Targeting NF-κB-Inducing Kinase (NIK) in Immunity, Inflammation, and Cancer


Abstract
:1. Introduction
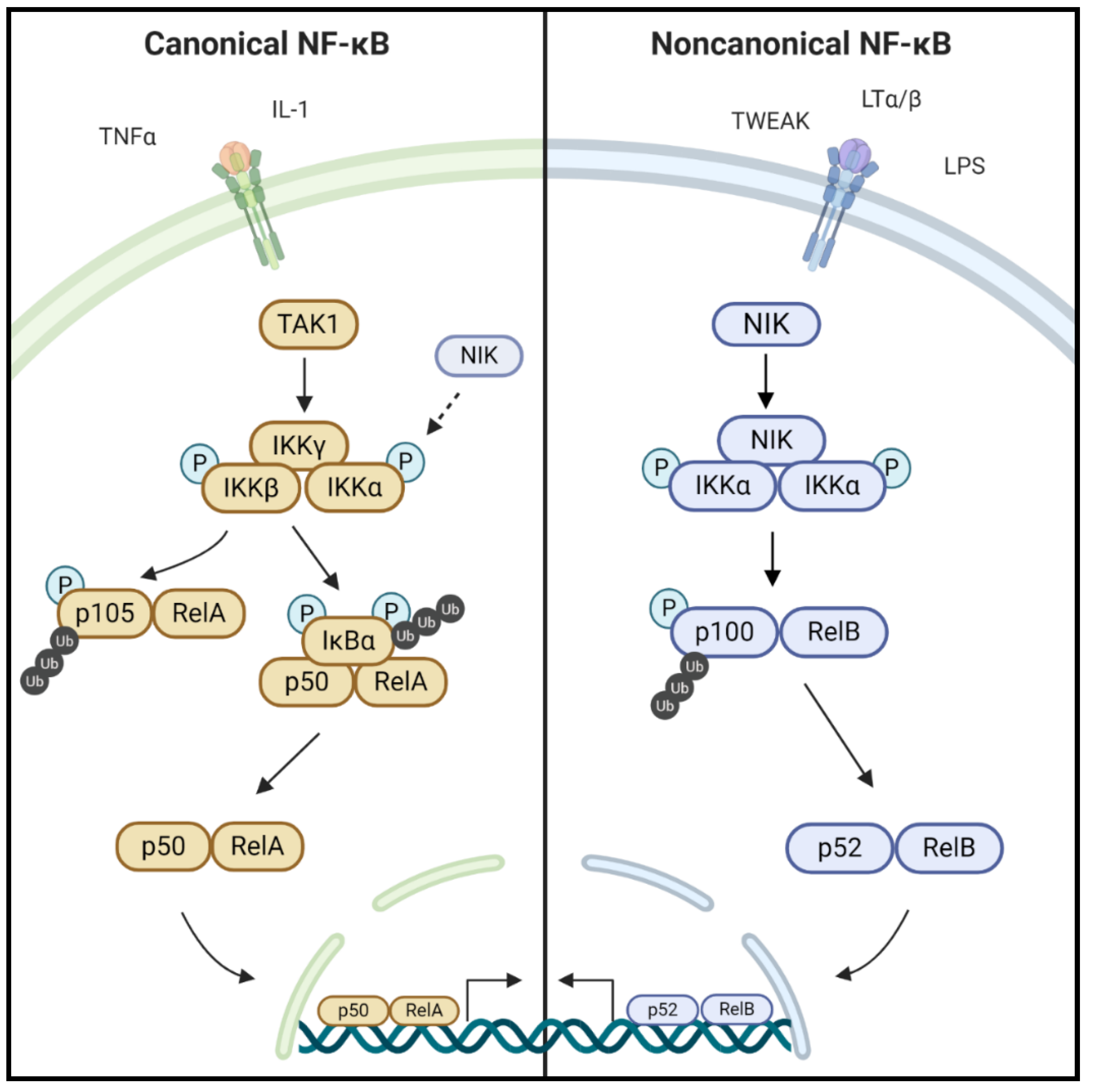
1.1. NF-κB Signaling
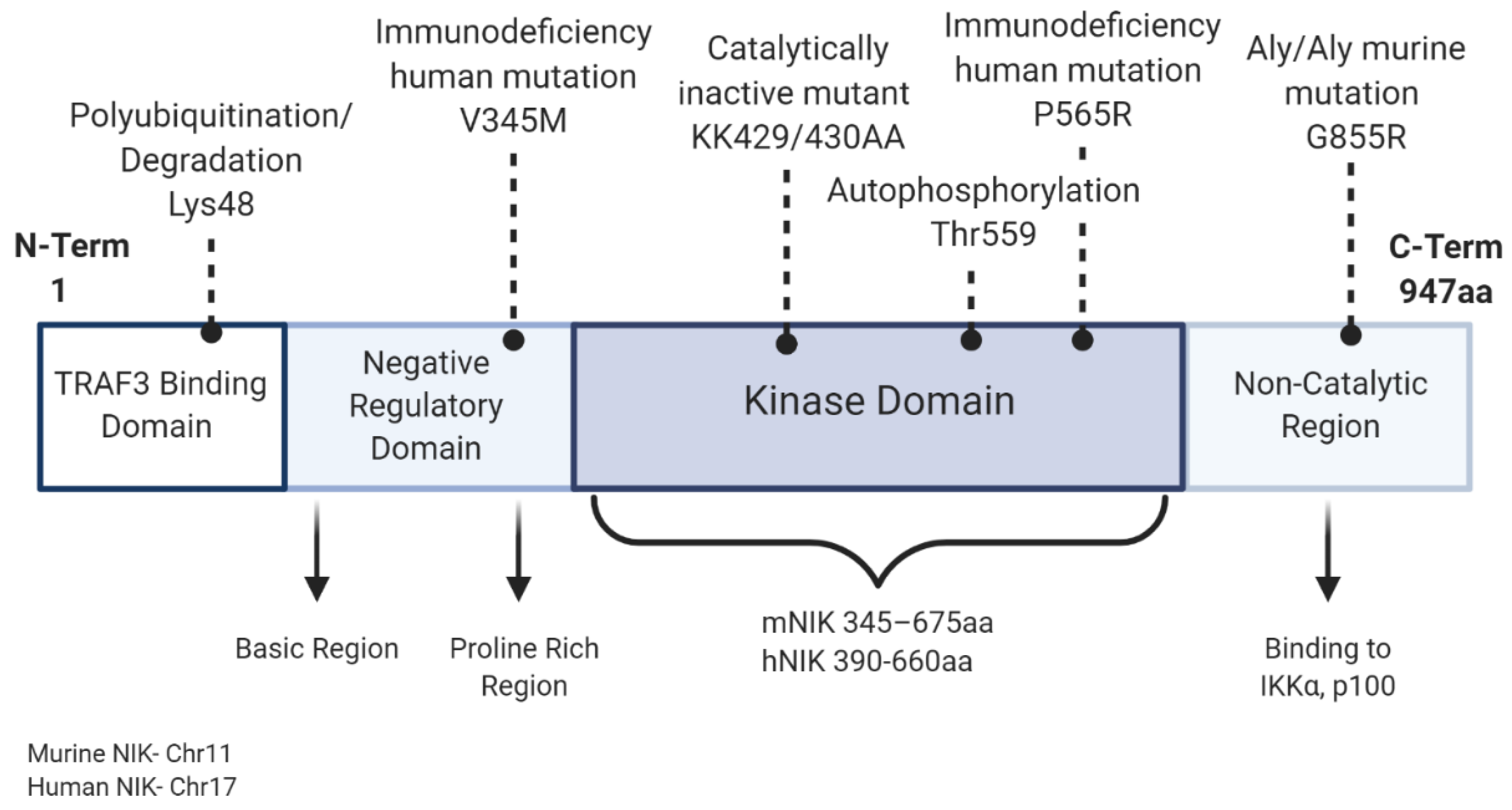
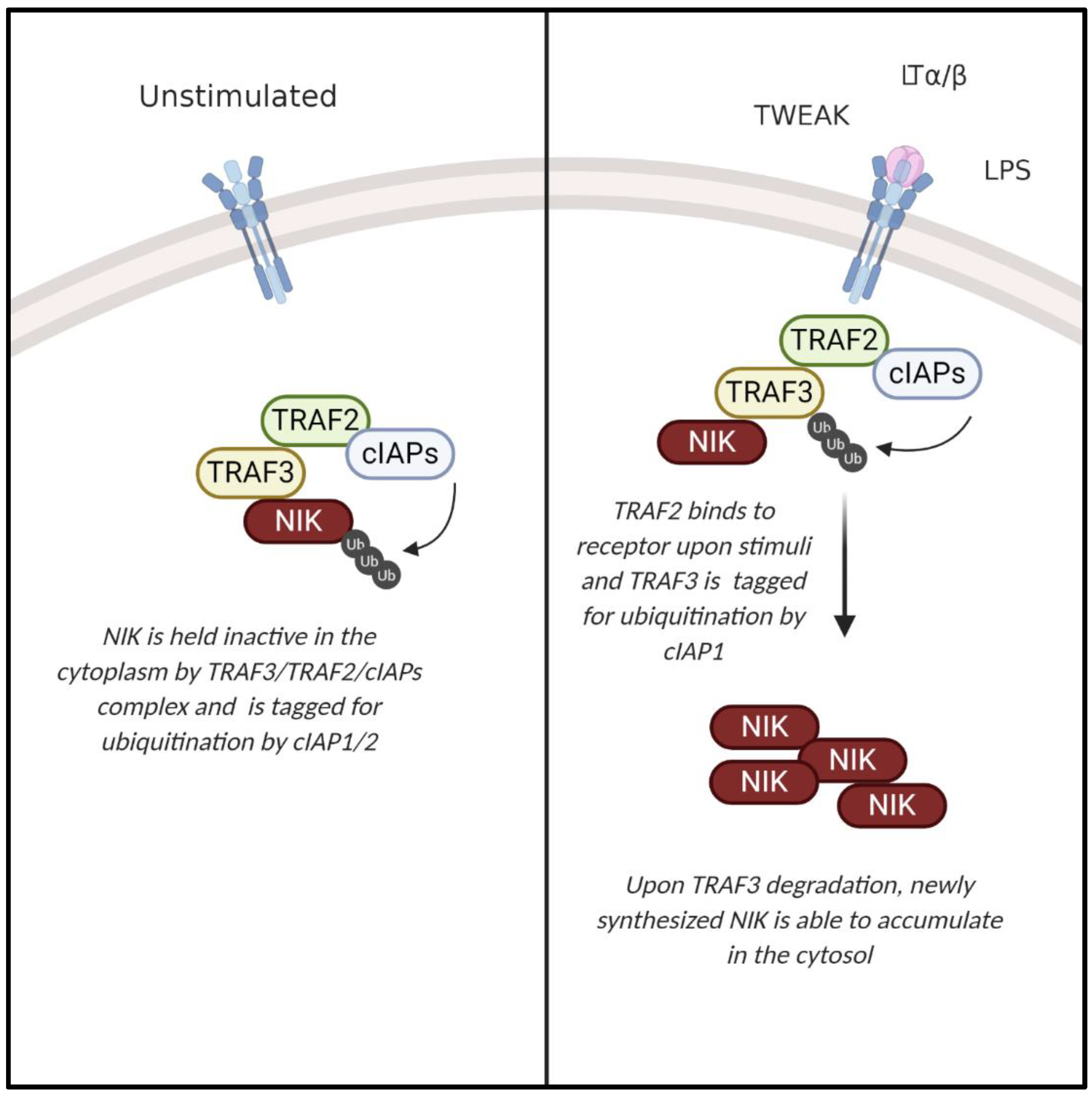
1.2. NIK and the Noncanonical NF-κB Pathway
2. NIK Regulation of Lymphoid Organogenesis, Immune Cell Development, and Hematopoiesis
2.1. Lymphoid Organogenesis and B-Cell Development
2.2. T-Cell Functions
2.3. Dendritic Cells
2.4. Macrophages
2.5. Hematopoietic Stem Cells (HSCs)
3. NIK in Immune, Inflammatory, and Metabolic Diseases
3.1. Immunodeficiency and Autoimmune Disorders
3.2. Systemic Lupus Erythematosus
3.3. Rheumatoid Arthritis
3.4. Bone Disorders
3.5. Aging and Cardiovascular Disease
3.6. Metabolic Disorders
4. Aberrant NIK Regulation in Cancer
4.1. Cancer Stem Cells
4.2. Leukemias and Lymphomas
4.3. Melanoma
4.4. Breast Cancer
4.5. Brain Cancer
4.6. Other Solid Tumors
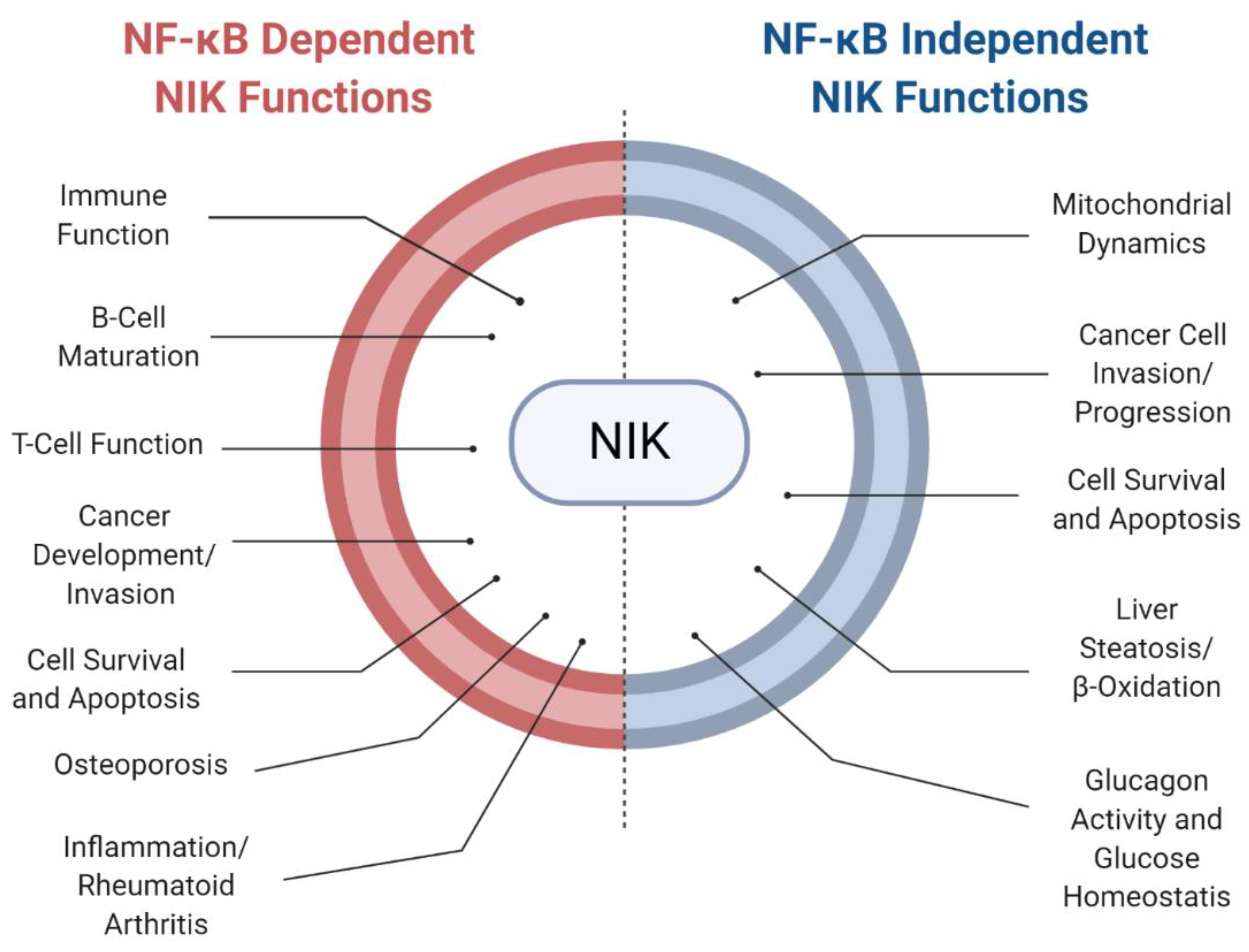
5. NF-κB-Independent Roles for NIK in Disease
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ALL | acute lymphoblastic leukemia |
| AML | acute myeloid leukemia |
| Aly | alymphoplasia |
| BAFF/BAFFR | B cell activating factor/B cell activating factor receptor |
| β TrCP | beta transducin repeats containing proteins |
| CDK4 | cyclin-dependent kinase 4 |
| cIAP1/2 | cellular inhibitor of apoptosis protein 1 and 2 |
| CML | chronic myeloid leukemia |
| Cpd33 | compound 33 |
| CREB | cAMP response element binding |
| CSC | cancer stem cells |
| DRP1 | dynamine-related protein 1 |
| ERK | extracellular signal regulated kinase |
| Eva1 | epithelial V-like antigen 1 |
| FKBP5 | FK506-binding protein 51 (FKBP51) |
| GBM | glioblastoma multiforme |
| GIC | GBM initiating cells |
| GVHD | graft versus host disease |
| GWAS | genome-wide association study |
| HL | Hodgkin lymphoma |
| hNIK | human NIK |
| IC50 | inhibitory concentration (half of maximal) |
| IGF-1R | insulin like growth factor 1 receptor |
| IBD | inflammatory bowel disease |
| IκBα | inhibitory kappa B alpha |
| IKKα | inhibitory kappa B kinase alpha (aka CHUK) |
| IKKβ | inhibitory kappa B kinase beta |
| IKKγ | inhibitory kappa B kinase gamma (aka NEMO) |
| IL | interleukin |
| INF-γ | interferon gamma |
| LCMV | lymphocytic choriomeningitis virus |
| LPS | lipopolysaccharide |
| LTα/β | lymphotoxin alpha/beta |
| MAP3K14 | mitogen activated protein kinase, kinase, kinase 14 (aka NIK) |
| MCL | Mantle cell lymphoma |
| MHC | major histocompatibility complex |
| MLL1 | mixed-lineage leukemia 1 |
| MMP9 | matrix metalloproteinase 9 |
| mNIK | mouse NIK |
| MT1-MMP | membrane-type matrix metalloproteinase |
| NFATc1 | nuclear factor of activated T-cells 1 |
| NF-κB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| NIK | NF-κB-inducing kinase |
| NRD | negative regulatory domain |
| OVX | ovariectomy |
| PD1 | programmed cell death protein 1 |
| PPARα | peroxisome proliferator-activated receptor alpha |
| RA | rheumatoid arthritis |
| RANK | receptor activator of NF-κB |
| RANKL | receptor activator of NF-κB ligand |
| RIP1 | receptor interacting kinase 1 |
| SIX | sine oculis homeobox homologue family transcription factors |
| SMAC | second mitochondria-derived activator of caspases |
| SMI1 | small molecule Inhibitor 1 |
| SPP | several species |
| TAK1 | transforming growth factor beta-activated kinase 1 |
| TBK1 | TANK-binding kinase 1 |
| TLR | Toll-like receptor |
| TNF | tumor necrosis factor |
| TNFSFR | tumor necrosis factor superfamily receptor |
| TRAF2/3 | TNF-receptor associated factor 2 and 3 |
| TWEAK | tumor necrosis factor-like weak inducer of apoptosis |
| VSV | vesicular stomatitis virus |
References
- Zhang, Q.; Lenardo, M.J.; Baltimore, D. 30 Years of NF-κB: A Blossoming of Relevance to Human Pathobiology. Cell 2017, 168, 37–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonizzi, G.; Karin, M. The two NF-κB activation pathways and their role in innate and adaptive immunity. Trends Immunol. 2004, 25, 280–288. [Google Scholar] [CrossRef] [PubMed]
- Gilmore, T.D. Introduction to NF-kappaB: Players, pathways, perspectives. Oncogene 2006, 25, 6680–6684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Häcker, H.; Karin, M. Regulation and function of IKK and IKK-related kinases. Sci. STKE Signal Transduct. Knowl. Environ. 2006, 2006, re13. [Google Scholar] [CrossRef]
- Israël, A. The IKK complex, a central regulator of NF-kappaB activation. Cold Spring Harb. Perspect. Biol. 2010, 2, a000158. [Google Scholar] [CrossRef] [Green Version]
- Salmerón, A.; Janzen, J.; Soneji, Y.; Bump, N.; Kamens, J.; Allen, H.; Ley, S.C. Direct Phosphorylation of NF-κB1 p105 by the IκB Kinase Complex on Serine 927 Is Essential for Signal-induced p105 Proteolysis. J. Biol. Chem. 2001, 276, 22215–22222. [Google Scholar] [CrossRef] [Green Version]
- Orian, A.; Gonen, H.; Bercovich, B.; Fajerman, I.; Eytan, E.; Iwai, K.; Schwartz, A.L.; Ciechanover, A. SCFb-TrCP ubiquitin ligase-mediated processing of NF-kB p105 requires phosphorylation of its C-terminus by IkB kinase. EMBO J. 2020, 19, 2580–2591. [Google Scholar] [CrossRef] [Green Version]
- Lin, L.; DeMartino, G.N.; Greene, W.C. Cotranslational biogenesis of NF-kappaB p50 by the 26S proteasome. Cell 1998, 92, 819–828. [Google Scholar] [CrossRef] [Green Version]
- Baldwin, A.S. The NF-kappa B and I kappa B proteins: New discoveries and insights. Annu. Rev. Immunol. 1996, 14, 649–683. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.C. The noncanonical NF-κB pathway. Immunol. Rev. 2012, 246, 125–140. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.C. The non-canonical NF-κB pathway in immunity and inflammation. Nat. Rev. Immunol. 2017, 17, 545–558. [Google Scholar] [CrossRef] [PubMed]
- Cildir, G.; Low, K.C.; Tergaonkar, V. Noncanonical NF-κB Signaling in Health and Disease. Trends Mol. Med. 2016, 22, 414–429. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Wu, L.; Wesche, H.; Arthur, C.D.; White, J.M.; Goeddel, D.V.; Schreiber, R.D. Defective lymphotoxin-beta receptor-induced NF-kappaB transcriptional activity in NIK-deficient mice. Sci. N. Y. 2001, 291, 2162–2165. [Google Scholar] [CrossRef] [PubMed]
- Shinkura, R.; Kitada, K.; Matsuda, F.; Tashiro, K.; Ikuta, K.; Suzuki, M.; Kogishi, K.; Serikawa, T.; Honjo, T. Alymphoplasia is caused by a point mutation in the mouse gene encoding Nf-kappa b-inducing kinase. Nat. Genet. 1999, 22, 74–77. [Google Scholar] [CrossRef]
- de Leon-Boenig, G.; Bowman, K.K.; Feng, J.A.; Crawford, T.; Everett, C.; Franke, Y.; Oh, A.; Stanley, M.; Staben, S.T.; Starovasnik, M.A.; et al. The crystal structure of the catalytic domain of the NF-κB inducing kinase reveals a narrow but flexible active site. Structure 2012, 20, 1704–1714. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Sudom, A.; Min, X.; Cao, Z.; Gao, X.; Ayres, M.; Lee, F.; Cao, P.; Johnstone, S.; Plotnikova, O.; et al. Structure of the nuclear factor κB-inducing kinase (NIK) kinase domain reveals a constitutively active conformation. J. Biol. Chem. 2012, 287, 27326–27334. [Google Scholar] [CrossRef] [Green Version]
- Qing, G.; Qu, Z.; Xiao, G. Stabilization of basally translated NF-kappaB-inducing kinase (NIK) protein functions as a molecular switch of processing of NF-kappaB2 p100. J. Biol. Chem. 2005, 280, 40578–40582. [Google Scholar] [CrossRef] [Green Version]
- Ling, L.; Cao, Z.; Goeddel, D.V. NF-kappaB-inducing kinase activates IKK-alpha by phosphorylation of Ser-176. Proc. Natl. Acad. Sci. USA 1998, 95, 3792–3797. [Google Scholar] [CrossRef] [Green Version]
- Heusch, M.; Lin, L.; Geleziunas, R.; Greene, W.C. The generation of nfkb2 p52: Mechanism and efficiency. Oncogene 1999, 18, 6201–6208. [Google Scholar] [CrossRef] [Green Version]
- Xiao, G.; Fong, A.; Sun, S.C. Induction of p100 Processing by NF-κB-inducing Kinase Involves Docking IκB Kinase α (IKKα) to p100 and IKKα-mediated Phosphorylation. J. Biol. Chem. 2004, 279, 30099–30105. [Google Scholar] [CrossRef] [Green Version]
- Gray, C.M.; Remouchamps, C.; McCorkell, K.A.; Solt, L.A.; Dejardin, E.; Orange, J.S.; May, M.J. Noncanonical NF-κB signaling is limited by classical NF-κB activity. Sci. Signal. 2014, 7, ra13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bram, R.J. TBK1 suppression of IgA in the NIK of time. Nat. Immunol. 2012, 13, 1027–1029. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Xiao, Y.; Chang, J.H.; Yu, J.; Hu, H.; Starr, R.; Brittain, G.C.; Chang, M.; Cheng, X.; Sun, S.C. The kinase TBK1 controls IgA class switching by negatively regulating noncanonical NF-κB signaling. Nat. Immunol. 2012, 13, 1101–1109. [Google Scholar] [CrossRef] [Green Version]
- Razani, B.; Zarnegar, B.; Ytterberg, A.J.; Shiba, T.; Dempsey, P.W.; Ware, C.F.; Loo, J.A.; Cheng, G. Negative Feedback in Noncanonical NF- B Signaling Modulates NIK Stability Through IKK -Mediated Phosphorylation. Sci. Signal. 2010, 3, ra41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, T.; Gao, Z.; Zhang, Y.; Fan, K.; Wang, F.; Li, Y.; Zhong, J.; Fan, H.Y.; Cao, Q.; Zhou, J.; et al. CRL4DCAF2 negatively regulates IL-23 production in dendritic cells and limits the development of psoriasis. J. Exp. Med. 2018, 215, 1999–2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyawaki, S.; Nakamura, Y.; Suzuka, H.; Koba, M.; Yasumizu, R.; Ikehara, S.; Shibata, Y. A new mutation, aly, that induces a generalized lack of lymph nodes accompanied by immunodeficiency in mice. Eur. J. Immunol. 1994, 24, 429–434. [Google Scholar] [CrossRef] [PubMed]
- Myles, A.; Cancro, M.P. The NIK of time for B cells. Eur. J. Immunol. 2016, 46, 547–551. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Wang, H.; Zhou, X.; Xie, X.; Chen, X.; Jie, Z.; Zou, Q.; Hu, H.; Zhu, L.; Cheng, X.; et al. Cell intrinsic role of NF-κB-inducing kinase in regulating T cell-mediated immune and autoimmune responses. Sci. Rep. 2016, 6, 22115. [Google Scholar] [CrossRef] [Green Version]
- Lacher, S.M.; Thurm, C.; Distler, U.; Mohebiany, A.N.; Israel, N.; Kitic, M.; Ebering, A.; Tang, Y.; Klein, M.; Wabnitz, G.H.; et al. NF-κB inducing kinase (NIK) is an essential post-transcriptional regulator of T-cell activation affecting F-actin dynamics and TCR signaling. J. Autoimmun. 2018. [Google Scholar] [CrossRef]
- Hamdan, T.A.; Bhat, H.; Cham, L.B.; Adomati, T.; Lang, J.; Li, F.; Murtaza, A.; Hardt, C.; Lang, P.A.; Duhan, V.; et al. Map3k14 as a Regulator of Innate and Adaptive Immune Response during Acute Viral Infection. Pathogens 2020, 9, 96. [Google Scholar] [CrossRef] [Green Version]
- Murray, S.E.; Polesso, F.; Rowe, A.M.; Basak, S.; Koguchi, Y.; Toren, K.G.; Hoffmann, A.; Parker, D.C. NF-κB–inducing kinase plays an essential T cell–intrinsic role in graft-versus-host disease and lethal autoimmunity in mice. J. Clin. Investig. 2011, 121, 4775. [Google Scholar] [CrossRef] [PubMed]
- Häcker, H.; Chi, L.; Rehg, J.E.; Redecke, V. NIK Prevents the Development of Hypereosinophilic Syndrome-like Disease in Mice Independent of IKKα Activation. J. Immunol. 2012, 188, 4602–4610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jie, Z.; Yang, J.Y.; Gu, M.; Wang, H.; Xie, X.; Li, Y.; Liu, T.; Zhu, L.; Shi, J.; Zhang, L.; et al. NIK signaling axis regulates dendritic cell function in intestinal immunity and homeostasis. Nat. Immunol. 2018, 19, 1224–1235. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, J.; Mair, F.; Greter, M.; Schmidt-Supprian, M.; Becher, B. NIK signaling in dendritic cells but not in T cells is required for the development of effector T cells and cell-mediated immune responses. J. Exp. Med. 2011, 208, 1917–1929. [Google Scholar] [CrossRef] [PubMed]
- Tamura, C.; Nakazawa, M.; Kasahara, M.; Hotta, C.; Yoshinari, M.; Sato, F.; Minami, M. Impaired function of dendritic cells in alymphoplasia (aly/aly) mice for expansion of CD25+CD4+ regulatory T cells. Autoimmunity 2006, 39, 445–453. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Mar, K.B.; Hanners, N.W.; Perelman, S.S.; Kanchwala, M.; Xing, C.; Schoggins, J.W.; Alto, N.M. A NIK–SIX signalling axis controls inflammation by targeted silencing of non-canonical NF-κB. Nature 2019, 22, 414–426. [Google Scholar] [CrossRef]
- González-Murillo, Á.; Fernández, L.; Baena, S.; Melen, G.J.; Sánchez, R.; Sánchez-Valdepeñas, C.; Segovia, J.C.; Liou, H.C.; Schmid, R.; Madero, L.; et al. The NFKB Inducing Kinase Modulates Hematopoiesis During Stress. Stem Cells 2015, 33, 2825–2837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willmann, K.L.; Klaver, S.; Doğu, F.; Santos-Valente, E.; Garncarz, W.; Bilic, I.; Mace, E.; Salzer, E.; Conde, C.D.; Sic, H.; et al. Biallelic loss-of-function mutation in NIK causes a primary immunodeficiency with multifaceted aberrant lymphoid immunity. Nat. Commun. 2014, 5, 5360. [Google Scholar] [CrossRef]
- Schlechter, N.; Glanzmann, B.; Hoal, E.G.; Schoeman, M.; Petersen, B.S.; Franke, A.; Lau, Y.L.; Urban, M.; van Helden, P.D.; Esser, M.M.; et al. Exome Sequencing Identifies a Novel MAP3K14 Mutation in Recessive Atypical Combined Immunodeficiency. Front. Immunol. 2017, 8, 1624. [Google Scholar] [CrossRef] [Green Version]
- Brightbill, H.D.; Suto, E.; Blaquiere, N.; Ramamoorthi, N.; Sujatha-Bhaskar, S.; Gogol, E.B.; Castanedo, G.M.; Jackson, B.T.; Kwon, Y.C.; Haller, S.; et al. NF-κB inducing kinase is a therapeutic target for systemic lupus erythematosus. Nat. Commun. 2018, 9, 179. [Google Scholar] [CrossRef]
- Noort, A.R.; Tak, P.P.; Tas, S.W. Non-canonical NF-κB signaling in rheumatoid arthritis: Dr Jekyll and Mr Hyde? Arthritis Res. Ther. 2015, 17, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kucharzewska, P.; Maracle, C.X.; Jeucken, K.C.M.; van Hamburg, J.P.; Israelsson, E.; Furber, M.; Tas, S.W.; Olsson, H.K. NIK–IKK complex interaction controls NF-κB-dependent inflammatory activation of endothelium in response to LTβR ligation. J. Cell Sci. 2019, 132, jcs225615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arya, R.; del Rincon, I.; Farook, V.S.; Restrepo, J.F.; Winnier, D.A.; Fourcaudot, M.J.; Battafarano, D.F.; de Almeida, M.; Kumar, S.; Curran, J.E.; et al. Genetic Variants Influencing Joint Damage in Mexican Americans and European Americans With Rheumatoid Arthritis: Genetic Variants Influencing Joint Damage. Genet. Epidemiol. 2015, 39, 678–688. [Google Scholar] [CrossRef] [PubMed]
- Noort, A.R.; van Zoest, K.P.M.; van Baarsen, L.G.; Maracle, C.X.; Helder, B.; Papazian, N.; Romera-Hernandez, M.; Tak, P.P.; Cupedo, T.; Tas, S.W. Tertiary Lymphoid Structures in Rheumatoid Arthritis. Am. J. Pathol. 2015, 185, 1935–1943. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Zhang, H.; Su, X.; Luo, Y.; Li, X.; Yu, C.; Xie, Q.; Xia, X.; He, G.; Yang, L. Therapeutic effects of a novel BAFF blocker on arthritis. Signal Transduct. Target. Ther. 2019, 4, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Chen, M.; Zhou, Y.; Tang, C.; Zhang, W.; Zhong, Y.; Chen, Y.; Zhou, H.; Sheng, L. NIK links inflammation to hepatic steatosis by suppressing PPARα in alcoholic liver disease. Theranostics 2020, 10, 3579–3593. [Google Scholar] [CrossRef]
- Ren, X.; Li, X.; Jia, L.; Chen, D.; Hou, H.; Rui, L.; Zhao, Y.; Chen, Z. A small-molecule inhibitor of NF-κB-inducing kinase (NIK) protects liver from toxin-induced inflammation, oxidative stress, and injury. FASEB J. 2017, 31, 711–718. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Li, X.; Su, M.B.; Gao, L.X.; Zhou, Y.B.; Yuan, B.; Lyu, X.; Yan, Z.; Hu, C.; Zhang, H.; et al. Discovery of a Potent and Selective NF-κB-Inducing Kinase (NIK) Inhibitor That Has Anti-inflammatory Effects in Vitro and in Vivo. J. Med. Chem. 2020, 63, 4388–4407. [Google Scholar] [CrossRef]
- Blaquiere, N.; Castanedo, G.M.; Burch, J.D.; Berezhkovskiy, L.M.; Brightbill, H.; Brown, S.; Chan, C.; Chiang, P.C.; Crawford, J.J.; Dong, T.; et al. Scaffold-Hopping Approach To Discover Potent, Selective, and Efficacious Inhibitors of NF-κB Inducing Kinase. J. Med. Chem. 2018, 61, 6801–6813. [Google Scholar] [CrossRef]
- Takakura, N.; Matsuda, M.; Khan, M.; Hiura, F.; Aoki, K.; Hirohashi, Y.; Mori, K.; Yasuda, H.; Hirata, M.; Kitamura, C.; et al. A novel inhibitor of NF-κB-inducing kinase prevents bone loss by inhibiting osteoclastic bone resorption in ovariectomized mice. Bone 2020, 135, 115316. [Google Scholar] [CrossRef]
- Cheng, G.; Mei, X.; Yan, Y.; Chen, J.; Zhang, B.; Li, J.; Dong, X.; Lin, N.; Zhou, Y. Identification of new NIK inhibitors by discriminatory analysis-based molecular docking and biological evaluation. Arch. Pharm. 2019, 352, 1800374. [Google Scholar] [CrossRef] [PubMed]
- Mortier, J.; Masereel, B.; Remouchamps, C.; Ganeff, C.; Piette, J.; Frederick, R. NF-kappaB inducing kinase (NIK) inhibitors: Identification of new scaffolds using virtual screening. Bioorg. Med. Chem. Lett. 2010, 20, 4515–4520. [Google Scholar] [CrossRef] [PubMed]
- Ranuncolo, S.M.; Pittaluga, S.; Evbuomwan, M.O.; Jaffe, E.S.; Lewis, B.A. Hodgkin lymphoma requires stabilized NIK and constitutive RelB expression for survival. Blood 2012, 120, 3756–3763. [Google Scholar] [CrossRef] [Green Version]
- Pippione, A.C.; Sainas, S.; Federico, A.; Lupino, E.; Piccinini, M.; Kubbutat, M.; Contreras, J.M.; Morice, C.; Barge, A.; Ducime, A.; et al. N-Acetyl-3-aminopyrazoles block the non-canonical NF-kB cascade by selectively inhibiting NIK †Electronic supplementary information (ESI) available: Additional biochemical data, chemistry, NMR characterization of final compounds, and biochemical protocols. MedChemComm 2018, 9, 963–968. [Google Scholar] [CrossRef] [PubMed]
- Takeda, T.; Tsubaki, M.; Sakamoto, K.; Ichimura, E.; Enomoto, A.; Suzuki, Y.; Itoh, T.; Imano, M.; Tanabe, G.; Muraoka, O.; et al. Mangiferin, a novel nuclear factor kappa B-inducing kinase inhibitor, suppresses metastasis and tumor growth in a mouse metastatic melanoma model. Toxicol. Appl. Pharmacol. 2016, 306, 105–112. [Google Scholar] [CrossRef]
- Gough, A.K.S.; Emery, P.; Holder, R.L.; Lilley, J.; Eyre, S. Generalised bone loss in patients with early rheumatoid arthritis. Lancet 1994, 344, 23–27. [Google Scholar] [CrossRef]
- Zeng, R.; Faccio, R.; Novack, D.V. Alternative NF-κB Regulates RANKL-Induced Osteoclast Differentiation and Mitochondrial Biogenesis via Independent Mechanisms. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2015, 30, 2287–2299. [Google Scholar] [CrossRef] [Green Version]
- Aya, K.; Alhawagri, M.; Hagen-Stapleton, A.; Kitaura, H.; Kanagawa, O.; Veis Novack, D. NF-κB–inducing kinase controls lymphocyte and osteoclast activities in inflammatory arthritis. J. Clin. Investig. 2005, 115, 1848–1854. [Google Scholar] [CrossRef]
- Yang, C.; McCoy, K.; Davis, J.L.; Schmidt-Supprian, M.; Sasaki, Y.; Faccio, R.; Novack, D.V. NIK Stabilization in Osteoclasts Results in Osteoporosis and Enhanced Inflammatory Osteolysis. PLoS ONE 2010, 5, e15383. [Google Scholar] [CrossRef]
- Sheng, L.; Zhou, Y.; Chen, Z.; Ren, D.; Cho, K.W.; Jiang, L.; Shen, H.; Sasaki, Y.; Rui, L. NF-κB–inducing kinase (NIK) promotes hyperglycemia and glucose intolerance in obesity by augmenting glucagon action. Nat. Med. 2012, 18, 943–949. [Google Scholar] [CrossRef] [Green Version]
- Malle, E.K.; Zammit, N.W.; Walters, S.N.; Koay, Y.C.; Wu, J.; Tan, B.M.; Villanueva, J.E.; Brink, R.; Loudovaris, T.; Cantley, J.; et al. Nuclear factor κB-inducing kinase activation as a mechanism of pancreatic β cell failure in obesity. J. Exp. Med. 2015, 212, 1239–1254. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Sheng, L.; Xiong, Y.; Shen, H.; Liu, Y.; Rui, L. Liver NF-κB-Inducing Kinase Promotes Liver Steatosis and Glucose Counterregulation in Male Mice With Obesity. Endocrinology 2017, 158, 1207–1216. [Google Scholar] [CrossRef] [PubMed]
- Russo, L.; Lumeng, C.N. Properties and functions of adipose tissue macrophages in obesity. Immunology 2018, 155, 407–417. [Google Scholar] [CrossRef] [PubMed]
- Dorrington, M.G.; Fraser, I.D.C. NF-κB Signaling in Macrophages: Dynamics, Crosstalk, and Signal Integration. Front. Immunol. 2019, 10, 705. [Google Scholar] [CrossRef]
- Zeng, T.; Zhou, J.; He, L.; Zheng, J.; Chen, L.; Wu, C.; Xia, W. Blocking Nuclear Factor-Kappa B Protects against Diet-Induced Hepatic Steatosis and Insulin Resistance in Mice. PLoS ONE 2016, 11, e0149677. [Google Scholar] [CrossRef]
- Hoesel, B.; Schmid, J.A. The complexity of NF-κB signaling in inflammation and cancer. Mol. Cancer 2013, 12, 86. [Google Scholar] [CrossRef] [Green Version]
- Bassères, D.S.; Baldwin, A.S. Nuclear factor-κB and inhibitor of κB kinase pathways in oncogenic initiation and progression. Oncogene 2006, 25, 6817–6830. [Google Scholar] [CrossRef] [Green Version]
- Taniguchi, K.; Karin, M. NF-κB, inflammation, immunity and cancer: Coming of age. Nat. Rev. Immunol. 2018, 18, 309–324. [Google Scholar] [CrossRef]
- Xia, Y.; Shen, S.; Verma, I.M. NF-κB, an active player in human cancers. Cancer Immunol. Res. 2014, 2, 823–830. [Google Scholar] [CrossRef] [Green Version]
- Maubach, G.; Feige, M.H.; Lim, M.C.C.; Naumann, M. NF-kappaB-inducing kinase in cancer. Biochim. Biophys. Acta BBA Rev. Cancer 2019, 1871, 40–49. [Google Scholar] [CrossRef]
- Kaltschmidt, B.; Greiner, J.F.W.; Kadhim, H.M.; Kaltschmidt, C. Subunit-Specific Role of NF-κB in Cancer. Biomedicines 2018, 6, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pattabiraman, D.R.; Weinberg, R.A. Tackling the cancer stem cells—What challenges do they pose? Nat. Rev. Drug Discov. 2014, 13, 497–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vazquez-Santillan, K.; Melendez-Zajgla, J.; Jimenez-Hernandez, L.E.; Gaytan-Cervantes, J.; Muñoz-Galindo, L.; Piña-Sanchez, P.; Martinez-Ruiz, G.; Torres, J.; Garcia-Lopez, P.; Gonzalez-Torres, C.; et al. NF-kappaΒ-inducing kinase regulates stem cell phenotype in breast cancer. Sci. Rep. 2016, 6, 37340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schramek, D.; Leibbrandt, A.; Sigl, V.; Kenner, L.; Pospisilik, J.A.; Lee, H.J.; Hanada, R.; Joshi, P.A.; Aliprantis, A.; Glimcher, L.; et al. Osteoclast differentiation factor RANKL controls development of progestin-driven mammary cancer. Nature 2010, 468, 98–102. [Google Scholar] [CrossRef] [Green Version]
- Ohtsu, N.; Nakatani, Y.; Yamashita, D.; Ohue, S.; Ohnishi, T.; Kondo, T. Eva1 Maintains the Stem-like Character of Glioblastoma-Initiating Cells by Activating the Noncanonical NF- B Signaling Pathway. Cancer Res. 2016, 76, 171–181. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Torres, C.; Gaytan-Cervantes, J.; Vazquez-Santillan, K.; Mandujano-Tinoco, E.A.; Ceballos-Cancino, G.; Garcia-Venzor, A.; Zampedri, C.; Sanchez-Maldonado, P.; Mojica-Espinosa, R.; Jimenez-Hernandez, L.E.; et al. NF-κB Participates in the Stem Cell Phenotype of Ovarian Cancer Cells. Arch. Med. Res. 2017, 48, 343–351. [Google Scholar] [CrossRef]
- Rahal, R.; Frick, M.; Romero, R.; Korn, J.M.; Kridel, R.; Chun Chan, F.; Meissner, B.; Bhang, H.; Ruddy, D.; Kauffmann, A.; et al. Pharmacological and genomic profiling identifies NF-κB-targeted treatment strategies for mantle cell lymphoma. Nat. Med. 2014, 20, 87–92. [Google Scholar] [CrossRef]
- Studencka-Turski, M.; Maubach, G.; Feige, M.H.; Naumann, M. Constitutive activation of nuclear factor kappa B-inducing kinase counteracts apoptosis in cells with rearranged mixed lineage leukemia gene. Leukemia 2018, 32, 2498–2501. [Google Scholar] [CrossRef]
- Xiu, Y.; Dong, Q.; Li, Q.; Li, F.; Borcherding, N.; Zhang, W.; Boyce, B.; Xue, H.; Zhao, C. Stabilization of NF-κB-Inducing Kinase Suppresses MLL-AF9-Induced Acute Myeloid Leukemia. Cell Rep. 2018, 22, 350–358. [Google Scholar] [CrossRef]
- Wu, H.; Tschopp, J.; Lin, S.C. Smac mimetics and TNFalpha: A dangerous liaison? Cell 2007, 131, 655–658. [Google Scholar] [CrossRef] [Green Version]
- Morrish, E.; Brumatti, G.; Silke, J. Future Therapeutic Directions for Smac-Mimetics. Cells 2020, 9, 406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, L.; Smith, D.C.; Wang, S. Small-Molecule SMAC Mimetics as New Cancer Therapeutics. Pharmacol. Ther. 2014, 144, 82–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lueck, S.C.; Russ, A.C.; Botzenhardt, U.; Schlenk, R.F.; Zobel, K.; Deshayes, K.; Vucic, D.; Döhner, H.; Döhner, K.; Fulda, S.; et al. Smac mimetic induces cell death in a large proportion of primary acute myeloid leukemia samples, which correlates with defined molecular markers. Oncotarget 2016, 7, 49539–49551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheung, H.H.; St Jean, M.; Beug, S.T.; Lejmi-Mrad, R.; LaCasse, E.; Baird, S.D.; Stojdl, D.F.; Screaton, R.A.; Korneluk, R.G. SMG1 and NIK regulate apoptosis induced by Smac mimetic compounds. Cell Death Dis. 2011, 2, e146. [Google Scholar] [CrossRef] [PubMed]
- Tchoghandjian, A.; Jennewein, C.; Eckhardt, I.; Rajalingam, K.; Fulda, S. Identification of non-canonical NF-κB signaling as a critical mediator of Smac mimetic-stimulated migration and invasion of glioblastoma cells. Cell Death Dis. 2013, 4, e564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garris, C.S.; Arlauckas, S.P.; Kohler, R.H.; Trefny, M.P.; Garren, S.; Piot, C.; Engblom, C.; Pfirschke, C.; Siwicki, M.; Gungabeesoon, J.; et al. Successful anti-PD-1 cancer immunotherapy requires T cell-dendritic cell crosstalk involving the cytokines IFN-γ and IL-12. Immunity 2018, 49, 1148–1161. [Google Scholar] [CrossRef] [Green Version]
- Pan, D.; Kobayashi, A.; Jiang, P.; de Andrade, L.F.; Tay, R.E.; Luoma, A.M.; Tsoucas, D.; Qiu, X.; Lim, K.; Rao, P.; et al. A major chromatin regulator determines resistance of tumor cells to T cell–mediated killing. Science 2018, 359, 770–775. [Google Scholar] [CrossRef] [Green Version]
- Peng, J.; Hamanishi, J.; Matsumura, N.; Abiko, K.; Murat, K.; Baba, T.; Yamaguchi, K.; Horikawa, N.; Hosoe, Y.; Murphy, S.K.; et al. Chemotherapy Induces Programmed Cell Death-Ligand 1 Overexpression via the Nuclear Factor- B to Foster an Immunosuppressive Tumor Microenvironment in Ovarian Cancer. Cancer Res. 2015, 75, 5034–5045. [Google Scholar] [CrossRef] [Green Version]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef]
- Dhawan, P.; Richmond, A. A novel NF-kappa B-inducing kinase-MAPK signaling pathway up-regulates NF-kappa B activity in melanoma cells. J. Biol. Chem. 2002, 277, 7920–7928. [Google Scholar] [CrossRef] [Green Version]
- Thu, Y.M.; Su, Y.; Yang, J.; Splittgerber, R.; Na, S.; Boyd, A.; Mosse, C.; Simons, C.; Richmond, A. NF-κB inducing kinase (NIK) modulates melanoma tumorigenesis by regulating expression of pro-survival factors through the β-catenin pathway. Oncogene 2012, 31, 2580–2592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Wang, Y.; Mao, Z.; Huang, D.; Zhou, J.; Wang, X. Expression of NF-κB-inducing kinase in breast carcinoma tissue and its clinical significance. Int. J. Clin. Exp. Pathol. 2015, 8, 14824–14829. [Google Scholar] [PubMed]
- Xu, Y.; Zhang, Z.; Zhang, L.; Zhang, C. Novel module and hub genes of distinctive breast cancer associated fibroblasts identified by weighted gene co-expression network analysis. Breast Cancer 2020, 27, 1017–1028. [Google Scholar] [CrossRef] [PubMed]
- Differential Impact of Classical and Non-Canonical NF-κB Pathway-Related Gene Expression on the Survival of Breast Cancer Patients. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6775609/ (accessed on 21 September 2020).
- Duran, C.L.; Lee, D.W.; Jung, J.U.; Ravi, S.; Pogue, C.B.; Toussaint, L.G.; Bayless, K.J.; Sitcheran, R. NIK regulates MT1-MMP activity and promotes glioma cell invasion independently of the canonical NF-κB pathway. Oncogenesis 2016, 5, e231. [Google Scholar] [CrossRef] [Green Version]
- Cherry, E.M.; Lee, D.W.; Jung, J.U.; Sitcheran, R. Tumor necrosis factor-like weak inducer of apoptosis (TWEAK) promotes glioma cell invasion through induction of NF-κB-inducing kinase (NIK) and noncanonical NF-κB signaling. Mol. Cancer 2015, 14, 9. [Google Scholar] [CrossRef] [Green Version]
- Jung, J.U.; Ravi, S.; Lee, D.W.; McFadden, K.; Kamradt, M.L.; Toussaint, L.G.; Sitcheran, R. NIK/MAP3K14 Regulates Mitochondrial Dynamics and Trafficking to Promote Cell Invasion. Curr. Biol. 2016, 26, 3288–3302. [Google Scholar] [CrossRef] [Green Version]
- Döppler, H.; Liou, G.Y.; Storz, P. Downregulation of TRAF2 Mediates NIK-Induced Pancreatic Cancer Cell Proliferation and Tumorigenicity. PLoS ONE 2013, 8, e53676. [Google Scholar] [CrossRef]
- Nishina, T.; Yamaguchi, N.; Gohda, J.; Semba, K.; Inoue, J. NIK is involved in constitutive activation of the alternative NF-κB pathway and proliferation of pancreatic cancer cells. Biochem. Biophys. Res. Commun. 2009, 388, 96–101. [Google Scholar] [CrossRef]
- Teng, H.; Xue, L.; Wang, Y.; Ding, X.; Li, J. Nuclear factor κB -inducing kinase is a diagnostic marker of gastric cancer. Medicine 2020, 99, e18864. [Google Scholar] [CrossRef]
- Boutaffala, L.; Bertrand, M.J.M.; Remouchamps, C.; Seleznik, G.; Reisinger, F.; Janas, M.; Bénézech, C.; Fernandes, M.T.; Marchetti, S.; Mair, F.; et al. NIK promotes tissue destruction independently of the alternative NF-κB pathway through TNFR1/RIP1-induced apoptosis. Cell Death Differ. 2015, 22, 2020–2033. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease | NIK Inhibitor | Therapeutic Effect |
|---|---|---|
Inflammatory/immune diseases:
| B022 | Inhibitor of NIK and subsequent liver inflammation and steatosis under alcoholic liver model [46]. Reduction in liver inflammation and damage due to toxin treatment of carbon tetrachloride [47]. |
| XT2 | NIK inhibitor that suppresses hepatocyte inflammation generated by toxin-induced liver injury by carbon tetrachloride, but to a lesser extent than B022 [48]. | |
| NIK SMI1 (small molecule inhibitor 1) | Favorable to inhibition of BAFF-induced B-cell survival [49]. Inhibits NIK in immune cells with improved survival rate in murine model of lupus [40]. | |
| Cpd33 (NIK Specific Inhibitor Compound 33) | Specifically inhibits noncanonical NF-κB pathway. Studied with in vitro RANKL activation and inhibition of downstream transcription factor NFATc1. Treatment with Cpd33 inhibits osteoclastogenesis in vitro and in murine OVX model. Cpd33 treatment also inhibited bone absorption ability in mature osteoclasts and overall prevented bone loss in murine model [50]. | |
Cancer:
| N-(3-(6-benzamido-3a,7a-dihydrobenzo[d]oxazol-2-yl)-phenyl)benzamide | NIK inhibitor with general IC50 of 48.9 μM and inhibition rate of about 56%. This is inhibitor was less efficient than B022 that had an IC50 of 9.9 nM. In SW1990 (pancreatic cancer cells), inhibitor had IC50 of 20.1 μM [51]. |
| 4H-isoquinoline-1,3-dione | Inhibits NIK activity by insertion to ATP-binding site. Has an IC50 of 51 μM and analogs are inhibitors for CDK4 and IGF-1R [52]. Treatment of Hodgkin lymphoma through inhibition of NIK/RelB [53]. | |
| N-Acetyl-3-aminopyrazoles | Selective inhibitor of NIK with IC50 of about 8.4 μM. Over 80% NF-κB inhibition in multiple myeloma cells at 25 μM, but low inhibition in MDA-MB-231 and SKBr3 cells even at 100 μM [54]. | |
| Mangiferin | A natural inhibitor of NF-κB kinases including NIK in a dose-dependent manner. Treatment with 100–200 mg/kg of mangiferin significantly inhibited melanoma tumor growth and metastasis [55]. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pflug, K.M.; Sitcheran, R. Targeting NF-κB-Inducing Kinase (NIK) in Immunity, Inflammation, and Cancer. Int. J. Mol. Sci. 2020, 21, 8470. https://doi.org/10.3390/ijms21228470
Pflug KM, Sitcheran R. Targeting NF-κB-Inducing Kinase (NIK) in Immunity, Inflammation, and Cancer. International Journal of Molecular Sciences. 2020; 21(22):8470. https://doi.org/10.3390/ijms21228470
Chicago/Turabian StylePflug, Kathryn M., and Raquel Sitcheran. 2020. "Targeting NF-κB-Inducing Kinase (NIK) in Immunity, Inflammation, and Cancer" International Journal of Molecular Sciences 21, no. 22: 8470. https://doi.org/10.3390/ijms21228470