholar] [CrossRef]
- Kim, D.K.; Nam, B.Y.; Li, J.J.; Park, J.T.; Lee, S.H.; Kim, D.H.; Kim, J.Y.; Kang, H.Y.; Han, S.H.; Yoo, T.H.; et al. Translationally controlled tumour protein is associated with podocyte hypertrophy in a mouse model of type 1 diabetes. Diabetologia 2012, 55, 1205–1217. [Google Scholar] [CrossRef] [Green Version]
- Guzman, J.; Jauregui, A.N.; Merscher-Gomez, S.; Maiguel, D.; Muresan, C.; Mitrofanova, A.; Diez-Sampedro, A.; Szust, J.; Yoo, T.H.; Villarreal, R.; et al. Podocyte-specific GLUT4-deficient mice have fewer and larger podocytes and are protected from diabetic nephropathy. Diabetes 2014, 63, 701–714. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Koh, A.; Jeong, H.; Kim, E.; Ha, T.S.; Saleem, M.A.; Ryu, S.H. C1-Ten is a PTPase of nephrin, regulating podocyte hypertrophy through mTORC1 activation. Sci. Rep. 2017, 7, 12346. [Google Scholar] [CrossRef] [Green Version]
- Ma, T.; Zhu, J.; Chen, X.; Zha, D.; Singhal, P.C.; Ding, G. High glucose induces autophagy in podocytes. Exp. Cell Res. 2013, 319, 779–789. [Google Scholar] [CrossRef] [Green Version]
- Azad, M.B.; Chen, Y.; Gibson, S.B. Regulation of autophagy by reactive oxygen species (ROS): implications for cancer progression and treatment. Antioxid Redox Signal. 2009, 11, 777–790. [Google Scholar] [CrossRef]
- Inoki, K.; Mori, H.; Wang, J.; Suzuki, T.; Hong, S.; Yoshida, S.; Blattner, S.M.; Ikenoue, T.; Ruegg, M.A.; Hall, M.N.; et al. mTORC1 activation in podocytes is a critical step in the development of diabetic nephropathy in mice. J. Clin. Invest. 2011, 121, 2181–2196. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Han, F.; Yang, Y.; Xie, Y.; Jiang, H.; Mao, Y.; Wang, H.; Wang, M.; Chen, R.; Yang, J.; et al. Evaluation of sphingolipid metabolism in renal cortex of rats with streptozotocin-induced diabetes and the effects of rapamycin. Nephrol. Dial. Transplant. 2011, 26, 1493–1502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, S.R.; Lim, J.H.; Kim, M.Y.; Kim, E.N.; Kim, Y.; Choi, B.S.; Kim, Y.S.; Kim, H.W.; Lim, K.M.; Kim, M.J.; et al. Adiponectin receptor agonist AdipoRon decreased ceramide, and lipotoxicity, and ameliorated diabetic nephropathy. Metabolism 2018, 85, 348–360. [Google Scholar] [CrossRef] [PubMed]
- Morita, Y.; Kurano, M.; Sakai, E.; Nishikawa, T.; Nishikawa, M.; Sawabe, M.; Aoki, J.; Yatomi, Y. Analysis of urinary sphingolipids using liquid chromatography-tandem mass spectrometry in diabetic nephropathy. J. Diabetes Investig. 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Gomez-Sintes, R.; Boya, P. Lysosomal membrane permeabilization and cell death. Traffic 2018, 19, 918–931. [Google Scholar] [CrossRef] [PubMed]
- Boya, P. Lysosomal function and dysfunction: mechanism and disease. Antioxid Redox Signal. 2012, 17, 766–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.J.; Gan, Y.; Huang, W.F.; Wu, H.L.; Zhang, X.Q.; Zheng, H.J.; Liu, H.F. Lysosome restoration to activate podocyte autophagy: a new therapeutic strategy for diabetic kidney disease. Cell Death Dis. 2019, 10, 806. [Google Scholar] [CrossRef] [Green Version]
- Adler, S.; Baker, P.J.; Johnson, R.J.; Ochi, R.F.; Pritzl, P.; Couser, W.G. Complement membrane attack complex stimulates production of reactive oxygen metabolites by cultured rat mesangial cells. J. Clin. Invest. 1986, 77, 762–767. [Google Scholar] [CrossRef]
- Zhou, L.L.; Cao, W.; Xie, C.; Tian, J.; Zhou, Z.; Zhou, Q.; Zhu, P.; Li, A.; Liu, Y.; Miyata, T.; et al. The receptor of advanced glycation end products plays a central role in advanced oxidation protein products-induced podocyte apoptosis. Kidney Int. 2012, 82, 759–770. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.J.; Shen, T.T.; Chen, R.H.; Wu, H.L.; Wang, Y.J.; Deng, J.K.; Chen, Q.H.; Pan, Q.; Huang Fu, C.M.; Tao, J.L.; et al. Autophagy-Lysosome Pathway in Renal Tubular Epithelial Cells Is Disrupted by Advanced Glycation End Products in Diabetic Nephropathy. J. Biol. Chem. 2015, 290, 20499–20510. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.J.; Xu, B.H.; Ye, L.; Liang, D.; Wu, H.L.; Zheng, Y.Y.; Deng, J.K.; Li, B.; Liu, H.F. Urinary proteins induce lysosomal membrane permeabilization and lysosomal dysfunction in renal tubular epithelial cells. Am. J. Physiol. Renal. Physiol. 2015, 308, F639–F649. [Google Scholar] [CrossRef] [Green Version]
- Johansson, A.C.; Appelqvist, H.; Nilsson, C.; Kagedal, K.; Roberg, K.; Ollinger, K. Regulation of apoptosis-associated lysosomal membrane permeabilization. Apoptosis 2010, 15, 527–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garsen, M.; Rops, A.L.; Dijkman, H.; Willemsen, B.; van Kuppevelt, T.H.; Russel, F.G.; Rabelink, T.J.; Berden, J.H.; Reinheckel, T.; van der Vlag, J. Cathepsin L is crucial for the development of early experimental diabetic nephropathy. Kidney Int. 2016, 90, 1012–1022. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, K.; Miyazawa, T.; Enya, T.; Miyazaki, K.; Okada, M.; Takemura, T. Cyclosporine A induced histological changes of Cathepsin L and CD2AP expression in renal glomeruli and tubules. Clin. Exp. Nephrol. 2017, 21, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Yaddanapudi, S.; Altintas, M.M.; Kistler, A.D.; Fernandez, I.; Moller, C.C.; Wei, C.; Peev, V.; Flesche, J.B.; Forst, A.L.; Li, J.; et al. CD2AP in mouse and human podocytes controls a proteolytic program that regulates cytoskeletal structure and cellular survival. J. Clin. Invest. 2011, 121, 3965–3980. [Google Scholar] [CrossRef] [Green Version]
- Sever, S.; Altintas, M.M.; Nankoe, S.R.; Moller, C.C.; Ko, D.; Wei, C.; Henderson, J.; del Re, E.C.; Hsing, L.; Erickson, A.; et al. Proteolytic processing of dynamin by cytoplasmic cathepsin L is a mechanism for proteinuric kidney disease. J. Clin. Invest. 2007, 117, 2095–2104. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Wei, Y.; Wang, X.; He, R. D-Ribosylated Tau forms globular aggregates with high cytotoxicity. Cell Mol. Life Sci. 2009, 66, 2559–2571. [Google Scholar] [CrossRef]
- Wei, Y.; Chen, L.; Chen, J.; Ge, L.; He, R.Q. Rapid glycation with D-ribose induces globular amyloid-like aggregations of BSA with high cytotoxicity to SH-SY5Y cells. BMC Cell Biol. 2009, 10, 10. [Google Scholar] [CrossRef] [Green Version]
- Hong, J.; Li, G.; Zhang, Q.; Ritter, J.; Li, W.; Li, P.L. D-Ribose Induces Podocyte NLRP3 Inflammasome Activation and Glomerular Injury via AGEs/RAGE Pathway. Front. Cell Dev. Biol. 2019, 7, 259. [Google Scholar] [CrossRef]
- Hong, J.; Bhat, O.M.; Li, G.; Dempsey, S.K.; Zhang, Q.; Ritter, J.K.; Li, W.; Li, P.L. Lysosomal regulation of extracellular vesicle excretion during d-ribose-induced NLRP3 inflammasome activation in podocytes. Biochim. Biophys Acta Mol. Cell Res. 2019, 1866, 849–860. [Google Scholar] [CrossRef]
- Nance, C.S.; Klein, C.J.; Banikazemi, M.; Dikman, S.H.; Phelps, R.G.; McArthur, J.C.; Rodriguez, M.; Desnick, R.J. Later-onset Fabry disease: an adult variant presenting with the cramp-fasciculation syndrome. Arch. Neurol. 2006, 63, 453–457. [Google Scholar] [CrossRef] [Green Version]
- Kruger, R.; Bruns, K.; Grunhage, S.; Rossmann, H.; Reinke, J.; Beck, M.; Lackner, K.J. Determination of globotriaosylceramide in plasma and urine by mass spectrometry. Clin. Chem. Lab. Med. 2010, 48, 189–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, E.; Mills, K.; Morris, P.; Vellodi, A.; Lee, P.; Waldek, S.; Winchester, B. Is globotriaosylceramide a useful biomarker in Fabry disease? Acta Paediatr. Suppl. 2005, 94, 51–54, discussion 37–58. [Google Scholar] [CrossRef] [PubMed]
- Askari, H.; Kaneski, C.R.; Semino-Mora, C.; Desai, P.; Ang, A.; Kleiner, D.E.; Perlee, L.T.; Quezado, M.; Spollen, L.E.; Wustman, B.A.; et al. Cellular and tissue localization of globotriaosylceramide in Fabry disease. Virchows Arch. 2007, 451, 823–834. [Google Scholar] [CrossRef] [PubMed]
- Alroy, J.; Sabnis, S.; Kopp, J.B. Renal pathology in Fabry disease. J. Am. Soc. Nephrol. 2002, 13 Suppl. 2, S134–S138. [Google Scholar] [CrossRef]
- Liebau, M.C.; Braun, F.; Hopker, K.; Weitbrecht, C.; Bartels, V.; Muller, R.U.; Brodesser, S.; Saleem, M.A.; Benzing, T.; Schermer, B.; et al. Dysregulated autophagy contributes to podocyte damage in Fabry’s disease. PLoS ONE 2013, 8, e63506. [Google Scholar] [CrossRef] [Green Version]
- Thurberg, B.L.; Rennke, H.; Colvin, R.B.; Dikman, S.; Gordon, R.E.; Collins, A.B.; Desnick, R.J.; O’Callaghan, M. Globotriaosylceramide accumulation in the Fabry kidney is cleared from multiple cell types after enzyme replacement therapy. Kidney Int. 2002, 62, 1933–1946. [Google Scholar] [CrossRef] [Green Version]
- Prabakaran, T.; Nielsen, R.; Larsen, J.V.; Sorensen, S.S.; Feldt-Rasmussen, U.; Saleem, M.A.; Petersen, C.M.; Verroust, P.J.; Christensen, E.I. Receptor-mediated endocytosis of alpha-galactosidase A in human podocytes in Fabry disease. PLoS ONE 2011, 6, e25065. [Google Scholar] [CrossRef] [Green Version]
- Reidy, K.; Kang, H.M.; Hostetter, T.; Susztak, K. Molecular mechanisms of diabetic kidney disease. J. Clin. Invest. 2014, 124, 2333–2340. [Google Scholar] [CrossRef]
- Ingram, A.J.; Krepinsky, J.C.; James, L.; Austin, R.C.; Tang, D.; Salapatek, A.M.; Thai, K.; Scholey, J.W. Activation of mesangial cell MAPK in response to homocysteine. Kidney Int. 2004, 66, 733–745. [Google Scholar] [CrossRef] [Green Version]
- Yamasaki, K.; Muto, J.; Taylor, K.R.; Cogen, A.L.; Audish, D.; Bertin, J.; Grant, E.P.; Coyle, A.J.; Misaghi, A.; Hoffman, H.M.; et al. NLRP3/cryopyrin is necessary for interleukin-1beta (IL-1beta) release in response to hyaluronan, an endogenous trigger of inflammation in response to injury. J. Biol. Chem. 2009, 284, 12762–12771. [Google Scholar] [CrossRef] [Green Version]
- Yi, F.; Zhang, A.Y.; Janscha, J.L.; Li, P.L.; Zou, A.P. Homocysteine activates NADH/NADPH oxidase through ceramide-stimulated Rac GTPase activity in rat mesangial cells. Kidney Int. 2004, 66, 1977–1987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, F.; Zhang, A.Y.; Li, N.; Muh, R.W.; Fillet, M.; Renert, A.F.; Li, P.L. Inhibition of ceramide-redox signaling pathway blocks glomerular injury in hyperhomocysteinemic rats. Kidney Int. 2006, 70, 88–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, F.; Chen, Q.Z.; Jin, S.; Li, P.L. Mechanism of homocysteine-induced Rac1/NADPH oxidase activation in mesangial cells: role of guanine nucleotide exchange factor Vav2. Cell Physiol. Biochem. 2007, 20, 909–918. [Google Scholar] [CrossRef] [PubMed]
- Boini, K.M.; Xia, M.; Li, C.; Zhang, C.; Payne, L.P.; Abais, J.M.; Poklis, J.L.; Hylemon, P.B.; Li, P.L. Acid sphingomyelinase gene deficiency ameliorates the hyperhomocysteinemia-induced glomerular injury in mice. Am. J. Pathol. 2011, 179, 2210–2219. [Google Scholar] [CrossRef] [PubMed]
- Boini, K.M.; Xia, M.; Abais, J.M.; Xu, M.; Li, C.X.; Li, P.L. Acid sphingomyelinase gene knockout ameliorates hyperhomocysteinemic glomerular injury in mice lacking cystathionine-beta-synthase. PLoS ONE 2012, 7, e45020. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Boini, K.M.; Xia, M.; Abais, J.M.; Li, X.; Liu, Q.; Li, P.L. Activation of Nod-like receptor protein 3 inflammasomes turns on podocyte injury and glomerular sclerosis in hyperhomocysteinemia. Hypertension 2012, 60, 154–162. [Google Scholar] [CrossRef] [PubMed]
- Abais, J.M.; Zhang, C.; Xia, M.; Liu, Q.; Gehr, T.W.; Boini, K.M.; Li, P.L. NADPH oxidase-mediated triggering of inflammasome activation in mouse podocytes and glomeruli during hyperhomocysteinemia. Antioxid Redox Signal. 2013, 18, 1537–1548. [Google Scholar] [CrossRef] [Green Version]
- Abais, J.M.; Xia, M.; Li, G.; Gehr, T.W.; Boini, K.M.; Li, P.L. Contribution of endogenously produced reactive oxygen species to the activation of podocyte NLRP3 inflammasomes in hyperhomocysteinemia. Free Radic. Biol. Med. 2014, 67, 211–220. [Google Scholar] [CrossRef] [Green Version]
- Boini, K.M.; Xia, M.; Koka, S.; Gehr, T.W.; Li, P.L. Instigation of NLRP3 inflammasome activation and glomerular injury in mice on the high fat diet: role of acid sphingomyelinase gene. Oncotarget 2016, 7, 19031–19044. [Google Scholar] [CrossRef] [Green Version]
- Boini, K.M.; Zhang, C.; Xia, M.; Han, W.Q.; Brimson, C.; Poklis, J.L.; Li, P.L. Visfatin-induced lipid raft redox signaling platforms and dysfunction in glomerular endothelial cells. Biochim. Biophys Acta 2010, 1801, 1294–1304. [Google Scholar] [CrossRef] [Green Version]
- Hall, J.E.; Henegar, J.R.; Dwyer, T.M.; Liu, J.; Da Silva, A.A.; Kuo, J.J.; Tallam, L. Is obesity a major cause of chronic kidney disease? Adv. Ren Replace Ther. 2004, 11, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Axelsson, J.; Heimburger, O.; Stenvinkel, P. Adipose tissue and inflammation in chronic kidney disease. Contrib. Nephrol. 2006, 151, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Ouchi, N.; Walsh, K. Adiponectin as an anti-inflammatory factor. Clin. Chim. Acta. 2007, 380, 24–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Yan, W.J.; Zhang, J.L.; Zhang, F.Y.; Gao, C.; Wang, Y.J.; Bond Law, W.; Tao, L. Adiponectin partially rescues high glucose/high fat-induced impairment of mitochondrial biogenesis and function in a PGC-1alpha dependent manner. Eur Rev. Med. Pharmacol. Sci. 2017, 21, 590–599. [Google Scholar]
- He, Y.; Zou, L.; Zhou, Y.; Hu, H.; Yao, R.; Jiang, Y.; Lau, W.B.; Yuan, T.; Huang, W.; Zeng, Z.; et al. Adiponectin ameliorates the apoptotic effects of paraquat on alveolar type cells via improvements in mitochondrial function. Mol. Med. Rep. 2016, 14, 746–752. [Google Scholar] [CrossRef] [Green Version]
- Lara-Castro, C.; Fu, Y.; Chung, B.H.; Garvey, W.T. Adiponectin and the metabolic syndrome: mechanisms mediating risk for metabolic and cardiovascular disease. Curr. Opin. Lipidol. 2007, 18, 263–270. [Google Scholar] [CrossRef]
- Fantuzzi, G. Adiponectin and inflammation: consensus and controversy. J. Allergy Clin. Immunol. 2008, 121, 326–330. [Google Scholar] [CrossRef]
- Li, G.; Zhang, Q.; Hong, J.; Ritter, J.K.; Li, P.L. Inhibition of pannexin-1 channel activity by adiponectin in podocytes: Role of acid ceramidase activation. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2018, 1863, 1246–1256. [Google Scholar] [CrossRef]
- Davis, S.; Nehus, E.; Inge, T.; Zhang, W.; Setchell, K.; Mitsnefes, M. Effect of bariatric surgery on urinary sphingolipids in adolescents with severe obesity. Surg. Obes. Relat Dis. 2018, 14, 446–451. [Google Scholar] [CrossRef]
- Wang, G.; Dinkins, M.; He, Q.; Zhu, G.; Poirier, C.; Campbell, A.; Mayer-Proschel, M.; Bieberich, E. Astrocytes secrete exosomes enriched with proapoptotic ceramide and prostate apoptosis response 4 (PAR-4): potential mechanism of apoptosis induction in Alzheimer disease (AD). J. Biol. Chem. 2012, 287, 21384–21395. [Google Scholar] [CrossRef] [Green Version]
- Kakazu, E.; Mauer, A.S.; Yin, M.; Malhi, H. Hepatocytes release ceramide-enriched pro-inflammatory extracellular vesicles in an IRE1alpha-dependent manner. J. Lipid Res. 2016, 57, 233–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Podbielska, M.; Szulc, Z.M.; Kurowska, E.; Hogan, E.L.; Bielawski, J.; Bielawska, A.; Bhat, N.R. Cytokine-induced release of ceramide-enriched exosomes as a mediator of cell death signaling in an oligodendroglioma cell line. J. Lipid Res. 2016, 57, 2028–2039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, G.; Huang, D.; Hong, J.; Bhat, O.M.; Yuan, X.; Li, P.L. Control of Lysosomal TRPML1 Channel Activity and Exosome Release by Acid Ceramidase in Mouse Podocytes. Am. J. Physiol. Cell Physiol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Wang, B.; Li, H.; Ling, L.; Niu, J.; Gu, Y. Glucagon-like peptide-1 analog prevents obesity-related glomerulopathy by inhibiting excessive autophagy in podocytes. Am. J. Physiol. Renal. Physiol. 2018, 314, F181–F189. [Google Scholar] [CrossRef]
- Eid, S.; Boutary, S.; Braych, K.; Sabra, R.; Massaad, C.; Hamdy, A.; Rashid, A.; Moodad, S.; Block, K.; Gorin, Y.; et al. mTORC2 Signaling Regulates Nox4-Induced Podocyte Depletion in Diabetes. Antioxid Redox. Signal. 2016, 25, 703–719. [Google Scholar] [CrossRef] [Green Version]
- Dunham, I.; Shimizu, N.; Roe, B.A.; Chissoe, S.; Hunt, A.R.; Collins, J.E.; Bruskiewich, R.; Beare, D.M.; Clamp, M.; Smink, L.J.; et al. The DNA sequence of human chromosome 22. Nature 1999, 402, 489–495. [Google Scholar] [CrossRef]
- Foster, M.C.; Coresh, J.; Fornage, M.; Astor, B.C.; Grams, M.; Franceschini, N.; Boerwinkle, E.; Parekh, R.S.; Kao, W.H. APOL1 variants associate with increased risk of CKD among African Americans. J. Am. Soc. Nephrol. 2013, 24, 1484–1491. [Google Scholar] [CrossRef] [Green Version]
- Friedman, D.J.; Kozlitina, J.; Genovese, G.; Jog, P.; Pollak, M.R. Population-based risk assessment of APOL1 on renal disease. J. Am. Soc. Nephrol. 2011, 22, 2098–2105. [Google Scholar] [CrossRef] [Green Version]
- Genovese, G.; Friedman, D.J.; Ross, M.D.; Lecordier, L.; Uzureau, P.; Freedman, B.I.; Bowden, D.W.; Langefeld, C.D.; Oleksyk, T.K.; Uscinski Knob, A.L.; et al. Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science 2010, 329, 841–845. [Google Scholar] [CrossRef] [Green Version]
- Tzur, S.; Rosset, S.; Shemer, R.; Yudkovsky, G.; Selig, S.; Tarekegn, A.; Bekele, E.; Bradman, N.; Wasser, W.G.; Behar, D.M.; et al. Missense mutations in the APOL1 gene are highly associated with end stage kidney disease risk previously attributed to the MYH9 gene. Human Genet. 2010, 128, 345–350. [Google Scholar] [CrossRef] [Green Version]
- Kopp, J.B.; Nelson, G.W.; Sampath, K.; Johnson, R.C.; Genovese, G.; An, P.; Friedman, D.; Briggs, W.; Dart, R.; Korbet, S.; et al. APOL1 genetic variants in focal segmental glomerulosclerosis and HIV-associated nephropathy. J. Am. Soc. Nephrol. 2011, 22, 2129–2137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quaggin, S.E.; George, A.L., Jr. Apolipoprotein l1 and the genetic basis for racial disparity in chronic kidney disease. J. Am. Soc. Nephrol. 2011, 22, 1955–1958. [Google Scholar] [CrossRef] [PubMed]
- Genovese, G.; Friedman, D.J.; Pollak, M.R. APOL1 variants and kidney disease in people of recent African ancestry. Nat. Rev. Nephrol. 2013, 9, 240–244. [Google Scholar] [CrossRef] [PubMed]
- Wasser, W.G.; Tzur, S.; Wolday, D.; Adu, D.; Baumstein, D.; Rosset, S.; Skorecki, K. Population genetics of chronic kidney disease: the evolving story of APOL1. J. Nephrol. 2012, 25, 603–618. [Google Scholar] [CrossRef]
- Madhavan, S.M.; O’Toole, J.F.; Konieczkowski, M.; Ganesan, S.; Bruggeman, L.A.; Sedor, J.R. APOL1 localization in normal kidney and nondiabetic kidney disease. J. Am. Soc. Nephrol. 2011, 22, 2119–2128. [Google Scholar] [CrossRef] [Green Version]
- Lan, X.; Jhaveri, A.; Cheng, K.; Wen, H.; Saleem, M.A.; Mathieson, P.W.; Mikulak, J.; Aviram, S.; Malhotra, A.; Skorecki, K.; et al. APOL1 risk variants enhance podocyte necrosis through compromising lysosomal membrane permeability. Am. J. Physiol. Renal. Physiol. 2014, 307, F326–F336. [Google Scholar] [CrossRef] [Green Version]
- Perez-Morga, D.; Vanhollebeke, B.; Paturiaux-Hanocq, F.; Nolan, D.P.; Lins, L.; Homble, F.; Vanhamme, L.; Tebabi, P.; Pays, A.; Poelvoorde, P.; et al. Apolipoprotein L-I promotes trypanosome lysis by forming pores in lysosomal membranes. Science 2005, 309, 469–472. [Google Scholar] [CrossRef] [Green Version]
- Beckerman, P.; Bi-Karchin, J.; Park, A.S.; Qiu, C.; Dummer, P.D.; Soomro, I.; Boustany-Kari, C.M.; Pullen, S.S.; Miner, J.H.; Hu, C.A.; et al. Transgenic expression of human APOL1 risk variants in podocytes induces kidney disease in mice. Nat. Med. 2017, 23, 429–438. [Google Scholar] [CrossRef]
- Mishra, A.; Ayasolla, K.; Kumar, V.; Lan, X.; Vashistha, H.; Aslam, R.; Hussain, A.; Chowdhary, S.; Marashi Shoshtari, S.; Paliwal, N.; et al. Modulation of apolipoprotein L1-microRNA-193a axis prevents podocyte dedifferentiation in high-glucose milieu. Am. J. Physiol. Renal. Physiol. 2018, 314, F832–F843. [Google Scholar] [CrossRef]
- Kumar, V.; Ayasolla, K.; Jha, A.; Mishra, A.; Vashistha, H.; Lan, X.; Qayyum, M.; Chinnapaka, S.; Purohit, R.; Mikulak, J.; et al. Disrupted apolipoprotein L1-miR193a axis dedifferentiates podocytes through autophagy blockade in an APOL1 risk milieu. Am. J. Physiol. Cell Physiol. 2019, 317, C209–C225. [Google Scholar] [CrossRef]
- Lambers Heerspink, H.J.; de Zeeuw, D. Novel drugs and intervention strategies for the treatment of chronic kidney disease. Br. J. Clin. Pharmacol. 2013, 76, 536–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, R.; Bo, H.; Villani, V.; Spencer, P.J.; Fu, P. The Inhibitory Effect of Rapamycin on Toll Like Receptor 4 and Interleukin 17 in the Early Stage of Rat Diabetic Nephropathy. Kidney Blood Press Res. 2016, 41, 55–69. [Google Scholar] [CrossRef] [PubMed]
- Velagapudi, C.; Bhandari, B.S.; Abboud-Werner, S.; Simone, S.; Abboud, H.E.; Habib, S.L. The tuberin/mTOR pathway promotes apoptosis of tubular epithelial cells in diabetes. J. Am. Soc. Nephrol. 2011, 22, 262–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosner, M.; Freilinger, A.; Hengstschlager, M. Akt regulates nuclear/cytoplasmic localization of tuberin. Oncogene 2007, 26, 521–531. [Google Scholar] [CrossRef] [Green Version]
- Allen, D.A.; Harwood, S.; Varagunam, M.; Raftery, M.J.; Yaqoob, M.M. High glucose-induced oxidative stress causes apoptosis in proximal tubular epithelial cells and is mediated by multiple caspases. FASEB J. 2003, 17, 908–910. [Google Scholar] [CrossRef]
- Mori, H.; Inoki, K.; Masutani, K.; Wakabayashi, Y.; Komai, K.; Nakagawa, R.; Guan, K.L.; Yoshimura, A. The mTOR pathway is highly activated in diabetic nephropathy and rapamycin has a strong therapeutic potential. Biochem. Biophys Res. Commun. 2009, 384, 471–475. [Google Scholar] [CrossRef]
- Verges, B.; Cariou, B. mTOR inhibitors and diabetes. Diabetes Res. Clin. Pract. 2015, 110, 101–108. [Google Scholar] [CrossRef]
- Viana, S.D.; Reis, F.; Alves, R. Therapeutic Use of mTOR Inhibitors in Renal Diseases: Advances, Drawbacks, and Challenges. Oxid Med. Cell Longev. 2018, 2018, 3693625. [Google Scholar] [CrossRef] [Green Version]
- Amer, H.; Cosio, F.G. Significance and management of proteinuria in kidney transplant recipients. J. Am. Soc. Nephrol. 2009, 20, 2490–2492. [Google Scholar] [CrossRef] [Green Version]
- Torras, J.; Herrero-Fresneda, I.; Gulias, O.; Flaquer, M.; Vidal, A.; Cruzado, J.M.; Lloberas, N.; Franquesa, M.; Grinyo, J.M. Rapamycin has dual opposing effects on proteinuric experimental nephropathies: is it a matter of podocyte damage? Nephrol. Dial. Transplant. 2009, 24, 3632–3640. [Google Scholar] [CrossRef] [Green Version]
- Sereno, J.; Parada, B.; Rodrigues-Santos, P.; Lopes, P.C.; Carvalho, E.; Vala, H.; Teixeira-Lemos, E.; Alves, R.; Figueiredo, A.; Mota, A.; et al. Serum and renal tissue markers of nephropathy in rats under immunosuppressive therapy: cyclosporine versus sirolimus. Transplant. Proc. 2013, 45, 1149–1156. [Google Scholar] [CrossRef] [PubMed]
- Sereno, J.; Nunes, S.; Rodrigues-Santos, P.; Vala, H.; Rocha-Pereira, P.; Fernandes, J.; Santos-Silva, A.; Teixeira, F.; Reis, F. Conversion to sirolimus ameliorates cyclosporine-induced nephropathy in the rat: focus on serum, urine, gene, and protein renal expression biomarkers. Biomed. Res. Int. 2014, 2014, 576929. [Google Scholar] [CrossRef] [PubMed]
- Sereno, J.; Vala, H.; Nunes, S.; Rocha-Pereira, P.; Carvalho, E.; Alves, R.; Teixeira, F.; Reis, F. Cyclosporine A-induced nephrotoxicity is ameliorated by dose reduction and conversion to sirolimus in the rat. J. Physiol. Pharmacol. 2015, 66, 285–299. [Google Scholar] [PubMed]
- Cheng, K.; Rai, P.; Plagov, A.; Lan, X.; Mathieson, P.W.; Saleem, M.A.; Husain, M.; Malhotra, A.; Singhal, P.C. Rapamycin-induced modulation of miRNA expression is associated with amelioration of HIV-associated nephropathy (HIVAN). Exp. Cell Res. 2013, 319, 2073–2080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, L.; Fan, Q.; Wang, X.; Li, L.; Lu, X.; Yue, Y.; Cao, X.; Liu, J.; Zhao, X.; Wang, L. Ursolic acid improves podocyte injury caused by high glucose. Nephrol. Dial. Transplant. 2017, 32, 1285–1293. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Li, Z.P.; Zhang, R.Q.; Zhang, H.M. Repression of miR-217 protects against high glucose-induced podocyte injury and insulin resistance by restoring PTEN-mediated autophagy pathway. Biochem. Biophys. Res. Commun. 2017, 483, 318–324. [Google Scholar] [CrossRef]
- Yamamoto, C.M.; Murakami, T.; Oakes, M.L.; Mitsuhashi, M.; Kelly, C.; Henry, R.R.; Sharma, K. Uromodulin mRNA from Urinary Extracellular Vesicles Correlate to Kidney Function Decline in Type 2 Diabetes Mellitus. Am. J. Nephrol. 2018, 47, 283–291. [Google Scholar] [CrossRef]
- Lv, L.L.; Cao, Y.H.; Pan, M.M.; Liu, H.; Tang, R.N.; Ma, K.L.; Chen, P.S.; Liu, B.C. CD2AP mRNA in urinary exosome as biomarker of kidney disease. Clin. Chim. Acta. 2014, 428, 26–31. [Google Scholar] [CrossRef]
- Feng, Y.; Lv, L.L.; Wu, W.J.; Li, Z.L.; Chen, J.; Ni, H.F.; Zhou, L.T.; Tang, T.T.; Wang, F.M.; Wang, B.; et al. Urinary Exosomes and Exosomal CCL2 mRNA as Biomarkers of Active Histologic Injury in IgA Nephropathy. Am. J. Pathol. 2018, 188, 2542–2552. [Google Scholar] [CrossRef] [Green Version]
- Lv, L.L.; Cao, Y.; Liu, D.; Xu, M.; Liu, H.; Tang, R.N.; Ma, K.L.; Liu, B.C. Isolation and quantification of microRNAs from urinary exosomes/microvesicles for biomarker discovery. Int. J. Biol. Sci. 2013, 9, 1021–1031. [Google Scholar] [CrossRef]
- Perez-Hernandez, J.; Olivares, D.; Forner, M.J.; Ortega, A.; Solaz, E.; Martinez, F.; Chaves, F.J.; Redon, J.; Cortes, R. Urinary exosome miR-146a is a potential marker of albuminuria in essential hypertension. J. Transl. Med. 2018, 16, 228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, L.L.; Cao, Y.H.; Ni, H.F.; Xu, M.; Liu, D.; Liu, H.; Chen, P.S.; Liu, B.C. MicroRNA-29c in urinary exosome/microvesicle as a biomarker of renal fibrosis. Am. J. Physiol. Renal. Physiol. 2013, 305, F1220–F1227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben-Dov, I.Z.; Tan, Y.C.; Morozov, P.; Wilson, P.D.; Rennert, H.; Blumenfeld, J.D.; Tuschl, T. Urine microRNA as potential biomarkers of autosomal dominant polycystic kidney disease progression: description of miRNA profiles at baseline. PLoS ONE 2014, 9, e86856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohan, A.; Singh, R.S.; Kumari, M.; Garg, D.; Upadhyay, A.; Ecelbarger, C.M.; Tripathy, S.; Tiwari, S. Urinary Exosomal microRNA-451-5p Is a Potential Early Biomarker of Diabetic Nephropathy in Rats. PLoS ONE 2016, 11, e0154055. [Google Scholar] [CrossRef] [Green Version]
- Ramezani, A.; Devaney, J.M.; Cohen, S.; Wing, M.R.; Scott, R.; Knoblach, S.; Singhal, R.; Howard, L.; Kopp, J.B.; Raj, D.S. Circulating and urinary microRNA profile in focal segmental glomerulosclerosis: a pilot study. Eur. J. Clin. Invest. 2015, 45, 394–404. [Google Scholar] [CrossRef] [Green Version]
- Jang, S.W.; Liu, X.; Chan, C.B.; Weinshenker, D.; Hall, R.A.; Xiao, G.; Ye, K. Amitriptyline is a TrkA and TrkB receptor agonist that promotes TrkA/TrkB heterodimerization and has potent neurotrophic activity. Chem. Biol. 2009, 16, 644–656. [Google Scholar] [CrossRef] [Green Version]
- Fu, L.; Wang, S.; Wang, X.; Wang, P.; Zheng, Y.; Yao, D.; Guo, M.; Zhang, L.; Ouyang, L. Crystal structure-based discovery of a novel synthesized PARP1 inhibitor (OL-1) with apoptosis-inducing mechanisms in triple-negative breast cancer. Sci. Rep. 2016, 6, 3. [Google Scholar] [CrossRef]
- Owens, M.J.; Morgan, W.N.; Plott, S.J.; Nemeroff, C.B. Neurotransmitter receptor and transporter binding profile of antidepressants and their metabolites. J. Pharmacol. Exp. Ther. 1997, 283, 1305–1322. [Google Scholar]
- Rauser, L.; Savage, J.E.; Meltzer, H.Y.; Roth, B.L. Inverse agonist actions of typical and atypical antipsychotic drugs at the human 5-hydroxytryptamine(2C) receptor. J. Pharmacol. Exp. Ther. 2001, 299, 83–89. [Google Scholar]
- Werling, L.L.; Keller, A.; Frank, J.G.; Nuwayhid, S.J. A comparison of the binding profiles of dextromethorphan, memantine, fluoxetine and amitriptyline: treatment of involuntary emotional expression disorder. Exp. Neurol. 2007, 207, 248–257. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, G.; Kidd, J.; Li, P.-L. Podocyte Lysosome Dysfunction in Chronic Glomerular Diseases. Int. J. Mol. Sci. 2020, 21, 1559. https://doi.org/10.3390/ijms21051559
Li G, Kidd J, Li P-L. Podocyte Lysosome Dysfunction in Chronic Glomerular Diseases. International Journal of Molecular Sciences. 2020; 21(5):1559. https://doi.org/10.3390/ijms21051559
Chicago/Turabian StyleLi, Guangbi, Jason Kidd, and Pin-Lan Li. 2020. "Podocyte Lysosome Dysfunction in Chronic Glomerular Diseases" International Journal of Molecular Sciences 21, no. 5: 1559. https://doi.org/10.3390/ijms21051559