Pre- and Neonatal Exposure to Lead (Pb) Induces Neuroinflammation in the Forebrain Cortex, Hippocampus and Cerebellum of Rat Pups

 , , , , and
, , , , and
Abstract
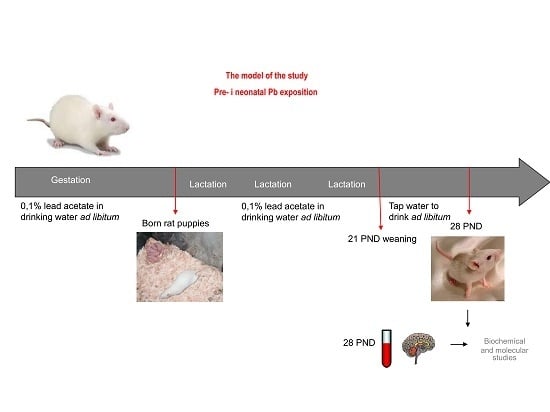
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Lead Concentration in Whole Blood and Brain Tissue
2.2. Exposure to Lead Increases the Level of Cytokines in the Brain
2.3. Lead-Induced Neuroinflammation Increases the Synthesis of Prostanoids
2.4. Increased Levels of Prostanoids in Lead-Induced Neuroinflammation are Accompanied by Increased Expression of Cyclooxygenases.
2.5. Exposure to Lead Increases NF-κB Concentration in the Brain
3. Discussion
3.1. Interleukin-1β
3.2. IL-6
3.3. TGF-β
3.4. NF-κB
3.5. Prostanoids
4. Materials and Methods
4.1. Reagents
4.2. Animals
4.3. Atomic Absorption Spectroscopy Pb Determination
4.4. Western Blotting Analysis of COX-1 and COX-2 Expression
4.5. Quantitative Real-Time PCR Analysis (qRT-PCR) of COX-1 and COX-2 mRNA
4.6. Measurements of Prostaglandin (PGE2) and Thromboxane B2 (TXB2) Concentrations by ELISA Method
4.7. Measurement of Interleukin 1β (IL-1β) and Interleukin 6 (IL-6) Concentration by ELISA Method
4.8. Measurement of Transforming Growth Factor-beta 1 (TGF-β1) and nuclear factor kappa B (NF-κB) concentration by ELISA method
4.9. Protein Assay
4.10. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Aβ | β-amyloid |
| ADHD | Attention-deficit hyperactivity disorder |
| ASD | Autism spectrum disorder |
| COX-1 | Cyclooxygenase 1 |
| COX-2 | Cyclooxygenase 2 |
| IL-1β | Interleukin 1β |
| IL-6 | Interleukin 6 |
| iNOS | Inducible nitric oxide synthase |
| LPS | Lipopolysaccharide |
| NF-κB | Nuclear factor κB |
| Pb | Lead |
| PbAc | Lead acetate |
| PGE2 | Prostaglandin E2 |
| TGF-β | Transforming growth factor-β |
| TXB2 | Thromboxane B2 |
References
- The Government of the United Kingdom. Environmental Protection: The Restriction of the Use of Certain Hazardous Substances in Electrical and Electronic Equipment Regulations; No. 2748; The Stationery Office Limited: London, UK, 2005. Available online: http://www.legislation.gov.uk/uksi/2005/2748/pdfs/uksi_ 20052748_en.pdf (accessed on 19 November 2019).
- The Government of the United Kingdom. Environmental Protection: The Restriction of the Use of Certain Hazardous Substances in Electrical and Electronic Equipment (Amendment) Regulations; No. 581; The Stationery Office Limited: London, UK, 2009. Available online: http://www.legislation.gov.uk/uksi/2009/581/pdfs/uksi_ 20090581_en.pdf (accessed on 19 November 2019).
- European Commission, Institute for Health and Consumer Protection Toxicology and Chemical Substances (& ECB). Opinion of the TC NES on the Environment Part of Industry Voluntary Risk Assessments on Lead and Lead Compounds; European Commission: Brussels, Belgium, 2008; Available online: https://echa.europa.eu/documents/10162/13630/tcnes_opinion_env_en.pdf (accessed on 19 November 2019).
- EFSA (European Food Safety Authority). Scientific opinion on lead in food. EFSA J. 2010, 8, 1570. [Google Scholar] [CrossRef]
- United Nations Environment Programme (UNEP). Leaded Petrol Phase-Out: Global Status as at March 2017. Available online: http://wedocs.unep.org/bitstream/handle/20.500.11822/17542/MapWorldLead_March%202017.pdf?sequence=1&isAllowed=y (accessed on 19 November 2019).
- ATSDR. Substance Priority List. Agency for Toxic Substances and Disease Registry. 2018. Available online: https://www.meritlabs.com/blog/2018/2/23/the-latest-atsdr-substance-priority-list-of-chemicals-and-elements-posing-the-most-significant-risk-at-npl-sites (accessed on 19 November 2019).
- Baranowska-Bosiacka, I.; Gutowska, I.; Marchetti, C.; Rutkowska, M.; Marchlewicz, M.; Kolasa, A.; Prokopowicz, A.; Wiernicki, I.; Piotrowska, K.; Baśkiewicz, M.; et al. Altered energy status of primary cerebellar granule neuronal cultures from rats exposed to lead in the pre- and neonatal period. Toxicology 2011, 280, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Baranowska-Bosiacka, I.; Gutowska, I.; Marchlewicz, M.; Marchetti, C.; Kurzawski, M.; Dziedziejko, V.; Kolasa, A.; Olszewska, M.; Rybicka, M.; Safranow, K.; et al. Disrupted pro- and antioxidative balance as a mechanism of neurotoxicity induced by perinatal exposure to lead. Brain Res. 2012, 1435, 56–71. [Google Scholar] [CrossRef] [PubMed]
- Baranowska-Bosiacka, I.; Strużyńska, L.; Gutowska, I.; Machalińska, A.; Kolasa, A.; Kłos, P.; Czapski, G.A.; Kurzawski, M.; Prokopowicz, A.; Marchlewicz, M.; et al. Perinatal exposure to lead induces morphological, ultrastructural and molecular alterations in the hippocampus. Toxicology 2013, 303, 187–200. [Google Scholar] [CrossRef] [PubMed]
- Gąssowska, M.; Baranowska-Bosiacka, I.; Moczydłowska, J.; Frontczak-Baniewicz, M.; Gewartowska, M.; Strużyńska, L.; Gutowska, I.; Chlubek, D.; Adamczyk, A. Perinatal exposure to lead (Pb) induces ultrastructural and molecular alterations in synapses of rat offspring. Toxicology 2016, 373, 13–29. [Google Scholar] [CrossRef] [PubMed]
- Gąssowska, M.; Baranowska-Bosiacka, I.; Moczydłowska, J.; Tarnowski, M.; Pilutin, A.; Gutowska, I.; Strużyńska, L.; Chlubek, D.; Adamczyk, A. Perinatal exposure to lead (Pb) promotes Tau phosphorylation in the rat brain in a GSK-3β and CDK5 dependent manner: Relevance to neurological disorders. Toxicology 2016, 347, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Baranowska-Bosiacka, I.; Chlubek, D. Biochemical mechanisms of neurotoxic lead activity. Postepy Biochem. 2006, 52, 320–329. [Google Scholar]
- Falkowska, A.; Gutowska, I.; Goschorska, M.; Nowacki, P.; Chlubek, D.; Baranowska-Bosiacka, I. Energy metabolism of the brain, including the cooperation between astrocytes and neurons, especially in the context of glycogen metabolism. Int. J. Mol. Sci. 2015, 16, 25959–25981. [Google Scholar] [CrossRef] [Green Version]
- Chibowska, K.; Baranowska-Bosiacka, I.; Falkowska, A.; Gutowska, I.; Goschorska, M.; Chlubek, D. Effect of lead (Pb) on inflammatory processes in the brain. Int. J. Mol. Sci. 2016, 17, E2140. [Google Scholar] [CrossRef] [Green Version]
- Metryka, E.; Chibowska, K.; Gutowska, I.; Falkowska, A.; Kupnicka, P.; Barczak, K.; Chlubek, D.; Baranowska-Bosiacka, I. Lead (Pb) exposure enhances expression of factors associated with inflammation. Int. J. Mol. Sci. 2018, 19, 1813. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, M.E.; Mack, C.M.; Lasley, S.M. The influence of developmental period of lead exposure on long-term potentiation in the adult rat dentate gyrus in vivo. Neurotoxicology 1999, 20, 57–69. [Google Scholar] [PubMed]
- Gilbert, M.E.; Lasley, S.M. Long-term consequences of developmental exposure to lead or polychlorinated biphenyls: Synaptic transmission and plasticity in the rodent CNS. Environ. Toxicol. Pharmacol. 2002, 12, 105–117. [Google Scholar] [CrossRef]
- Verina, T.; Rohde, C.A.; Guilarte, T.R. Environmental lead exposure during early life alters granule cell neurogenesis and morphology in the hippocampus of young adult rats. Neuroscience 2007, 145, 1037–1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Saleh, I.; Nester, M.; Mashhour, A.; Moncari, L.; Shinwari, N.; Mohamed, G.-D.; Rabah, A. Prenatal and postnatal lead exposure and early cognitive development: Longitudinal study in Saudi Arabia. J. Environ. Pathol. Toxicol. Oncol. 2009, 28, 283–302. [Google Scholar] [CrossRef]
- Sioen, I.; Den Hond, E.; Nelen, V.; Van de Mieroop, E.; Croes, K.; Van Larebeke, N.; Nawrot, T.S.; Schoeters, G. Prenatal exposure to environmental contaminants and behavioural problems at age 7–8years. Environ. Int. 2013, 59, 225–231. [Google Scholar] [CrossRef]
- Nicolescu, R.; Petcu, C.; Cordeanu, A.; Fabritius, K.; Schlumpf, M.; Krebs, R.; Krämer, U.; Winneke, G. Environmental exposure to lead, but not other neurotoxic metals, relates to core elements of ADHD in Romanian children: Performance and questionnaire data. Environ. Res. 2010, 110, 476–483. [Google Scholar] [CrossRef]
- Hong, S.B.; Im, M.H.; Kim, J.W.; Park, E.J.; Shin, M.S.; Kim, B.N.; Yoo, H.J.; Cho, I.H.; Bhang, S.Y.; Hong, Y.C.; et al. Environmental lead exposure and attention deficit/hyperactivity disorder symptom domains in a community sample of South Korean school-age children. Environ. Health Perspect. 2015, 123, 271–276. [Google Scholar] [CrossRef]
- Blaurock-Busch, E.; Amin, O.R.; Rabah, T. Heavy metals and trace elements in hair and urine of a sample of Arab children with autistic spectrum disorder. Maedica 2011, 6, 247–257. [Google Scholar]
- El-Ansary, A.; Bjørklund, G.; Tinkov, A.A.; Skalny, A.V.; Al Dera, H. Relationship between selenium, lead, and mercury in red blood cells of Saudi autistic children. Metab. Brain Dis. 2017, 32, 1073–1080. [Google Scholar] [CrossRef]
- Smith, M.R.; Yevoo, P.; Sadahiro, M.; Austin, C.; Amarasiriwardena, C.; Awawda, M.; Arora, M.; Dudley, J.T.; Morishita, H. Integrative bioinformatics identifies postnatal lead (Pb) exposure disrupts developmental cortical plasticity. Sci. Rep. 2018, 8, 16388. [Google Scholar] [CrossRef]
- Wu, J.; Liu, D.J.; Shou, X.J.; Zhang, J.S.; Meng, F.C.; Liu, Y.Q.; Han, S.P.; Zhang, R.; Jia, J.Z.; Wang, J.Y.; et al. Chinese children with autism: A multiple chemical elements profile in erythrocytes. Autism Res. 2018, 11, 834–845. [Google Scholar] [CrossRef] [PubMed]
- Baghurst, P.A.; McMichael, A.J.; Wigg, N.R.; Vimpani, G.V.; Robertson, E.F.; Roberts, R.J.; Tong, S.L. Environmental exposure to lead and children’s intelligence at the age of seven years. The Port Pirie Cohort Study. N. Engl. J. Med. 1992, 327, 1279–1284. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.; Dietrich, K.N.; Ware, J.H.; Radcliffe, J.; Rogan, W.J. IQ and blood lead from 2 to 7 years of age: Are the effects in older children the residual of high blood lead concentrations in 2-year-olds? Environ. Health Perspect. 2005, 113, 597–601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Advisory Committee on Childhood Lead Poisoning Prevention. Interpreting and managing blood lead levels <10 µg/dL in children and reducing childhood exposures to lead: Recommendations of CDC’s advisory committee on childhood lead poisoning prevention. MMWR Recomm. Rep. 2007, 56, 1–16. [Google Scholar]
- CDC (Centers for Disease Control and Prevention). Sources of Lead. 2012. Available online: http://www.cdc.gov/nceh/lead/tips/sources.htm (accessed on 19 November 2019).
- Chiodo, L.M.; Jacobson, S.W.; Jacobson, J.L. Neurodevelopmental effects of postnatal lead exposure at very low levels. Neurotoxicol. Teratol. 2004, 26, 359–371. [Google Scholar] [CrossRef]
- Lanphear, B.P.; Hornung, R.; Khoury, J.; Yolton, K.; Baghurst, P.; Bellinger, D.C.; Canfield, R.L.; Dietrich, K.N.; Bornschein, R.; Greene, T.; et al. Low-level environmental lead exposure and children’s intellectual function: An international pooled analysis. Environ. Health Perspect. 2005, 113, 894–899. [Google Scholar] [CrossRef]
- Miranda, M.L.; Kim, D.; Galeano, M.A.; Paul, C.J.; Hull, A.P.; Morgan, S.P. The relationship between early childhood blood lead levels and performance on end-of-grade tests. Environ. Health Perspect. 2007, 115, 1242–1247. [Google Scholar] [CrossRef]
- Geier, D.A.; Kern, J.K.; Geier, M.R. Blood lead levels and learning disabilities: A cross-sectional study of the 2003–2004 national health and nutrition examination survey (NHANES). Int. J. Environ. Res. Public Health 2017, 14, 1202. [Google Scholar] [CrossRef] [Green Version]
- Advisory Committee on Childhood Lead Poisoning Prevention. Report of the Advisory Committee on Childhood Lead Poisoning Prevention of the Centers for Disease Control and Prevention Low Level Lead Exposure Harms Children: A Renewed Call for Primary Prevention; CDC: Atlanta, GA, USA, 2012. Available online: http://www.cdc.gov/nceh/lead/acclpp/final_document_030712.pdf (accessed on 19 November 2019).
- Listos, J.; Baranowska-Bosiacka, I.; Talarek, S.; Listos, P.; Orzelska, J.; Fidecka, S.; Gutowska, I.; Kolasa, A.; Rybicka, M.; Chlubek, D. The effect of perinatal lead exposure on dopamine receptor D2 expression in morphine dependent rats. Toxicology 2013, 310, 73–83. [Google Scholar] [CrossRef]
- Baranowska-Bosiacka, I.; Listos, J.; Gutowska, I.; Machoy-Mokrzyńska, A.; Kolasa-Wołosiuk, A.; Tarnowski, M.; Puchałowicz, K.; Prokopowicz, A.; Talarek, S.; Listos, P.; et al. Effects of perinatal exposure to lead (Pb) on purine receptor expression in the brain and gliosis in rats tolerant to morphine analgesia. Toxicology 2016, 339, 19–33. [Google Scholar] [CrossRef]
- Schneider, J.S.; Anderson, D.W.; Wade, T.V.; Smith, M.G.; Leibrandt, P.; Zuck, L.; Lidsky, T.I. Inhibition of progenitor cell proliferation in the dentate gyrus of rats following post-weaning lead exposure. Neurotoxicology 2005, 26, 141–145. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, S.; Flores-Montoya, M.G.; Sobin, C. Early chronic exposure to low-level lead alters total hippocampal microglia in pre-adolescent mice. Toxicol. Lett. 2019, 302, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Davidovics, Z.; DiCicco-Bloom, E. Moderate lead exposure elicits neurotrophic effects in cerebral cortical precursor cells in culture. J. Neurosci. Res. 2005, 80, 817–825. [Google Scholar] [CrossRef]
- Mousa, A.M.; Al-Fadhli, A.S.; Rao, M.S.; Kilarkaje, N. Gestational lead exposure induces developmental abnormalities and up-regulates apoptosis of fetal cerebellar cells in rats. Drug Chem. Toxicol. 2015, 38, 73–83. [Google Scholar] [CrossRef]
- Ahmad, F.; Salahuddin, M.; Alamoudi, W.; Acharya, S. Dysfunction of cortical synapse-specific mitochondria in developing rats exposed to lead and its amelioration by ascorbate supplementation. Neuropsychiatr. Dis. Treat. 2018, 14, 813–824. [Google Scholar] [CrossRef] [Green Version]
- Baranowska-Bosiacka, I.; Falkowska, A.; Gutowska, I.; Gąssowska, M.; Kolasa-Wołosiuk, A.; Tarnowski, M.; Chibowska, K.; Goschorska, M.; Lubkowska, A.; Chlubek, D. Glycogen metabolism in brain and neurons—Astrocytes metabolic cooperation can be altered by pre- and neonatal lead (Pb) exposure. Toxicology 2017, 390, 146–158. [Google Scholar] [CrossRef]
- Ekdahl, C.T.; Claasen, J.H.; Bonde, S.; Kokaia, Z.; Lindvall, O. Inflammation is detrimental for neurogenesis in adult brain. Proc. Natl. Acad. Sci. USA 2003, 100, 13632–13637. [Google Scholar] [CrossRef] [Green Version]
- Mouihate, A. TLR4-mediated brain inflammation halts neurogenesis: Impact of hormonal replacement therapy. Front. Cell Neurosci. 2014, 8, 146. [Google Scholar] [CrossRef] [Green Version]
- Shiow, L.R.; Favrais, G.; Schirmer, L.; Schang, A.L.; Cipriani, S.; Andres, C.; Wright, J.N.; Nobuta, H.; Fleiss, B.; Gressens, P.; et al. Reactive astrocyte COX2-PGE2 production inhibits oligodendrocyte maturation in neonatal white matter injury. Glia 2017, 65, 2024–2037. [Google Scholar] [CrossRef]
- Cai, Z.; Pan, Z.L.; Pang, Y.; Evans, O.B.; Rhodes, P.G. Cytokine induction in fetal rat brains and brain injury in neonatal rats after maternal lipopolysaccharide administration. Pediatr. Res. 2000, 47, 64–72. [Google Scholar] [CrossRef] [Green Version]
- Cai, Z.; Lin, S.; Pang, Y.; Rhodes, P.G. Brain injury induced by intracerebral injection of interleukin-1β and tumor necrosis factor-α in the neonatal rat. Pediatr. Res. 2004, 56, 377–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rousset, C.I.; Chalon, S.; Cantagrel, S.; Bodard, S.; Andres, C.; Gressens, P.; Saliba, E. Maternal exposure to LPS induces hypomyelination in the internal capsule and programmed cell death in the deep gray matter in newborn rats. Pediatr. Res. 2006, 59, 428–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Penta, A.; Moreno, B.; Reix, S.; Fernandez-Diez, B.; Villanueva, M.; Errea, O.; Escala, N.; Vandenbroeck, K.; Comella, J.X.; Villoslada, P. Oxidative stress and proinflammatory cytokines contribute to demyelination and axonal damage in a cerebellar culture model of neuroinflammation. PLoS ONE 2013, 8, e54722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duncan, J.R.; Cock, M.L.; Scheerlinck, J.P.; Westcott, K.T.; McLean, C.; Harding, R.; Rees, S.M. White matter injury after repeated endotoxin exposure in the preterm ovine fetus. Pediatr. Res. 2002, 52, 941–949. [Google Scholar] [CrossRef] [PubMed]
- Bastos, G.N.; Moriya, T.; Inui, F.; Katura, T.; Nakahata, N. Involvement of cyclooxygenase-2 in lipopolysaccharide-induced impairment of the newborn cell survival in the adult mouse dentate gyrus. Neuroscience 2008, 155, 454–462. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.M.; Hong, J.S. Why neurodegenerative diseases are progressive: Uncontrolled inflammation drives disease progression. Trends Immunol. 2008, 29, 357–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, L.W.; Kaizaki, A.; Tien, L.T.; Pang, Y.; Tanaka, S.; Numazawa, S.; Bhatt, A.J.; Cai, Z. Celecoxib attenuates systemic lipopolysaccharide-induced brain inflammation and white matter injury in the neonatal rats. Neuroscience 2013, 240, 27–38. [Google Scholar] [CrossRef] [Green Version]
- Smith, M.R.; Burman, P.; Sadahiro, M.; Kidd, B.A.; Dudley, J.T.; Morishita, H. Integrative analysis of disease signatures shows inflammation disrupts juvenile experience-dependent cortical plasticity. eNeuro 2017, 3. [Google Scholar] [CrossRef] [Green Version]
- Hsiao, E.Y.; McBride, S.W.; Chow, J.; Mazmanian, S.K.; Patterson, P.H. Modeling an autism risk factor in mice leads to permanent immune dysregulation. Proc. Natl. Acad. Sci. USA 2012, 109, 12776–12781. [Google Scholar] [CrossRef] [Green Version]
- Wong, C.T.; Ussyshkin, N.; Ahmad, E.; Rai-Bhogal, R.; Li, H.; Crawford, D.A. Prostaglandin E2 promotes neural proliferation and differentiation and regulates Wnt target gene expression. J. Neurosci. Res. 2016, 94, 759–775. [Google Scholar] [CrossRef]
- Struzynska, L.; Dabrowska-Bouta, B.; Koza, K.; Sulkowski, G. Inflammation-like glial response in lead-exposed immature rat brain. Toxicol. Sci. 2007, 95, 156–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramesh, G.T.; Manna, S.K.; Aggarwal, B.B.; Jadhav, A.L. Lead exposure activates nuclear factor κB, activator protein-1, c-Jun N-terminal kinase and caspases in the rat brain. Toxicol. Lett. 2001, 123, 195–207. [Google Scholar] [CrossRef]
- Liu, M.C.; Liu, X.Q.; Wang, W.; Shen, X.F.; Che, H.L.; Guo, Y.Y.; Zhao, M.G.; Chen, J.Y.; Luo, W.J. Involvement of microglia activation in the lead induced long-term potentiation impairment. PLoS ONE 2012, 7, e43924. [Google Scholar] [CrossRef] [PubMed]
- Kumawat, K.L.; Kaushik, D.K.; Goswami, P.; Basu, A. Acute exposure to lead acetate activates microglia and induces subsequent bystander neuronal death via caspase-3 activation. Neurotoxicology 2014, 41, 143–153. [Google Scholar] [CrossRef]
- Liu, J.T.; Chen, B.Y.; Zhang, J.Q.; Kuang, F.; Chen, L.W. Lead exposure induced microgliosis and astrogliosis in hippocampus of young mice potentially by triggering TLR4-MyD88-NFκB signaling cascades. Toxicol. Lett. 2015, 239, 97–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raphael, I.; Nalawade, S.; Eagar, T.N.; Forsthuber, T.G. T cell subsets and their signature cytokines in autoimmune and inflammatory diseases. Cytokine 2015, 74, 5–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Browne, T.C.; McQuillan, K.; McManus, R.M.; O’Reilly, J.A.; Mills, K.H.; Lynch, M.A. IFN-γ Production by amyloid β-specific Th1 cells promotes microglial activation and increases plaque burden in a mouse model of Alzheimer’s disease. J. Immunol. 2013, 190, 2241–2251. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.D.; Jin, J.J.; Maxwell, J.A.; Fukuchi, K. Enhancing Th2 immune responses against amyloid protein by a DNA prime-adenovirus boost regimen for Alzheimer’s disease. Immunol. Lett. 2007, 112, 30–38. [Google Scholar] [CrossRef] [Green Version]
- Kong, W.; Yen, J.H.; Ganea, D. Docosahexaenoic acid prevents dendritic cell maturation, inhibits antigen-specific Th1/Th17 differentiation and suppresses experimental autoimmune encephalomyelitis. Brain Behav. Immun. 2011, 25, 872–882. [Google Scholar] [CrossRef] [Green Version]
- Luo, Y.; Zhou, Y.; Xiao, W.; Liang, Z.; Dai, J.; Weng, X.; Wu, X. Interleukin-33 ameliorates ischemic brain injury in experimental stroke through promoting Th2 response and suppressing Th17 response. Brain Res. 2015, 1597, 86–94. [Google Scholar] [CrossRef]
- Dolati, S.; Ahmadi, M.; Khalili, M.; Taheraghdam, A.A.; Siahmansouri, H.; Babaloo, Z.; Aghebati-Maleki, L.; Jadidi-Niaragh, F.; Younesi, V.; Yousefi, M. Peripheral Th17/Treg imbalance in elderly patients with ischemic stroke. Neurol. Sci. 2018, 39, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Liu, X.; Zhang, P.; Qiao, M.; Li, H.; Li, X.; Zhang, H.; Yu, Z. The effects of early life lead exposure on the expression of interleukin (IL) 1β, IL-6, and glial fibrillary acidic protein in the hippocampus of mouse pups. Hum. Exp. Toxicol. 2015, 34, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Liu, F.; Song, L.; Zhang, P.; Qiao, M.; Zhao, Q.; Li, W. The effects of early life Pb exposure on the expression of IL1-β, TNF-α and Aβ in cerebral cortex of mouse pups. J. Trace Elem. Med. Biol. 2014, 28, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Kim, B.J.; Kim, Y.B.; Chung, P.W.; Moon, H.S.; Suh, B.C.; Yoon, W.T.; Jin, D.K.; Park, Y.S.; Lee, Y.T.; et al. IL-1β induction and IL-6 suppression are associated with aggravated neuronal damage in a lipopolysaccharide-pretreated kainic acid-induced rat pup seizure model. Neuroimmunomodulation 2012, 19, 319–325. [Google Scholar] [CrossRef]
- Pang, Y.; Tien, L.T.; Zhu, H.; Shen, J.; Wright, C.F.; Jones, T.K.; Mamoon, S.A.; Bhatt, A.J.; Cai, Z.; Fan, L.W. Interleukin-1 receptor antagonist reduces neonatal lipopolysaccharide-induced long-lasting neurobehavioral deficits and dopaminergic neuronal injury in adult rats. Int. J. Mol. Sci. 2015, 16, 8635–8654. [Google Scholar] [CrossRef]
- De Rivero Vaccari, J.P.; Lotocki, G.; Alonso, O.F.; Bramlett, H.M.; Dietrich, W.D.; Keane, R.W. Therapeutic neutralization of the NLRP1 inflammasome reduces the innate immune response and improves histopathology after traumatic brain injury. J. Cereb. Blood Flow Metab. 2009, 29, 1251–1261. [Google Scholar] [CrossRef] [Green Version]
- Schielke, G.P.; Yang, G.Y.; Shivers, B.D.; Betz, A.L. Reduced ischemic brain injury in interleukin-1β converting enzyme-deficient mice. J. Cereb. Blood Flow Metab. 1998, 18, 180–185. [Google Scholar] [CrossRef] [Green Version]
- Emsley, H.C.; Smith, C.J.; Georgiou, R.F.; Vail, A.; Hopkins, S.J.; Rothwell, N.J.; Tyrrell, P.J. Acute stroke investigators. A randomised phase II study of interleukin-1 receptor antagonist in acute stroke patients. J. Neurol. Neurosurg. Psychiatry 2005, 76, 1366–1372. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Hovanesian, V.; Naqvi, S.; Lim, Y.P.; Tucker, R.; Donahue, J.E.; Stopa, E.G.; Stonestreet, B.S. Systemic infusions of anti-interleukin-1β neutralizing antibodies reduce short-term brain injury after cerebral ischemia in the ovine fetus. Brain Behav. Immun. 2018, 67, 24–35. [Google Scholar] [CrossRef]
- Clausen, F.; Hånell, A.; Björk, M.; Hillered, L.; Mir, A.K.; Gram, H.; Marklund, N. Neutralization of interleukin-1β modifies the inflammatory response and improves histological and cognitive outcome following traumatic brain injury in mice. Eur. J. Neurosci. 2009, 30, 385–396. [Google Scholar] [CrossRef]
- Herx, L.M.; Rivest, S.; Yong, V.W. Central nervous system-initiated inflammation and neurotrophism in trauma: IL-1β is required for the production of ciliary neurotrophic factor. J. Immunol. 2000, 165, 2232–2239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herx, L.M.; Yong, V.W. Interleukin-1β is required for the early evolution of reactive astrogliosis following CNS lesion. J. Neuropathol. Exp. Neurol. 2001, 60, 961–971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, L.W.; Mitchell, H.J.; Tien, L.T.; Rhodes, P.G.; Cai, Z. Interleukin-1β-induced brain injury in the neonatal rat can be ameliorated by α-phenyl-n-tert-butyl-nitrone. Exp. Neurol 2009, 220, 143–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Girard, S.; Sébire, H.; Brochu, M.E.; Briota, S.; Sarret, P.; Sébire, G. Postnatal administration of IL-1Ra exerts neuroprotective effects following perinatal inflammation and/or hypoxic-ischemic injuries. Brain Behav. Immun. 2012, 26, 1331–1339. [Google Scholar] [CrossRef] [Green Version]
- Holmin, S.; Mathiesen, T. Intracerebral administration of interleukin-1β and induction of inflammation, apoptosis, and vasogenic edema. J. Neurosurg. 2000, 92, 108–120. [Google Scholar] [CrossRef] [Green Version]
- Lan, K.M.; Tien, L.T.; Pang, Y.; Bhatt, A.J.; Fan, L.W. IL-1 receptor antagonist attenuates neonatal lipopolysaccharide-induced long-lasting learning impairment and hippocampal injury in adult rats. Toxicol. Lett. 2015, 234, 30–39. [Google Scholar] [CrossRef] [Green Version]
- Kasten-Jolly, J.; Heo, Y.; Lawrence, D.A. Central nervous system cytokine gene expression: Modulation by lead. J. Biochem. Mol. Toxicol. 2011, 25, 41–54. [Google Scholar] [CrossRef] [Green Version]
- Spooren, A.; Kolmus, K.; Laureys, G.; Clinckers, R.; De Keyser, J.; Haegeman, G.; Gerlo, S. Interleukin-6, a mental cytokine. Brain Res. Rev. 2011, 67, 157–183. [Google Scholar] [CrossRef]
- Sparkman, N.L.; Buchanan, J.B.; Heyen, J.R.; Chen, J.; Beverly, J.L.; Johnson, R.W. Interleukin-6 facilitates lipopolysaccharide-induced disruption in working memory and expression of other proinflammatory cytokines in hippocampal neuronal cell layers. J. Neurosci. 2006, 26, 10709–10716. [Google Scholar] [CrossRef] [Green Version]
- Pang, Y.; Fan, L.W.; Zheng, B.; Cai, Z.; Rhodes, P.G. Role of interleukin-6 in lipopolysaccharide-induced brain injury and behavioral dysfunction in neonatal rats. Neuroscience 2006, 141, 745–755. [Google Scholar] [CrossRef]
- Burton, M.D.; Rytych, J.L.; Freund, G.G.; Johnson, R.W. Central inhibition of interleukin-6 trans-signaling during peripheral infection reduced neuroinflammation and sickness in aged mice. Brain Behav. Immun. 2013, 30, 66–72. [Google Scholar] [CrossRef] [Green Version]
- Basu, A.; Krady, J.K.; Enterline, J.R.; Levison, S.W. Transforming growth factor beta1 prevents IL-1β-induced microglial activation, whereas TNFα- and IL-6-stimulated activation are not antagonized. Glia 2002, 40, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Li, M.O.; Wan, Y.Y.; Sanjabi, S.; Robertson, A.K.; Flavell, R.A. Transforming growth factor-β regulation of immune responses. Annu. Rev. Immunol. 2006, 24, 99–146. [Google Scholar] [CrossRef] [PubMed]
- Spittau, B.; Wullkopf, L.; Zhou, X.; Rilka, J.; Pfeifer, D.; Krieglstein, K. Endogenous transforming growth factor-β promotes quiescence of primary microglia in vitro. Glia 2013, 61, 287–300. [Google Scholar] [CrossRef]
- Henrich-Noack, P.; Prehn, J.H.; Krieglstein, J. TGF-β 1 protects hippocampal neurons against degeneration caused by transient global ischemia. Dose-response relationship and potential neuroprotective mechanisms. Stroke 1996, 27, 1609–1615. [Google Scholar] [CrossRef]
- Ren, R.F.; Flanders, K.C. Transforming growth factors-beta protect primary rat hippocampal neuronal cultures from degeneration induced by β-amyloid peptide. Brain Res. 1996, 732, 16–24. [Google Scholar] [CrossRef]
- Chen, J.H.; Ke, K.F.; Lu, J.H.; Qiu, Y.H.; Peng, Y.P. Protection of TGF-β1 against neuroinflammation and neurodegeneration in Aβ1-42-induced Alzheimer’s disease model rats. PLoS ONE 2015, 10, e0116549. [Google Scholar] [CrossRef] [Green Version]
- Prehn, J.H.; Peruche, B.; Unsicker, K.; Krieglstein, J. Isoform-specific effects of transforming growth factors-β on degeneration of primary neuronal cultures induced by cytotoxic hypoxia or glutamate. J. Neurochem. 1993, 60, 1665–1672. [Google Scholar] [CrossRef]
- Boche, D.; Cunningham, C.; Gauldie, J.; Perry, V.H. Transforming growth factor-β1-mediated neuroprotection against excitotoxic injury in vivo. J. Cereb. Blood Flow Metab. 2003, 23, 1174–1182. [Google Scholar] [CrossRef] [Green Version]
- Lee, P.W.; Yang, Y.; Racke, M.K.; Lovett-Racke, A.E. Analysis of TGF-β1 and TGF-β3 as regulators of encephalitogenic Th17 cells: Implications for multiple sclerosis. Brain Behav. Immun. 2015, 46, 44–49. [Google Scholar] [CrossRef] [Green Version]
- Severin, M.E.; Lee, P.W.; Liu, Y.; Selhorst, A.J.; Gormley, M.G.; Pei, W.; Yang, Y.; Guerau-de-Arellano, M.; Racke, M.K.; Lovett-Racke, A.E. MicroRNAs targeting TGFβ signalling underlie the regulatory T cell defect in multiple sclerosis. Brain 2016, 139, 1747–1761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamaguchi, M.; Muramatsu, R.; Fujimura, H.; Mochizuki, H.; Kataoka, H.; Yamashita, T. Circulating transforming growth factor-β1 facilitates remyelination in the adult central nervous system. eLife 2019, 8, e41869. [Google Scholar] [CrossRef] [PubMed]
- Lindholm, D.; Castrén, E.; Kiefer, R.; Zafra, F.; Thoenen, H. Transforming growth factor-β1 in the rat brain: Increase after injury and inhibition of astrocyte proliferation. J. Cell Biol. 1992, 117, 395–400. [Google Scholar] [CrossRef] [PubMed]
- Krupinski, J.; Kumar, P.; Kumar, S.; Kaluza, J. Increased expression of TGF-β1 in brain tissue after ischemic stroke in humans. Stroke 1996, 27, 852–857. [Google Scholar] [CrossRef]
- Islam, A.; Choudhury, M.E.; Kigami, Y.; Utsunomiya, R.; Matsumoto, S.; Watanabe, H.; Kumon, Y.; Kunieda, T.; Yano, H.; Tanaka, J. Sustained anti-inflammatory effects of TGF-β1 on microglia/macrophages. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 721–734. [Google Scholar] [CrossRef]
- Tomoda, T.; Shirasawa, T.; Yahagi, Y.I.; Ishii, K.; Takagi, H.; Furiya, Y.; Arai, K.I.; Mori, H.; Muramatsu, M.A. Transforming growth factor-β is a survival factor for neonate cortical neurons: Coincident expression of type I receptors in developing cerebral cortices. Dev. Biol. 1996, 179, 79–90. [Google Scholar] [CrossRef]
- Falk, S.; Wurdak, H.; Ittner, L.M.; Ille, F.; Sumara, G.; Schmid, M.T.; Draganova, K.; Lang, K.S.; Paratore, C.; Leveen, P.; et al. Brain area-specific effect of TGF-β signaling on Wnt-dependent neural stem cell expansion. Cell Stem Cell 2008, 2, 472–483. [Google Scholar] [CrossRef] [Green Version]
- Stipursky, J.; Francis, D.; Dezonne, R.S.; Bérgamo de Araújo, A.P.; Souza, L.; Moraes, C.A.; Alcantara Gomes, F.C. TGF-β1 promotes cerebral cortex radial glia-astrocyte differentiation in vivo. Front. Cell Neurosci. 2014, 8, 393. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.Z.; Shan, C.J.; Bullock, L.; Baker, L.; Rajanna, B. Pb2+ reduces PKCs and NF-κB in vitro. Cell Biol. Toxicol. 2006, 22, 189–198. [Google Scholar] [CrossRef]
- Chen, C.; Edelstein, L.C.; Gélinas, C. The Rel/NF-κB family directly activates expression of the apoptosis inhibitor Bcl-xL. Mol. Cell Biol. 2000, 20, 2687–2695. [Google Scholar] [CrossRef]
- Korbecki, J.; Baranowska-Bosiacka, I.; Gutowska, I.; Chlubek, D. Cyclooxygenase pathways. Acta Biochim. Pol. 2014, 61, 639–649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tumbaga, P.; Beharry, K.; Akmal, Y.; Federico, C.; Modanlou, H.D. Biochemical changes in prostanoids and cerebral expression of cyclooxygenase (COX)-1 and COX-2 during morphine sulfate infusion in the newborn piglet. Prostaglandins Lipid Mediat. 1999, 58, 273–284. [Google Scholar] [CrossRef]
- Choi, S.H.; Langenbach, R.; Bosetti, F. Cyclooxygenase-1 and -2 enzymes differentially regulate the brain upstream NF-κB pathway and downstream enzymes involved in prostaglandin biosynthesis. J. Neurochem. 2006, 98, 801–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doré, S.; Otsuka, T.; Mito, T.; Sugo, N.; Hand, T.; Wu, L.; Hurn, P.D.; Traystman, R.J.; Andreasson, K. Neuronal overexpression of cyclooxygenase-2 increases cerebral infarction. Ann. Neurol. 2003, 54, 155–162. [Google Scholar] [CrossRef]
- Hétu, P.O.; Riendeau, D. Cyclo-oxygenase-2 contributes to constitutive prostanoid production in rat kidney and brain. Biochem. J. 2005, 391, 561–566. [Google Scholar] [CrossRef]
- Liang, X.; Wu, L.; Hand, T.; Andreasson, K. Prostaglandin D2 mediates neuronal protection via the DP1 receptor. J. Neurochem. 2005, 92, 477–486. [Google Scholar] [CrossRef]
- Yang, W.; Yan, A.; Zhang, T.; Shao, J.; Liu, T.; Yang, X.; Xia, W.; Fu, Y. Thromboxane A2 receptor stimulation enhances microglial interleukin-1β and NO biosynthesis mediated by the activation of ERK pathway. Front. Aging Neurosci. 2016, 8, 8. [Google Scholar] [CrossRef] [Green Version]
- Yan, A.; Zhang, T.; Yang, X.; Shao, J.; Fu, N.; Shen, F.; Fu, Y.; Xia, W. Thromboxane A2 receptor antagonist SQ29548 reduces ischemic stroke-induced microglia/macrophages activation and enrichment, and ameliorates brain injury. Sci. Rep. 2016, 6, 35885. [Google Scholar] [CrossRef]
- Yan, A.; Cai, G.; Xia, W.; Fu, Y. Thromboxane A2 receptor antagonist SQ29548 suppresses the LPS-induced release of inflammatory cytokines in BV2 microglia cells via suppressing MAPK and NF-κB signaling pathways. Mol. Med. Rep. 2017, 16, 2491–2496. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Hu, J.; Gao, X.; Liang, H.; Yu, H.; Liu, S.; Liu, Z. Hyperglycemia via activation of thromboxane A2 receptor impairs the integrity and function of blood-brain barrier in microvascular endothelial cells. Oncotarget 2017, 8, 30030–30038. [Google Scholar] [CrossRef] [Green Version]
- McCullough, L.; Wu, L.; Haughey, N.; Liang, X.; Hand, T.; Wang, Q.; Breyer, R.M.; Andreasson, K. Neuroprotective function of the PGE2 EP2 receptor in cerebral ischemia. J. Neurosci. 2004, 24, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Niwa, K.; Araki, E.; Morham, S.G.; Ross, M.E.; Iadecola, C. Cyclooxygenase-2 contributes to functional hyperemia in whisker-barrel cortex. J. Neurosci. 2000, 20, 763–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Bazan, N.G. Endogenous PGE2 regulates membrane excitability and synaptic transmission in hippocampal CA1 pyramidal neurons. J. Neurophysiol. 2005, 93, 929–941. [Google Scholar] [CrossRef] [PubMed]
- Waschbisch, A.; Fiebich, B.L.; Akundi, R.S.; Schmitz, M.L.; Hoozemans, J.J.; Candelario-Jalil, E.; Virtainen, N.; Veerhuis, R.; Slawik, H.; Yrjänheikki, J.; et al. Interleukin-1β-induced expression of the prostaglandin E2-receptor subtype EP3 in U373 astrocytoma cells depends on protein kinase C and nuclear factor-κB. J. Neurochem. 2006, 96, 680–693. [Google Scholar] [CrossRef]
- Ikeda-Matsuo, Y.; Tanji, H.; Ota, A.; Hirayama, Y.; Uematsu, S.; Akira, S.; Sasaki, Y. Microsomal prostaglandin E synthase-1 contributes to ischaemic excitotoxicity through prostaglandin E2 EP3 receptors. Br. J. Pharmacol. 2010, 160, 847–859. [Google Scholar] [CrossRef] [Green Version]
- Takemiya, T.; Matsumura, K.; Sugiura, H.; Yasuda, S.; Uematsu, S.; Akira, S.; Yamagata, K. Endothelial microsomal prostaglandin E synthase-1 facilitates neurotoxicity by elevating astrocytic Ca2+ levels. Neurochem. Int. 2011, 58, 489–496. [Google Scholar] [CrossRef]
- Kawano, T.; Anrather, J.; Zhou, P.; Park, L.; Wang, G.; Frys, K.A.; Kunz, A.; Cho, S.; Orio, M.; Iadecola, C. Prostaglandin E2 EP1 receptors: Downstream effectors of COX-2 neurotoxicity. Nat. Med. 2006, 12, 225–229. [Google Scholar] [CrossRef]
- Kang, H.G.; Jeong, S.H.; Cho, M.R.; Cho, J.H.; Bischoff, K. Time-dependent changes in lead and δ-aminolevulinic acid after subchronic lead exposure in rats. Hum. Exp. Toxicol. 2009, 28, 647–654. [Google Scholar] [CrossRef]
- Xu, J.; Yan, C.H.; Yu, X.G.; Shen, X.M.; Gao, Y.; Yu, X.D.; Wu, S.H.; Shen, X. Effects of the lead exposure on expression of mGluR gene in developed hippocampus. Zhonghua Yi Xue Za Zhi 2005, 85, 705–707. [Google Scholar]
- Yant, L.J.; Ran, Q.; Rao, L.; Van Remmen, H.; Shibatani, T.; Belter, J.G.; Motta, L.; Richardson, A.; Prolla, T.A. The selenoprotein GPX4 is essential for mouse development and protects from radiation and oxidative damage insults. Free Radic. Biol. Med. 2003, 34, 496–502. [Google Scholar] [CrossRef]
- Brenner, A.J.; Harris, E.D. A quantitative test for copper using bicinchoninic acid. Anal. Biochem. 1995, 226, 80–84. [Google Scholar] [CrossRef] [PubMed]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chibowska, K.; Korbecki, J.; Gutowska, I.; Metryka, E.; Tarnowski, M.; Goschorska, M.; Barczak, K.; Chlubek, D.; Baranowska-Bosiacka, I. Pre- and Neonatal Exposure to Lead (Pb) Induces Neuroinflammation in the Forebrain Cortex, Hippocampus and Cerebellum of Rat Pups. Int. J. Mol. Sci. 2020, 21, 1083. https://doi.org/10.3390/ijms21031083
Chibowska K, Korbecki J, Gutowska I, Metryka E, Tarnowski M, Goschorska M, Barczak K, Chlubek D, Baranowska-Bosiacka I. Pre- and Neonatal Exposure to Lead (Pb) Induces Neuroinflammation in the Forebrain Cortex, Hippocampus and Cerebellum of Rat Pups. International Journal of Molecular Sciences. 2020; 21(3):1083. https://doi.org/10.3390/ijms21031083
Chicago/Turabian StyleChibowska, Karina, Jan Korbecki, Izabela Gutowska, Emilia Metryka, Maciej Tarnowski, Marta Goschorska, Katarzyna Barczak, Dariusz Chlubek, and Irena Baranowska-Bosiacka. 2020. "Pre- and Neonatal Exposure to Lead (Pb) Induces Neuroinflammation in the Forebrain Cortex, Hippocampus and Cerebellum of Rat Pups" International Journal of Molecular Sciences 21, no. 3: 1083. https://doi.org/10.3390/ijms21031083