GM2 Gangliosidoses: Clinical Features, Pathophysiological Aspects, and Current Therapies

 ,
,  ,
,  and
and
Abstract
:1. Introduction
2. Gangliosides: Structure and Physiological Role
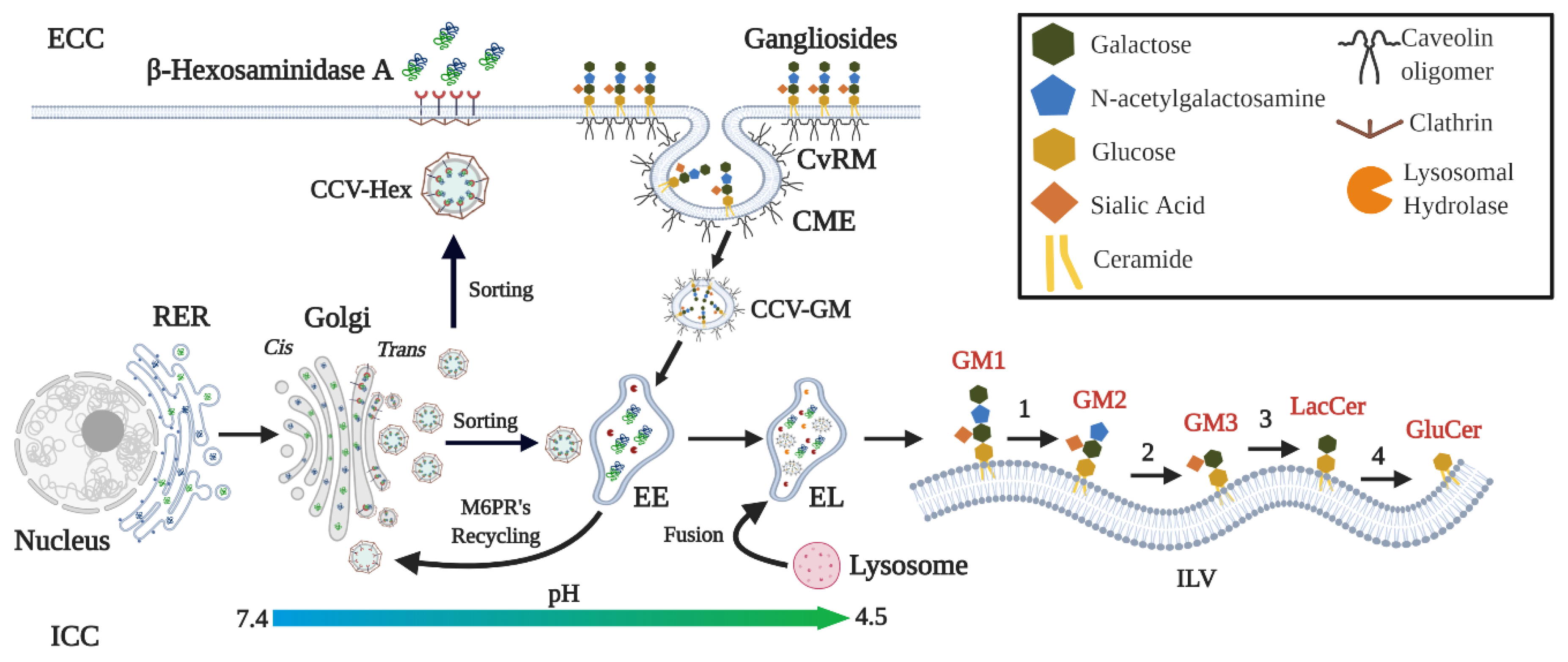
3. β-Hexosaminidases: Synthesis, Transport, and Catalytic Functions
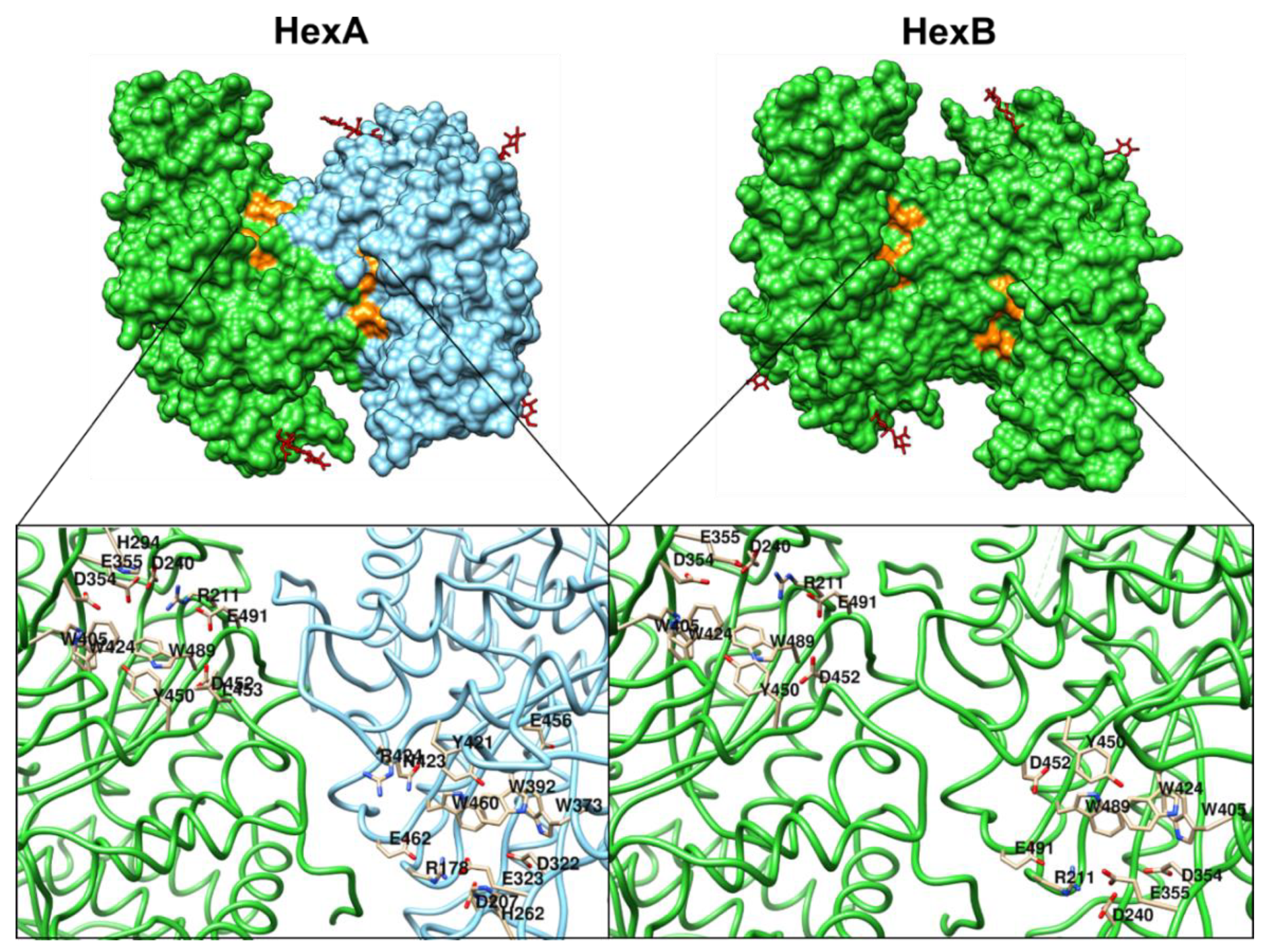
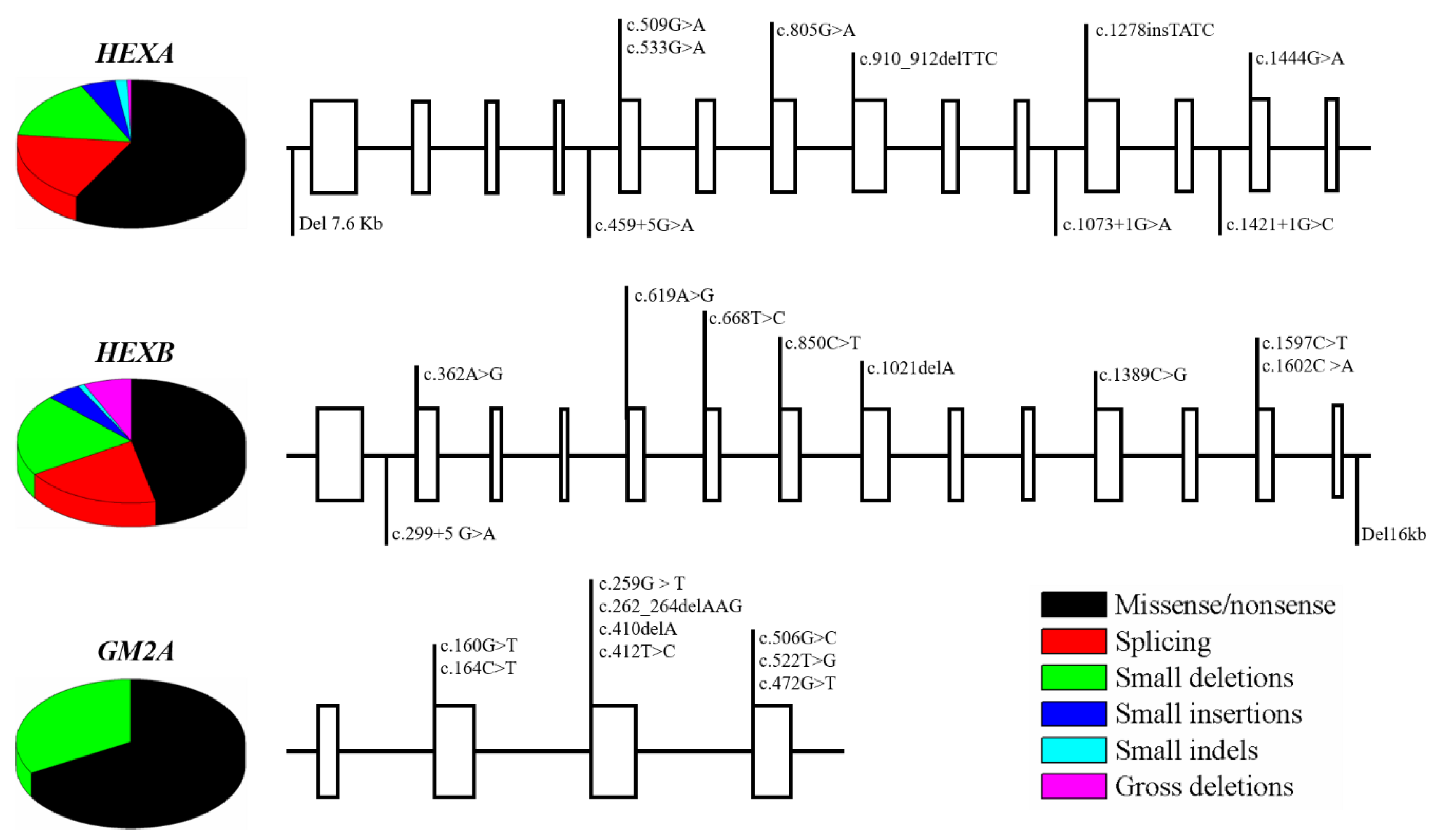
4. Mutations of β-Hexosaminidases A and B, and GM2 Activator Protein
5. Clinical Presentations and Biochemical Correlations of GM2 Gangliosidoses
Diagnosis of GM2 Gangliosidose
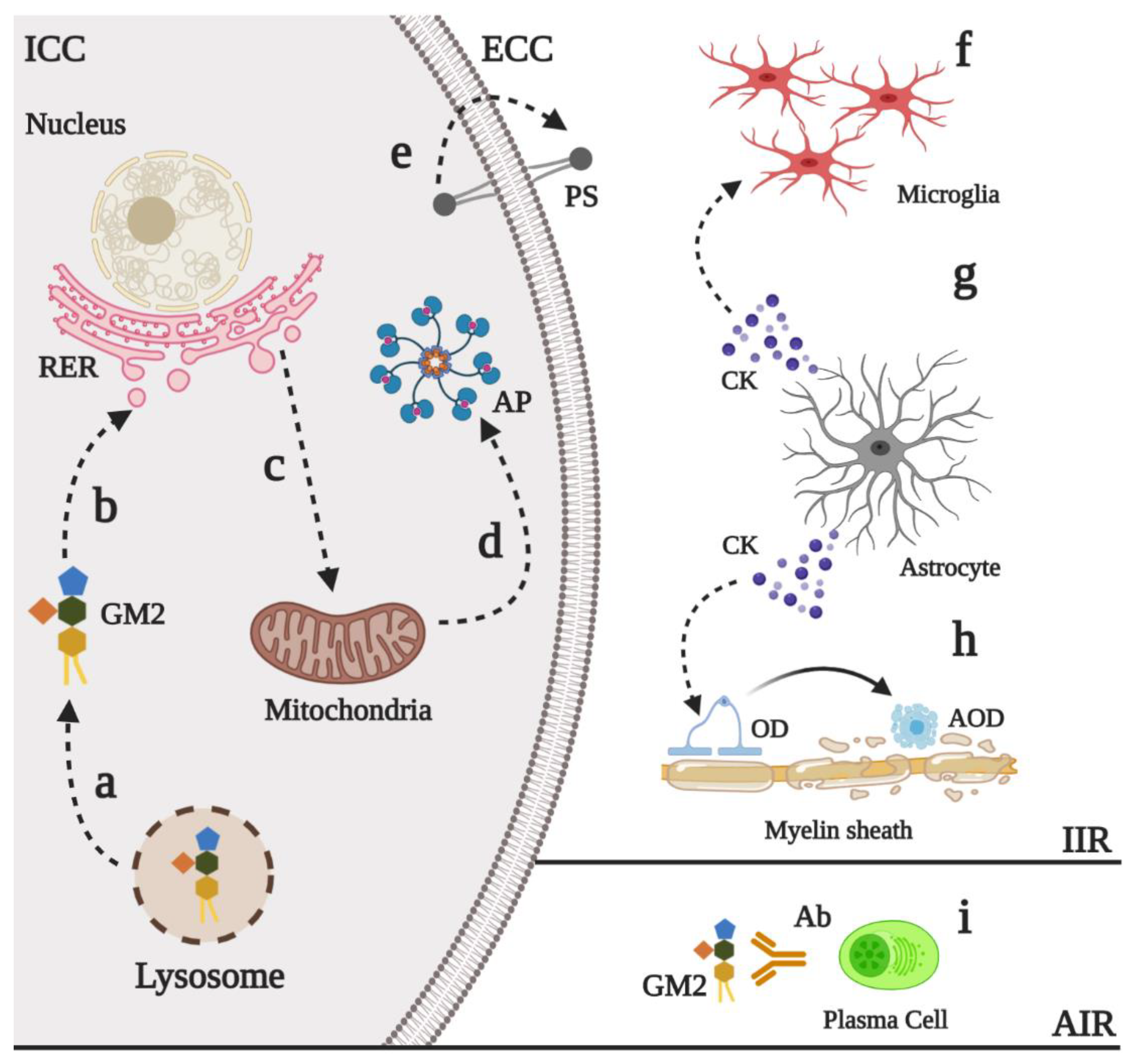
6. Physiopathology of GM2 Gangliosidoses
6.1. Neurodevelopment Process
6.2. Neural Death and Neuroinflammation
7. Current Proposals for the Treatment of GM2 Gangliosidoses
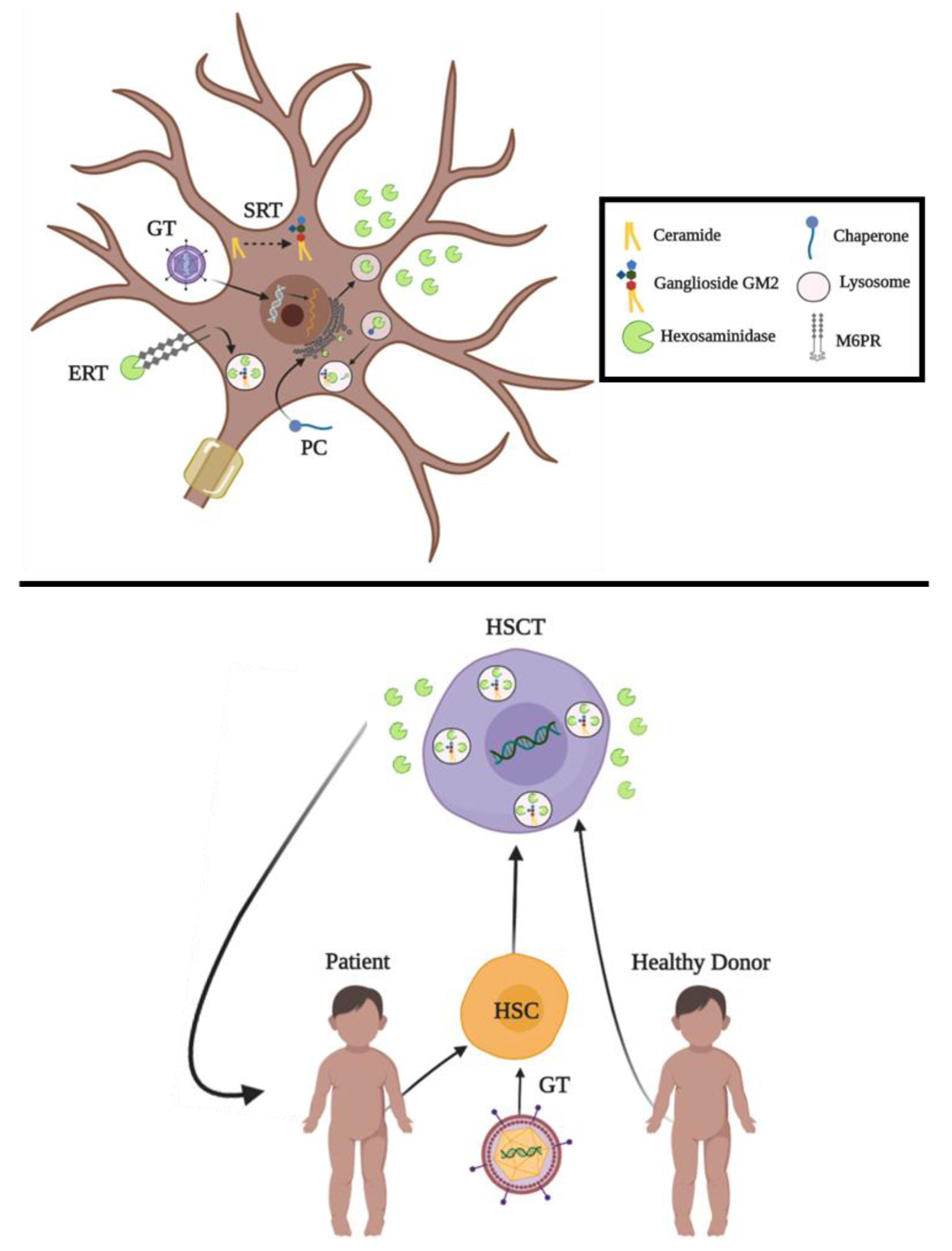
7.1. Enzyme Replacement Therapy
7.2. Hematopoietic Stem Cell Transplantation
7.3. Pharmacological Chaperones
7.4. Substrate Reduction Therapy
7.5. Gene Therapy
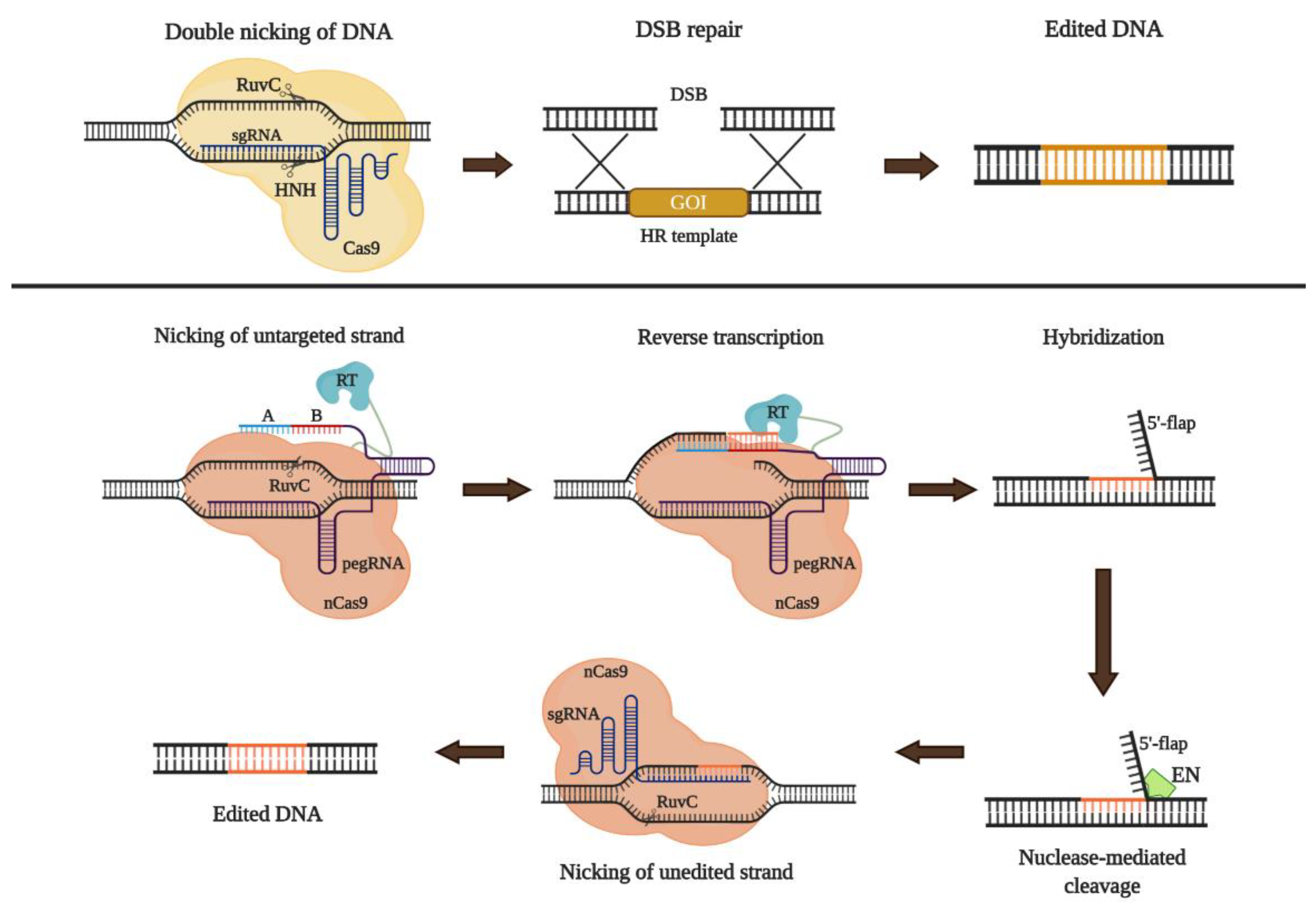
7.6. CRISPR/Cas9-Based Gene Therapy
8. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AAV | Adeno-associated virus |
| Ab | Antibodies |
| AIR | Adaptative immune response |
| AOD | Apoptotic oligodendrocytes |
| AP | Apoptosis |
| BBB | Blood–brain barrier |
| Cas9 | CRISPR-associated protein 9 |
| CK | Cytokines |
| CNS | Central Nervous System |
| CRISPR | Clustered Regularly Interspaced Short Palindromic Repeats |
| DMDP | 2,5-dideoxy-2,5-imino-D-mannitol |
| DMSO | Dimethyl sulfoxide |
| DOAJ | Directory of open access journals |
| DSB | Double-strand break |
| ECC | Extracellular compartment |
| EE | Early endosome |
| ERT | Enzyme Replacement Therapy |
| FDA | Food and Drug Administration |
| GAG | Glycosaminoglycans |
| GCS | Glucosylceramide synthase |
| GluCer | Glucosylceramide |
| GM2-AP | GM2-activator protein |
| GT | Gene therapy |
| GVHD | Graft-versus-host disease |
| HDR | Homologous direct repair |
| Hex | Hexosaminidase |
| HLA | human leukocyte antigen |
| HSCT | Hematopoietic Stem Cell Transplantation |
| ICC | Intracellular compartment |
| IIR | Innate immune response |
| iPS | Induced pluripotent stem |
| LacCer | Lactoceramide |
| LD | Linear dichroism |
| LSD | Lysosomal Storage Disorders |
| M6PR | Mannose-6 phosphate receptor |
| MDPI | Multidisciplinary Digital Publishing Institute |
| MPS | Mucopolysaccharidosis |
| MUG | 4-methlyumbelliferyl-N-acetylglucosaminide |
| MUGS | 4-methylumbelliferyl-beta-D-N-acetyl-glucosamine-6-sulfate |
| NHEJ | Non-homologous end joining |
| OD | Oligodendrocytes |
| PC | Pharmacological Chaperones |
| PERK | PKR-like endoplasmic reticulum kinase |
| PGRN | Progranulin |
| PS | Phosphatidylserine |
| PYR | Pyrimethamine |
| RER | Rough endoplasmic reticulum |
| SD | Sandhoff disease |
| SRT | Substrate Reduction Therapy |
| TGN | Trans-Golgi network |
| TSD | Tay–Sachs disease |
| UCB | Umbilical cordon blood |
References
- Sandhoff, K.; Harzer, K. Gangliosides and Gangliosidoses: Principles of Molecular and Metabolic Pathogenesis. J. Neurosci. 2013, 33, 10195–10208. [Google Scholar] [CrossRef] [PubMed]
- Schnaar, R.L. The Biology of Gangliosides. Adv. Carbohydr. Chem. Biochem. 2019, 76, 113–148. [Google Scholar] [PubMed]
- Ledeen, R.; Wu, G. Gangliosides of the Nervous System. Methods Mol. Biol. 2018, 1804, 19–55. [Google Scholar] [PubMed]
- Cachon-Gonzalez, M.B.; Zaccariotto, E.; Cox, T.M. Genetics and Therapies for GM2 Gangliosidosis. Curr. Gene Ther. 2018, 18, 68–89. [Google Scholar] [CrossRef]
- Virgolini, M.J.; Feliziani, C.; Cambiasso, M.J.; Lopez, P.H.; Bollo, M. Neurite Atrophy and Apoptosis Mediated by PERK Signaling after Accumulation of GM2-Ganglioside. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 225–239. [Google Scholar] [CrossRef]
- Masingue, M.; Dufour, L.; Lenglet, T.; Saleille, L.; Goizet, C.; Ayrignac, X.; Ory-Magne, F.; Barth, M.; Lamari, F.; Mandia, D.; et al. Natural History of Adult Patients with GM2 Gangliosidosis. Ann. Neurol. 2020, 87, 609–617. [Google Scholar] [CrossRef]
- Jain, A.; Kohli, A.; Sachan, D. Infantile Sandhoff’s Disease with Peripheral Neuropathy. Pediatr. Neurol. 2010, 42, 459–461. [Google Scholar] [CrossRef]
- Venugopalan, P.; Joshi, S.N. Cardiac Involvement in Infantile Sandhoff Disease. J. Paediatr. Child Health 2002, 38, 98–100. [Google Scholar] [CrossRef]
- Hall, P.; Minnich, S.; Teigen, C.; Raymond, K. Diagnosing Lysosomal Storage Disorders: The GM2 Gangliosidoses. Curr. Protoc. Hum. Genet. 2014, 83, 1–17. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, H.; Kornreich, R.; Yu, C. Prenatal Diagnosis of Tay-Sachs Disease. Methods Mol. Biol. 2019, 1885, 233–250. [Google Scholar] [CrossRef]
- Lawson, C.A.; Martin, D.R. Animal Models of GM2 Gangliosidosis: Utility and Limitations. Appl. Clin. Genet. 2016, 9, 111–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seyrantepe, V.; Demir, S.A.; Timur, Z.K.; Von Gerichten, J.; Marsching, C.; Erdemli, E.; Oztas, E.; Takahashi, K.; Yamaguchi, K.; Ates, N.; et al. Murine Sialidase Neu3 Facilitates GM2 Degradation and Bypass in Mouse Model of Tay-Sachs Disease. Exp. Neurol. 2018, 299, 26–41. [Google Scholar] [CrossRef] [PubMed]
- Concolino, D.; Deodato, F.; Parini, R. Enzyme Replacement Therapy: Efficacy and Limitations. Ital. J. Pediatr. 2018, 44, 120. [Google Scholar] [CrossRef] [PubMed]
- Giugliani, R.; Vairo, F.; Kubaski, F.; Poswar, F.; Riegel, M.; Baldo, G.; Saute, J.A. Neurological Manifestations of Lysosomal Disorders and Emerging Therapies Targeting the CNS. Lancet Child Adolesc. Health 2018, 2, 56–68. [Google Scholar] [CrossRef]
- Leal, A.F.; Espejo-Mojica, A.J.; Sánchez, O.F.; Ramírez, C.M.; Reyes, L.H.; Cruz, J.C.; Alméciga-Díaz, C.J. Lysosomal Storage Diseases: Current Therapies and Future Alternatives. J. Mol. Med. 2020, 1–16, in press. [Google Scholar] [CrossRef]
- Yu, R.K.; Tsai, Y.T.; Ariga, T.; Yanagisawa, M. Structures, Biosynthesis, and Functions of Gangliosides—An Overview. J. Oleo Sci. 2011, 60, 537–544. [Google Scholar] [CrossRef] [Green Version]
- Prokazova, N.V.; Samovilova, N.N.; Gracheva, E.V.; Golovanova, N.K. Ganglioside GM3 and its Biological Functions. Biochemistry 2009, 74, 235–249. [Google Scholar] [CrossRef]
- Aureli, M.; Mauri, L.; Ciampa, M.G.; Prinetti, A.; Toffano, G.; Secchieri, C.; Sonnino, S. GM1 Ganglioside: Past Studies and Future Potential. Mol. Neurobiol. 2016, 53, 1824–1842. [Google Scholar] [CrossRef]
- Riboni, L.; Malesci, A.; Gaini, S.M.; Sonnino, S.; Ghidoni, R.; Tettamanti, G. Ganglioside Pattern of Normal Human Brain, from Samples Obtained at Surgery. A Study Especially Referred to Alkali Labile Species. J. Biochem. 1984, 96, 1943–1946. [Google Scholar] [CrossRef]
- Zeller, C.B.; Marchase, R.B. Gangliosides as Modulators of Cell Function. Am. J. Physiol. 1992, 262, C1341–C1355. [Google Scholar] [CrossRef]
- Sonnino, S.; Chiricozzi, E.; Grassi, S.; Mauri, L.; Prioni, S.; Prinetti, A. Gangliosides in Membrane Organization. Prog. Mol. Biol. Transl. Sci. 2018, 156, 83–120. [Google Scholar] [CrossRef] [PubMed]
- Lopez, P.H.H.; Báez, B.B. Gangliosides in Axon Stability and Regeneration. Prog. Mol. Biol. Transl. Sci. 2018, 156, 383–412. [Google Scholar] [CrossRef] [PubMed]
- Todeschini, A.; Hakomori, S.I. Functional Role of Glycosphingolipids and Gangliosides in Control of Cell Adhesion, Motility, and Growth, through Glycosynaptic Microdomains. Biochim. Biophys. Acta Gen. Subj. 2008, 1780, 421–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groux-Degroote, S.; Rodríguez-Walker, M.; Dewald, J.H.; Daniotti, J.L.; Delannoy, P. Gangliosides in Cancer Cell Signaling. Prog. Mol. Biol. Transl. Sci. 2018, 156, 197–227. [Google Scholar] [CrossRef] [PubMed]
- Rubovitch, V.; Zilberstein, Y.; Chapman, J.; Schreiber, S.; Pick, C.G. Restoring GM1 Ganglioside Expression Ameliorates Axonal Outgrowth Inhibition and Cognitive Impairments Induced by Blast Traumatic Brain Injury. Sci. Rep. 2017, 7, 41269. [Google Scholar] [CrossRef] [Green Version]
- Kolter, T. Ganglioside Biochemistry. ISRN Biochem. 2012, 2012, 506160. [Google Scholar] [CrossRef] [Green Version]
- Lutz, M.S.; Jaskiewicz, E.; Darling, D.S.; Furukawa, K.; Young, W.W. Cloned Beta 1,4 N-Acetylgalactosaminyltransferase Synthesizes GA2 as Well as Gangliosides GM2 and GD2. GM3 Synthesis has Priority over GA2 Synthesis for Utilization of Lactosylceramide Substrate In Vivo. J. Biol. Chem. 1994, 269, 29227–29231. [Google Scholar]
- Mahuran, D.J. Biochemical Consequences of Mutations Causing the GM2 Gangliosidoses. Biochim. Biophys. Acta 1999, 1455, 105–138. [Google Scholar] [CrossRef] [Green Version]
- Hasilik, A.; Neufeld, E.F. Biosynthesis of Lysosomal Enzymes in Fibroblasts. Phosphorylation of Mannose Residues. J. Biol. Chem. 1980, 255, 4946–4950. [Google Scholar]
- Proia, R.L.; Soravia, E. Organization of the Gene Encoding the Human Beta-Hexosaminidase Alpha-Chain. J. Biol. Chem. 1987, 262, 5677–5681. [Google Scholar]
- Little, L.E.; Lau, M.M.; Quon, D.V.; Fowler, A.V.; Neufeld, E.F. Proteolytic Processing of the Alpha-Chain of the Lysosomal Enzyme, Beta-Hexosaminidase, in Normal Human Fibroblasts. J. Biol. Chem. 1988, 263, 4288–4292. [Google Scholar]
- Quon, D.V.; Proia, R.L.; Fowler, A.V.; Bleibaum, J.; Neufeld, E.F. Proteolytic Processing of the Beta-Subunit of the Lysosomal Enzyme, Beta-Hexosaminidase, in Normal Human Fibroblasts. J. Biol. Chem. 1989, 264, 3380–3384. [Google Scholar] [PubMed]
- Lemieux, M.J.; Mark, B.L.; Cherney, M.M.; Withers, S.G.; Mahuran, D.J.; James, M.N. Crystallographic Structure of Human Beta-Hexosaminidase A: Interpretation of Tay-Sachs Mutations and Loss of GM2 Ganglioside Hydrolysis. J. Mol. Biol. 2006, 359, 913–929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Dowd, B.F.; Cumming, D.A.; Gravel, R.A.; Mahuran, D. Oligosaccharide Structure and Amino Acid Sequence of the Major Glycopeptides of Mature Human Beta-Hexosaminidase. Biochemistry 1988, 27, 5216–5226. [Google Scholar] [CrossRef] [PubMed]
- Weitz, G.; Proia, R.L. Analysis of the Glycosylation and Phosphorylation of the Alpha-Subunit of the Lysosomal Enzyme, Beta-Hexosaminidase A, by Site-Directed Mutagenesis. J. Biol. Chem. 1992, 267, 10039–10044. [Google Scholar] [PubMed]
- Proia, R.L.; d’Azzo, A.; Neufeld, E.F. Association of Alpha- And Beta-Subunits during the Biosynthesis of Beta-Hexosaminidase in Cultured Human Fibroblasts. J. Biol. Chem. 1984, 259, 3350–3354. [Google Scholar] [PubMed]
- Saftig, P.; Klumperman, J. Lysosome Biogenesis and Lysosomal Membrane Proteins: Trafficking Meets Function. Nat. Rev. Mol. Cell Biol. 2009, 10, 623–635. [Google Scholar] [CrossRef]
- Sandhoff, R.; Sandhoff, K. Emerging Concepts of Ganglioside Metabolism. FEBS Lett. 2018, 592, 3835–3864. [Google Scholar] [CrossRef] [Green Version]
- Mencarelli, S.; Cavalieri, C.; Magini, A.; Tancini, B.; Basso, L.; Lemansky, P.; Hasilik, A.; Li, Y.T.; Chigorno, V.; Orlacchio, A.; et al. Identification of Plasma Membrane Associated Mature Beta-Hexosaminidase A, Active Towards GM2 Ganglioside, in Human Fibroblasts. FEBS Lett. 2005, 579, 5501–5506. [Google Scholar] [CrossRef] [Green Version]
- Tropak, M.B.; Yonekawa, S.; Karumuthil-Melethil, S.; Thompson, P.; Wakarchuk, W.; Gray, S.J.; Walia, J.S.; Mark, B.L.; Mahuran, D. Construction of a Hybrid Β-Hexosaminidase Subunit Capable of Forming Stable Homodimers that Hydrolyze GM2 Ganglioside In Vivo. Mol. Ther. Methods Clin. Dev. 2016, 3, 15057. [Google Scholar] [CrossRef]
- Lahey, H.G.; Webber, C.J.; Golebiowski, D.; Izzo, C.M.; Horn, E.; Taghian, T.; Rodriguez, P.; Batista, A.R.; Ellis, L.E.; Hwang, M.; et al. Pronounced Therapeutic Benefit of a Single Bidirectional AAV Vector Administered Systemically in Sandhoff Mice. Mol. Ther. 2020. [Google Scholar] [CrossRef]
- Maier, T.; Strater, N.; Schuette, C.G.; Klingenstein, R.; Sandhoff, K.; Saenger, W. The X-Ray Crystal Structure of Human Beta-Hexosaminidase B Provides New Insights into Sandhoff Disease. J. Mol. Biol. 2003, 328, 669–681. [Google Scholar] [CrossRef]
- Schröder, M.; Klima, H.; Nakano, T.; Kwon, H.; Quintern, L.E.; Gärtner, S.; Suzuki, K.; Sandhoff, K. Isolation of a cDNA Encoding the Human GM2 Activator Protein. FEBS Lett. 1989, 251, 197–200. [Google Scholar] [CrossRef] [Green Version]
- Wendeler, M.; Hoernschemeyer, J.; Hoffmann, D.; Kolter, T.; Schwarzmann, G.; Sandhoff, K. Photoaffinity Labelling of the Human GM2-Activator Protein. Mechanistic Insight into Ganglioside GM2 Degradation. Eur. J. Biochem. 2004, 271, 614–627. [Google Scholar] [CrossRef]
- Braulke, T.; Bonifacino, J.S. Sorting of Lysosomal Proteins. Biochim. Biophys. Acta 2009, 1793, 605–614. [Google Scholar] [CrossRef] [Green Version]
- Kleiman, F.E.; de Kremer, R.D.; de Ramirez, A.O.; Gravel, R.A.; Argaraña, C.E. Sandhoff disease in Argentina: High Frequency of a Splice Site Mutation in the HEXB Gene and Correlation between Enzyme and DNA-Based Tests for Heterozygote Detection. Hum. Genet. 1994, 94, 279–282. [Google Scholar] [CrossRef]
- Zampieri, S.; Cattarossi, S.; Oller Ramirez, A.M.; Rosano, C.; Lourenco, C.M.; Passon, N.; Moroni, I.; Uziel, G.; Pettinari, A.; Stanzial, F.; et al. Sequence and Copy Number Analyses of HEXB Gene in Patients Affected by Sandhoff Disease: Functional Characterization of 9 Novel Sequence Variants. PLoS ONE 2012, 7, e41516. [Google Scholar] [CrossRef]
- Sheth, J.; Datar, C.; Mistri, M.; Bhavsar, R.; Sheth, F.; Shah, K. GM2 Gangliosidosis AB Variant: Novel Mutation from INDIA—A Case Report with A Review. BMC Pediatr. 2016, 16, 88. [Google Scholar] [CrossRef] [Green Version]
- Martins, C.; Brunel-Guitton, C.; Lortie, A.; Gauvin, F.; Morales, C.R.; Mitchell, G.A.; Pshezhetsky, A.V. Atypical Juvenile Presentation of G M2 Gangliosidosis AB in a Patient Compound-Heterozygote for c.259G ≫ T and c.164C ≫ T Mutations in the GM2A Gene. Mol. Genet. Metab. Rep. 2017, 11, 24–29. [Google Scholar] [CrossRef]
- Bley, A.E.; Giannikopoulos, O.A.; Hayden, D.; Kubilus, K.; Tifft, C.J.; Eichler, F.S. Natural History of Infantile G(M2) Gangliosidosis. Pediatrics 2011, 128, e1233–e1241. [Google Scholar] [CrossRef]
- Maegawa, G.H.; Stockley, T.; Tropak, M.; Banwell, B.; Blaser, S.; Kok, F.; Giugliani, R.; Mahuran, D.; Clarke, J.T. The Natural History of Juvenile Or Subacute GM2 Gangliosidosis: 21 New Cases and Literature Review of 134 Previously Reported. Pediatrics 2006, 118, e1550–e1562. [Google Scholar] [CrossRef] [Green Version]
- Gushulak, L.; Hemming, R.; Martin, D.; Seyrantepe, V.; Pshezhetsky, A.; Triggs-Raine, B. Hyaluronidase 1 and β-Hexosaminidase have Redundant Functions in Hyaluronan and Chondroitin Sulfate Degradation. J. Biol. Chem. 2012, 287, 16689–16697. [Google Scholar] [CrossRef] [Green Version]
- Okada, S.; O’Brien, J.S. Tay-Sachs Disease: Generalized Absence of a Beta-D-N-Acetylhexosaminidase Component. Science 1969, 165, 698–700. [Google Scholar] [CrossRef] [PubMed]
- Gort, L.; de Olano, N.; Macías-Vidal, J.; Coll, M.A.; Group, S.G.W. GM2 Gangliosidoses in Spain: Analysis of the HEXA and HEXB Genes in 34 Tay-Sachs and 14 Sandhoff Patients. Gene 2012, 506, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Er, E.; Canda, E.; Yazıcı, H.; Eraslan, C.; Sozmen, E.; Ucar, S.; Coker, M. An Evalution of the Demographic and Clinical Characterictics of Patients with GM2 Gangliosidosis. J. Pediatr. Res. 2018, 5, 4. [Google Scholar] [CrossRef]
- Karimzadeh, P.; Jafari, N.; Nejad Biglari, H.; Jabbeh Dari, S.; Ahmad Abadi, F.; Alaee, M.R.; Nemati, H.; Saket, S.; Tonekaboni, S.H.; Taghdiri, M.M.; et al. GM2-Gangliosidosis (Sandhoff and Tay Sachs disease): Diagnosis and Neuroimaging Findings (An Iranian Pediatric Case Series). Iran. J. Child. Neurol. 2014, 8, 55–60. [Google Scholar]
- Neudorfer, O.; Pastores, G.M.; Zeng, B.J.; Gianutsos, J.; Zaroff, C.M.; Kolodny, E.H. Late-onset Tay-Sachs Disease: Phenotypic Characterization and Genotypic Correlations in 21 Affected Patients. Genet. Med. 2005, 7, 119–123. [Google Scholar] [CrossRef] [Green Version]
- Bellettato, C.; Tomanin, R.; Rigon, L.; Zanetti, A.; Volpi, N.; Scarpa, M. Glycosaminoglycans: Biosynthesis, Degradation, and Related Lysosomal Storage Disorders. In Mucopolysaccharidoses Update (2 Volume Set); Tomatsu, S., Lavery, C., Giugliani, R., Harmatz, P., Scarpa, M., Węgrzyn, G., Orii, T., Eds.; Nova Science Publishers, Inc.: Hauppauge, NY, USA, 2018; Volume I, pp. 115–142. [Google Scholar]
- Sandhoff, K. My Journey into the World of Sphingolipids and Sphingolipidoses. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2012, 88, 554–582. [Google Scholar] [CrossRef] [Green Version]
- Adam, M.P.; Ardinger, H.H.; Pagon, R.A.; Wallace, S.E.; Bean, L.J.H.; Stephens, K.; Amemiya, A. Hexosaminidase a Deficiency. In GeneReviews; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Leinekugel, P.; Michel, S.; Conzelmann, E.; Sandhoff, K. Quantitative correlation between the Residual Activity of Beta-Hexosaminidase A and Arylsulfatase A and the Severity of the Resulting Lysosomal Storage disEase. Hum. Genet. 1992, 88, 513–523. [Google Scholar] [CrossRef]
- Yamaguchi, A.; Katsuyama, K.; Nagahama, K.; Takai, T.; Aoki, I.; Yamanaka, S. Possible Role of Autoantibodies in the Pathophysiology of GM2 Gangliosidoses. J. Clin. Invest. 2004, 113, 200–208. [Google Scholar] [CrossRef]
- Parker, H.; Bigger, B.W. The Role of Innate Immunity in Mucopolysaccharide Diseases. J. Neurochem. 2019, 148, 639–651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ballabio, A. The Awesome Lysosome. EMBO Mol. Med. 2016, 8, 73–76. [Google Scholar] [CrossRef] [PubMed]
- Darios, F.; Stevanin, G. Impairment of Lysosome Function and Autophagy in Rare Neurodegenerative Diseases. J. Mol. Biol. 2020, 432, 2714–2734. [Google Scholar] [CrossRef]
- Yim, W.W.; Mizushima, N. Lysosome Biology in Autophagy. Cell Discov. 2020, 6, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Setia, H.; Muotri, A.R. Brain Organoids as a Model System for Human Neurodevelopment and Disease. Semin. Cell Dev. Biol. 2019, 95, 93–97. [Google Scholar] [CrossRef]
- Allende, M.L.; Cook, E.K.; Larman, B.C.; Nugent, A.; Brady, J.M.; Golebiowski, D.; Sena-Esteves, M.; Tifft, C.J.; Proia, R.L. Cerebral Organoids Derived from Sandhoff Disease-Induced Pluripotent Stem Cells Exhibit Impaired Neurodifferentiation. J. Lipid Res. 2018, 59, 550–563. [Google Scholar] [CrossRef] [Green Version]
- Kuil, L.E.; López Martí, A.; Carreras Mascaro, A.; van den Bosch, J.C.; van den Berg, P.; van der Linde, H.C.; Schoonderwoerd, K.; Ruijter, G.J.G.; van Ham, T.J. Hexb Enzyme Deficiency Leads to Lysosomal Abnormalities in Radial Glia and Microglia in Zebrafish Brain Development. Glia 2019, 67, 1705–1718. [Google Scholar] [CrossRef] [Green Version]
- Beattie, R.; Hippenmeyer, S. Mechanisms of Radial Glia Progenitor Cell Lineage Progression. FEBS Lett. 2017, 591, 3993–4008. [Google Scholar] [CrossRef] [Green Version]
- Sargeant, T.J.; Drage, D.J.; Wang, S.; Apostolakis, A.A.; Cox, T.M.; Cachón-González, M.B. Characterization of Inducible Models of Tay-Sachs and Related Disease. PLoS Genet. 2012, 8, e1002943. [Google Scholar] [CrossRef]
- Huang, J.Q.; Trasler, J.M.; Igdoura, S.; Michaud, J.; Hanal, N.; Gravel, R.A. Apoptotic Cell Death in Mouse Models of GM2 Gangliosidosis and Observations on Human Tay-Sachs and Sandhoff Diseases. Hum. Mol. Genet. 1997, 6, 1879–1885. [Google Scholar] [CrossRef] [Green Version]
- Wada, R.; Tifft, C.J.; Proia, R.L. Microglial Activation Precedes Acute Neurodegeneration in Sandhoff Disease and is Suppressed by Bone Marrow Transplantation. Proc. Natl. Acad. Sci. USA 2000, 97, 10954–10959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sargeant, T.J.; Wang, S.; Bradley, J.; Smith, N.J.; Raha, A.A.; McNair, R.; Ziegler, R.J.; Cheng, S.H.; Cox, T.M.; Cachón-González, M.B. Adeno-Associated Virus-Mediated Expression of Β-Hexosaminidase Prevents Neuronal Loss in the Sandhoff Mouse Brain. Hum. Mol. Genet. 2011, 20, 4371–4380. [Google Scholar] [CrossRef] [Green Version]
- Ginzburg, L.; Li, S.C.; Li, Y.T.; Futerman, A.H. An Exposed Carboxyl Group on Sialic Acid is Essential for Gangliosides to Inhibit Calcium Uptake via the Sarco/Endoplasmic Reticulum Ca2+-ATPase: Relevance to Gangliosidoses. J. Neurochem. 2008, 104, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Tian, M.; Ding, C.; Yu, S. The C/EBP Homologous Protein (CHOP) Transcription Factor Functions in Endoplasmic Reticulum Stress-Induced Apoptosis and Microbial Infection. Front. Immunol. 2018, 9, 3083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witting, A.; Müller, P.; Herrmann, A.; Kettenmann, H.; Nolte, C. Phagocytic clearance of apoptotic neurons by Microglia/Brain Macrophages In Vitro: Involvement of Lectin-, Integrin-, and Phosphatidylserine-Mediated Recognition. J. Neurochem. 2000, 75, 1060–1070. [Google Scholar] [CrossRef] [PubMed]
- Nonaka, S.; Nakanishi, H. Microglial Clearance of Focal Apoptotic Synapses. Neurosci. Lett. 2019, 707, 134317. [Google Scholar] [CrossRef]
- Bradbury, A.M.; Peterson, T.A.; Gross, A.L.; Wells, S.Z.; McCurdy, V.J.; Wolfe, K.G.; Dennis, J.C.; Brunson, B.L.; Gray-Edwards, H.; Randle, A.N.; et al. AAV-Mediated Gene Delivery Attenuates Neuroinflammation in Feline Sandhoff Disease. Neuroscience 2017, 340, 117–125. [Google Scholar] [CrossRef] [Green Version]
- Jeyakumar, M.; Thomas, R.; Elliot-Smith, E.; Smith, D.A.; van der Spoel, A.C.; d’Azzo, A.; Perry, V.H.; Butters, T.D.; Dwek, R.A.; Platt, F.M. Central Nervous System Inflammation is a Hallmark of Pathogenesis in Mouse Models of Gm1 and Gm2 Gangliosidosis. Brain 2003, 126, 974–987. [Google Scholar] [CrossRef] [Green Version]
- Gray-Edwards, H.L.; Randle, A.N.; Maitland, S.A.; Benatti, H.R.; Hubbard, S.M.; Canning, P.F.; Vogel, M.B.; Brunson, B.L.; Hwang, M.; Ellis, L.E.; et al. Adeno-Associated Virus Gene Therapy in a Sheep Model of Tay-Sachs Disease. Hum. Gene Ther. 2018, 29, 312–326. [Google Scholar] [CrossRef]
- Ogawa, Y.; Furusawa, E.; Saitoh, T.; Sugimoto, H.; Omori, T.; Shimizu, S.; Kondo, H.; Yamazaki, M.; Sakuraba, H.; Oishi, K. Inhibition of Astrocytic Adenosine Receptor A. Neurobiol. Dis. 2018, 118, 142–154. [Google Scholar] [CrossRef]
- Ogawa, Y.; Sano, T.; Irisa, M.; Kodama, T.; Saito, T.; Furusawa, E.; Kaizu, K.; Yanagi, Y.; Tsukimura, T.; Togawa, T.; et al. FcRγ-Dependent Immune Activation Initiates Astrogliosis During the Asymptomatic Phase of Sandhoff Disease Model Mice. Sci. Rep. 2017, 7, 40518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domingues, H.S.; Portugal, C.C.; Socodato, R.; Relvas, J.B. Oligodendrocyte, Astrocyte, and Microglia Crosstalk in Myelin Development, Damage, and Repair. Front. Cell Dev. Biol. 2016, 4, 71. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Barres, B.A. Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity 2017, 46, 957–967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haberland, C.; Brunngraber, E.; Witting, L.; Brown, B. The White Matter in G M2 Gangliosidosis. A Comparative Histopathological and Biochemical Study. Acta Neuropathol. 1973, 24, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Tavasoli, A.R.; Parvaneh, N.; Ashrafi, M.R.; Rezaei, Z.; Zschocke, J.; Rostami, P. Clinical Presentation and Outcome in Infantile Sandhoff Disease: A Case Series of 25 Patients from Iranian Neurometabolic Bioregistry with Five Novel Mutations. Orphanet. J. Rare Dis. 2018, 13, 130. [Google Scholar] [CrossRef]
- Boado, R.J.; Lu, J.Z.; Hui, E.K.; Lin, H.; Pardridge, W.M. Bi-Functional IgG-Lysosomal Enzyme Fusion Proteins for Brain Drug Delivery. Sci. Rep. 2019, 9, 18632. [Google Scholar] [CrossRef] [Green Version]
- Boado, R.J.; Hui, E.K.; Lu, J.Z.; Pardridge, W.M. Glycemic Control and Chronic Dosing of Rhesus Monkeys with a Fusion Protein of Iduronidase and a Monoclonal Antibody against the Human Insulin Receptor. Drug. Metab. Dispos. 2012, 40, 2021–2025. [Google Scholar] [CrossRef] [Green Version]
- Boado, R.J.; Ka-Wai Hui, E.; Zhiqiang Lu, J.; Pardridge, W.M. Insulin Receptor Antibody-Iduronate 2-Sulfatase Fusion Protein: Pharmacokinetics, Anti-Drug Antibody, and Safety Pharmacology in Rhesus Monkeys. Biotechnol. Bioeng. 2014, 111, 2317–2325. [Google Scholar] [CrossRef] [Green Version]
- Boado, R.J.; Lu, J.Z.; Hui, E.K.; Pardridge, W.M. Insulin Receptor Antibody-Sulfamidase Fusion Protein Penetrates the Primate Blood-Brain Barrier and Reduces Glycosoaminoglycans in Sanfilippo Type A Cells. Mol. Pharm. 2014, 11, 2928–2934. [Google Scholar] [CrossRef] [Green Version]
- Boado, R.J.; Lu, J.Z.; Hui, E.K.; Lin, H.; Pardridge, W.M. Insulin Receptor Antibody-α-N-Acetylglucosaminidase Fusion Protein Penetrates the Primate Blood-Brain Barrier and Reduces Glycosoaminoglycans in Sanfilippo Type B Fibroblasts. Mol. Pharm. 2016, 13, 1385–1392. [Google Scholar] [CrossRef]
- Beard, H.; Luck, A.J.; Hassiotis, S.; King, B.; Trim, P.J.; Snel, M.F.; Hopwood, J.J.; Hemsley, K.M. Determination of the Role of Injection Site on the Efficacy of Intra-CSF Enzyme Replacement Therapy in MPS IIIA Mice. Mol. Genet. Metab. 2015, 115, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Schulz, A.; Ajayi, T.; Specchio, N.; de Los Reyes, E.; Gissen, P.; Ballon, D.; Dyke, J.P.; Cahan, H.; Slasor, P.; Jacoby, D.; et al. Study of Intraventricular Cerliponase Alfa for CLN2 Disease. N. Engl. J. Med. 2018, 378, 1898–1907. [Google Scholar] [CrossRef] [PubMed]
- Li, M. Enzyme Replacement Therapy: A Review and Its Role in Treating Lysosomal Storage Diseases. Pediatr. Ann. 2018, 47, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Garbade, S.F.; Zielonka, M.; Mechler, K.; Kolker, S.; Hoffmann, G.F.; Staufner, C.; Mengel, E.; Ries, M. FDA Orphan Drug Designations for Lysosomal Storage Disorders—A Cross-Sectional Analysis. PLoS ONE 2020, 15, e0230898. [Google Scholar] [CrossRef]
- Johnson, W.G.; Desnick, R.J.; Long, D.M.; Sharp, H.L.; Krivit, W.; Brady, B.; Brady, R.O. Intravenous Injection of Purified Hexosaminidase a into a Patient with Tay-Sachs Disease. Birth Defects Orig. Artic. Ser. 1973, 9, 120–124. [Google Scholar]
- Matsuoka, K.; Tsuji, D.; Aikawa, S.; Matsuzawa, F.; Sakuraba, H.; Itoh, K. Introduction of an N-glycan Sequon into HEXA Enhances Human Beta-Hexosaminidase Cellular Uptake in a Model of Sandhoff Disease. Mol. Ther. 2010, 18, 1519–1526. [Google Scholar] [CrossRef] [Green Version]
- Tsuji, D.; Akeboshi, H.; Matsuoka, K.; Yasuoka, H.; Miyasaki, E.; Kasahara, Y.; Kawashima, I.; Chiba, Y.; Jigami, Y.; Taki, T.; et al. Highly Phosphomannosylated Enzyme Replacement Therapy for GM2 Gangliosidosis. Ann. Neurol. 2011, 69, 691–701. [Google Scholar] [CrossRef]
- Matsuoka, K.; Tamura, T.; Tsuji, D.; Dohzono, Y.; Kitakaze, K.; Ohno, K.; Saito, S.; Sakuraba, H.; Itoh, K. Therapeutic Potential of Intracerebroventricular Replacement of Modified Human β-Hexosaminidase B for GM2 Gangliosidosis. Mol. Ther. 2011, 19, 1017–1024. [Google Scholar] [CrossRef]
- Espejo-Mojica, A.J.; Mosquera, A.; Rodrfguez-Lopez, A.; Diaz, D.; Beltran, L.; Hernandez, F.L.; Alméciga-Diaz, C.J.; Barrera, L.A. Characterization of Recombinant Human Lysosomal Beta-Hexosaminidases Produced in the Methylotrophic Yeast Pichia Pastoris. Univ. Scientiarum 2016, 21, 195–217. [Google Scholar] [CrossRef] [Green Version]
- Espejo-Mojica, A.J.; Almeciga-Diaz, C.J.; Rodriguez, A.; Mosquera, A.; Diaz, D.; Beltran, L.; Diaz, S.; Pimentel, N.; Moreno, J.; Sanchez, J.; et al. Human Recombinant Lysosomal Enzymes Produced in Microorganisms. Mol. Genet. Metab. 2015, 116, 13–23. [Google Scholar] [CrossRef]
- Almeciga-Diaz, C.J.; Espejo-Mojica, A.; Rodriguez-López, A.; Losada, C.; Sanchez, J.; Ramírez, A.M.; Pimentel, N.; Beltran, L.M.; Diaz, D.; Barrera, L.A. Cell Uptake Evaluation of Human Recombinant Lysosomal Enzymes Produced in Pichia Pastoris. Mol. Genet. Metab. 2016, 117, S17–S18. [Google Scholar] [CrossRef]
- Vu, M.; Li, R.; Baskfield, A.; Lu, B.; Farkhondeh, A.; Gorshkov, K.; Motabar, O.; Beers, J.; Chen, G.; Zou, J.; et al. Neural Stem Cells for Disease Modeling and Evaluation of Therapeutics for Tay-Sachs Disease. Orphanet J. Rare Dis. 2018, 13, 152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pulido, Z.; Cifuentes, J.; Benincore, E.; Garzon, R.; Castellanos, M.C.; Leal, A.; Cruz, J.C.; Almeciga-Diaz, C.J.; Espejo-Mojica, A.J. Recombinant Hexosaminidases Conjugated to Magnetite Nanoparticles: Alternative Therapeutic Treatment Routes in GM2 Fibroblasts. Mol. Genet. Metab. 2020, 129, S132–S133. [Google Scholar] [CrossRef]
- Biffi, A. Hematopoietic Stem Cell Gene Therapy for Storage Disease: Current and New Indications. Mol. Ther. 2017, 25, 1155–1162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawamoto, K.; Chen, H.H.; Almeciga-Diaz, C.J.; Mason, R.W.; Tomatsu, S. Gene therapy for Mucopolysaccharidoses. Mol. Genet. Metab. 2018, 123, 59–68. [Google Scholar] [CrossRef]
- Hatzimichael, E.; Tuthill, M. Hematopoietic Stem Cell Transplantation. Stem. Cells Cloning 2010, 3, 105–117. [Google Scholar] [CrossRef] [Green Version]
- Hobbs, J.R.; Hugh-Jones, K.; Barrett, A.J.; Byrom, N.; Chambers, D.; Henry, K.; James, D.C.; Lucas, C.F.; Rogers, T.R.; Benson, P.F.; et al. Reversal of Clinical Features of Hurler’s Disease and Biochemical Improvement after Treatment by Bone-Marrow Transplantation. Lancet 1981, 2, 709–712. [Google Scholar] [CrossRef]
- Visigalli, I.; Delai, S.; Politi, L.S.; Di Domenico, C.; Cerri, F.; Mrak, E.; D’Isa, R.; Ungaro, D.; Stok, M.; Sanvito, F.; et al. Gene Therapy Augments the Efficacy of Hematopoietic Cell Transplantation and Fully Corrects Mucopolysaccharidosis Type I Phenotype in the Mouse Model. Blood 2010, 116, 5130–5139. [Google Scholar] [CrossRef]
- Gleitz, H.F.; Liao, A.Y.; Cook, J.R.; Rowlston, S.F.; Forte, G.M.; D’Souza, Z.; O’Leary, C.; Holley, R.J.; Bigger, B.W. Brain-targeted Stem Cell Gene Therapy Corrects Mucopolysaccharidosis Type Ii via Multiple Mechanisms. EMBO Mol. Med. 2018, 10, e8730. [Google Scholar] [CrossRef]
- Somaraju, U.R.; Tadepalli, K. Hematopoietic Stem Cell Transplantation for Gaucher Disease. Cochrane Database Syst. Rev. 2017, 10, CD006974. [Google Scholar] [CrossRef]
- Jacobs, J.F.; Willemsen, M.A.; Groot-Loonen, J.J.; Wevers, R.A.; Hoogerbrugge, P.M. Allogeneic BMT followed by Substrate Reduction Therapy in a Child With Subacute Tay-Sachs Disease. Bone Marrow Transpl. 2005, 36, 925–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prasad, V.K.; Mendizabal, A.; Parikh, S.H.; Szabolcs, P.; Driscoll, T.A.; Page, K.; Lakshminarayanan, S.; Allison, J.; Wood, S.; Semmel, D.; et al. Unrelated Donor Umbilical Cord Blood Transplantation for Inherited Metabolic Disorders in 159 Pediatric Patients from a Single Center: Influence of Cellular Composition of the Graft on Transplantation Outcomes. Blood 2008, 112, 2979–2989. [Google Scholar] [CrossRef] [PubMed]
- Ornaghi, F.; Sala, D.; Tedeschi, F.; Maffia, M.C.; Bazzucchi, M.; Morena, F.; Valsecchi, M.; Aureli, M.; Martino, S.; Gritti, A. Novel Bicistronic Lentiviral Vectors Correct Β-Hexosaminidase Deficiency in Neural and Hematopoietic Stem Cells and Progeny: Implications for In Vivo And Ex Vivo Gene Therapy of GM2 Gangliosidosis. Neurobiol. Dis. 2020, 134, 104667. [Google Scholar] [CrossRef] [PubMed]
- Madden, K.; Chabot-Richards, D. HLA Testing in the Molecular Diagnostic Laboratory. Virchows Arch. 2019, 474, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Wang, M.; Duan, S.; Franco, P.J.; Kenty, J.H.; Hedrick, P.; Xia, Y.; Allen, A.; Ferreira, L.M.R.; Strominger, J.L.; et al. Generation of Hypoimmunogenic Human Pluripotent Stem Cells. Proc. Natl. Acad. Sci. USA 2019, 116, 10441–10446. [Google Scholar] [CrossRef] [Green Version]
- Deuse, T.; Hu, X.; Gravina, A.; Wang, D.; Tediashvili, G.; De, C.; Thayer, W.O.; Wahl, A.; Garcia, J.V.; Reichenspurner, H.; et al. Hypoimmunogenic Derivatives of Induced Pluripotent Stem Cells Evade Immune Rejection in Fully Immunocompetent Allogeneic Recipients. Nat. Biotechnol. 2019, 37, 252–258. [Google Scholar] [CrossRef]
- Biffi, A. Gene Therapy for Lysosomal Storage Disorders: A Good Start. Hum. Mol. Genet. 2016, 25, R65–R75. [Google Scholar] [CrossRef] [Green Version]
- Arakawa, T.; Ejima, D.; Kita, Y.; Tsumoto, K. Small Molecule Pharmacological Chaperones: From Thermodynamic Stabilization to Pharmaceutical Drugs. Biochim. Biophys. Acta 2006, 1764, 1677–1687. [Google Scholar] [CrossRef]
- Cohen, F.E.; Kelly, J.W. Therapeutic Approaches to Protein-Misfolding Diseases. Nature 2003, 426, 905–909. [Google Scholar] [CrossRef]
- Losada Díaz, J.C.; Cepeda del Castillo, J.; Rodriguez-López, E.A.; Alméciga-Díaz, C.J. Advances in the Development of Pharmacological Chaperones for the Mucopolysaccharidoses. Int. J. Mol. Sci. 2020, 21, 232. [Google Scholar] [CrossRef] [Green Version]
- Boyd, R.E.; Lee, G.; Rybczynski, P.; Benjamin, E.R.; Khanna, R.; Wustman, B.A.; Valenzano, K.J. Pharmacological Chaperones as Therapeutics for Lysosomal Storage Diseases. J. Med. Chem. 2013, 56, 2705–2725. [Google Scholar] [CrossRef] [PubMed]
- Liguori, L.; Monticelli, M.; Allocca, M.; Hay Mele, B.; Lukas, J.; Cubellis, M.V.; Andreotti, G. Pharmacological Chaperones: A Therapeutic Approach for Diseases Caused by Destabilizing Missense Mutations. Int. J. Mol. Sci. 2020, 21, 489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortez, L.; Sim, V. The Therapeutic Potential of Chemical Chaperones in Protein Folding Diseases. Prion 2014, 8, 197–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maegawa, G.H.; Tropak, M.; Buttner, J.; Stockley, T.; Kok, F.; Clarke, J.T.; Mahuran, D.J. Pyrimethamine as a Potential Pharmacological Chaperone for Late-Onset Forms of GM2 Gangliosidosis. J. Biol. Chem. 2007, 282, 9150–9161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bateman, K.S.; Cherney, M.M.; Mahuran, D.J.; Tropak, M.; James, M.N. Crystal structure of β-Hexosaminidase B in Complex with Pyrimethamine, a Potential Pharmacological Chaperone. J. Med. Chem. 2011, 54, 1421–1429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiricozzi, E.; Niemir, N.; Aureli, M.; Magini, A.; Loberto, N.; Prinetti, A.; Bassi, R.; Polchi, A.; Emiliani, C.; Caillaud, C.; et al. Chaperone Therapy for GM2 Gangliosidosis: Effects of Pyrimethamine on Β-Hexosaminidase Activity in Sandhoff Fibroblasts. Mol. Neurobiol. 2014, 50, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Clarke, J.T.; Mahuran, D.J.; Sathe, S.; Kolodny, E.H.; Rigat, B.A.; Raiman, J.A.; Tropak, M.B. An Open-Label Phase I/II Clinical Trial of Pyrimethamine for the Treatment of Patients Affected with Chronic GM2 Gangliosidosis (Tay-Sachs or Sandhoff variants). Mol. Genet. Metab. 2011, 102, 6–12. [Google Scholar] [CrossRef] [Green Version]
- Osher, E.; Fattal-Valevski, A.; Sagie, L.; Urshanski, N.; Sagiv, N.; Peleg, L.; Lerman-Sagie, T.; Zimran, A.; Elstein, D.; Navon, R.; et al. Effect of Cyclic, Low dose Pyrimethamine Treatment in Patients With Late Onset Tay Sachs: An Open Label, Extended Pilot Study. Orphanet J. Rare Dis. 2015, 10, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Kato, A.; Nakagome, I.; Nakagawa, S.; Kinami, K.; Adachi, I.; Jenkinson, S.F.; Désiré, J.; Blériot, Y.; Nash, R.J.; Fleet, G.W.J.; et al. In silico Analyses Of Essential Interactions of Iminosugars with the Hex A Active Site and Evaluation of their Pharmacological Chaperone Effects for Tay-Sachs Disease. Org. Biomol. Chem. 2017, 15, 9297–9304. [Google Scholar] [CrossRef]
- Chen, Y.; Jian, J.; Hettinghouse, A.; Zhao, X.; Setchell, K.; Sun, Y.; Liu, C. Progranulin Associates with Hexosaminidase A and Ameliorates GM2 ganglioside Accumulation and Lysosomal Storage in Tay-Sachs Disease. J. Mol. Med. 2018, 96, 1359–1373. [Google Scholar] [CrossRef]
- Jian, J.; Tian, Q.Y.; Hettinghouse, A.; Zhao, S.; Liu, H.; Wei, J.; Grunig, G.; Zhang, W.; Setchell, K.D.R.; Sun, Y.; et al. Progranulin Recruits HSP70 to β-Glucocerebrosidase and is Therapeutic against Gaucher Disease. EBioMedicine 2016, 13, 212–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Platt, F.M.; Jeyakumar, M. Substrate Reduction Therapy. Acta Paediatr. 2008, 97, 88–93. [Google Scholar] [CrossRef] [PubMed]
- Platt, F.M.; Neises, G.R.; Dwek, R.A.; Butters, T.D. N-butyldeoxynojirimycin is a Novel Inhibitor of Glycolipid Biosynthesis. J. Biol. Chem. 1994, 269, 8362–8365. [Google Scholar] [PubMed]
- Pastores, G.M.; Barnett, N.L.; Kolodny, E.H. An Open-Label, Noncomparative Study of Miglustat in Type I Gaucher Disease: Efficacy and Tolerability over 24 Months of Treatment. Clin. Ther. 2005, 27, 1215–1227. [Google Scholar] [CrossRef] [PubMed]
- Zervas, M.; Somers, K.L.; Thrall, M.A.; Walkley, S.U. Critical Role for Glycosphingolipids in Niemann-Pick Disease Type C. Curr. Biol. 2001, 11, 1283–1287. [Google Scholar] [CrossRef] [Green Version]
- Elliot-Smith, E.; Speak, A.O.; Lloyd-Evans, E.; Smith, D.A.; van der Spoel, A.C.; Jeyakumar, M.; Butters, T.D.; Dwek, R.A.; d’Azzo, A.; Platt, F.M. Beneficial Effects of Substrate Reduction Therapy in a Mouse Model of GM1 Gangliosidosis. Mol. Genet. Metab. 2008, 94, 204–211. [Google Scholar] [CrossRef]
- Platt, F.M.; Jeyakumar, M.; Andersson, U.; Heare, T.; Dwek, R.A.; Butters, T.D. Substrate Reduction Therapy in Mouse Models of the Glycosphingolipidoses. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2003, 358, 947–954. [Google Scholar] [CrossRef] [Green Version]
- Jeyakumar, M.; Butters, T.D.; Cortina-Borja, M.; Hunnam, V.; Proia, R.L.; Perry, V.H.; Dwek, R.A.; Platt, F.M. Delayed Symptom Onset and Increased Life Expectancy in Sandhoff Disease Mice Treated with N-Butyldeoxynojirimycin. Proc. Natl. Acad. Sci. USA 1999, 96, 6388–6393. [Google Scholar] [CrossRef] [Green Version]
- Platt, F.M.; Neises, G.R.; Reinkensmeier, G.; Townsend, M.J.; Perry, V.H.; Proia, R.L.; Winchester, B.; Dwek, R.A.; Butters, T.D. Prevention of Lysosomal Storage in Tay-Sachs Mice Treated with N-Butyldeoxynojirimycin. Science 1997, 276, 428–431. [Google Scholar] [CrossRef]
- Maegawa, G.H.; Banwell, B.L.; Blaser, S.; Sorge, G.; Toplak, M.; Ackerley, C.; Hawkins, C.; Hayes, J.; Clarke, J.T. Substrate Reduction Therapy in Juvenile GM2 Gangliosidosis. Mol. Genet. Metab. 2009, 98, 215–224. [Google Scholar] [CrossRef]
- Masciullo, M.; Santoro, M.; Modoni, A.; Ricci, E.; Guitton, J.; Tonali, P.; Silvestri, G. Substrate Reduction Therapy with Miglustat in Chronic GM2 Gangliosidosis Type SANDHOFF: Results of a 3-Year Follow-Up. J. Inherit. Metab. Dis. 2010, 33, S355–S361. [Google Scholar] [CrossRef] [PubMed]
- Ashe, K.M.; Bangari, D.; Li, L.; Cabrera-Salazar, M.A.; Bercury, S.D.; Nietupski, J.B.; Cooper, C.G.; Aerts, J.M.; Lee, E.R.; Copeland, D.P.; et al. Iminosugar-Based Inhibitors of Glucosylceramide Synthase Increase Brain Glycosphingolipids and Survival in a Mouse Model of Sandhoff Disease. PLoS ONE 2011, 6, e21758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glass, C.K.; Saijo, K.; Winner, B.; Marchetto, M.C.; Gage, F.H. Mechanisms Underlying Inflammation in Neurodegeneration. Cell 2010, 140, 918–934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arthur, J.R.; Wilson, M.W.; Larsen, S.D.; Rockwell, H.E.; Shayman, J.A.; Seyfried, T.N. Ethylenedioxy-PIP2 Oxalate Reduces Ganglioside Storage in Juvenile Sandhoff Disease Mice. Neurochem. Res. 2013, 38, 866–875. [Google Scholar] [CrossRef]
- Ohashi, T. Gene Therapy for Lysosomal Storage Diseases And Peroxisomal Diseases. J. Hum. Genet. 2019, 64, 139–143. [Google Scholar] [CrossRef]
- Schneller, J.L.; Lee, C.M.; Bao, G.; Venditti, C.P. Genome Editing for Inborn Errors of Metabolism: Advancing towards the Clinic. BMC Med. 2017, 15, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Solovyeva, V.V.; Shaimardanova, A.A.; Chulpanova, D.S.; Kitaeva, K.V.; Chakrabarti, L.; Rizvanov, A.A. New Approaches to Tay-Sachs Disease Therapy. Front. Physiol. 2018, 9, 1663. [Google Scholar] [CrossRef]
- Rabinowitz, J.; Chan, Y.K.; Samulski, R.J. Adeno-Associated Virus (AAV) Versus Immune Response. Viruses 2019, 11, 102. [Google Scholar] [CrossRef] [Green Version]
- Mays, L.E.; Wilson, J.M. The Complex and Evolving Story of T Cell Activation to AAV Vector-Encoded Transgene Products. Mol. Ther. 2011, 19, 16–27. [Google Scholar] [CrossRef]
- Barnes, C.; Scheideler, O.; Schaffer, D. Engineering the AAV Capsid to Evade Immune responses. Curr. Opin. Biotechnol. 2019, 60, 99–103. [Google Scholar] [CrossRef]
- Cachón-González, M.B.; Wang, S.Z.; McNair, R.; Bradley, J.; Lunn, D.; Ziegler, R.; Cheng, S.H.; Cox, T.M. Gene Transfer Corrects Acute GM2 Gangliosidosis—Potential Therapeutic Contribution of Perivascular Enzyme Flow. Mol. Ther. 2012, 20, 1489–1500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osmon, K.J.; Woodley, E.; Thompson, P.; Ong, K.; Karumuthil-Melethil, S.; Keimel, J.G.; Mark, B.L.; Mahuran, D.; Gray, S.J.; Walia, J.S. Systemic Gene Transfer of a Hexosaminidase Variant Using an scAAV9.47 Vector Corrects GM2 Gangliosidosis in Sandhoff Mice. Hum. Gene Ther. 2016, 27, 497–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, F.; Doudna, J.A. CRISPR-Cas9 Structures and Mechanisms. Annu. Rev. Biophys. 2017, 46, 505–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sung, P. Introduction to the Thematic Minireview Series: DNA double-strand break repair and pathway choice. J. Biol. Chem. 2018, 293, 10500–10501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, L.O.W.; O’Brien, A.R.; Bauer, D.C. The Current State and Future of CRISPR-Cas9 gRNA Design Tools. Front. Pharmacol. 2018, 9, 749. [Google Scholar] [CrossRef]
- Zhang, H.X.; Zhang, Y.; Yin, H. Genome Editing with mRNA Encoding ZFN, TALEN, and Cas9. Mol. Ther. 2019, 27, 735–746. [Google Scholar] [CrossRef] [Green Version]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Rockwell, H.E.; McCurdy, V.J.; Eaton, S.C.; Wilson, D.U.; Johnson, A.K.; Randle, A.N.; Bradbury, A.M.; Gray-Edwards, H.L.; Baker, H.J.; Hudson, J.A.; et al. AAV-Mediated Gene Delivery in a Feline Model of Sandhoff Disease Corrects Lysosomal Storage in the Central Nervous System. ASN Neuro 2015, 7, 1–13. [Google Scholar] [CrossRef]
- Golebiowski, D.; van der Bom, I.M.J.; Kwon, C.S.; Miller, A.D.; Petrosky, K.; Bradbury, A.M.; Maitland, S.; Kühn, A.L.; Bishop, N.; Curran, E.; et al. Direct Intracranial Injection of AAVrh8 Encoding Monkey β-N-Acetylhexosaminidase Causes Neurotoxicity in the Primate Brain. Hum. Gene Ther. 2017, 28, 510–522. [Google Scholar] [CrossRef]
- Arfi, A.; Bourgoin, C.; Basso, L.; Emiliani, C.; Tancini, B.; Chigorno, V.; Li, Y.T.; Orlacchio, A.; Poenaru, L.; Sonnino, S.; et al. Bicistronic Lentiviral Vector Corrects Beta-Hexosaminidase Deficiency in Transduced and Cross-Corrected Human Sandhoff Fibroblasts. Neurobiol. Dis. 2005, 20, 583–593. [Google Scholar] [CrossRef]
- Woodley, E.; Osmon, K.J.L.; Thompson, P.; Richmond, C.; Chen, Z.; Gray, S.J.; Walia, J.S. Efficacy of a Bicistronic Vector for Correction of Sandhoff Disease in a Mouse Model. Mol. Ther. Methods Clin. Dev. 2019, 12, 47–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, B.X.; Loh, S.J.H.; Chan, W.K.; Soh, B.S. In Vivo Genome Editing as a Therapeutic Approach. Int. J. Mol. Sci. 2018, 19, 2721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishii, A.; Kurosawa, A.; Saito, S.; Adachi, N. Analysis of the Role of Homology Arms in Gene-Targeting Vectors in Human Cells. PLoS ONE 2014, 9, e108236. [Google Scholar] [CrossRef] [PubMed]
- Kanca, O.; Zirin, J.; Garcia-Marques, J.; Knight, S.M.; Yang-Zhou, D.; Amador, G.; Chung, H.; Zuo, Z.; Ma, L.; He, Y.; et al. An Efficient CRISPR-Based Strategy to Insert Small and Large Fragments of DNA Using Short Homology Arms. Elife 2019, 8, e51539. [Google Scholar] [CrossRef]
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-Replace Genome Editing without Double-Strand Breaks or Donor DNA. Nature 2019, 576, 149–157. [Google Scholar] [CrossRef]
- Keijzers, G.; Bohr, V.A.; Rasmussen, L.J. Human exonuclease 1 (EXO1) Activity Characterization and its Function on Flap Structures. Biosci. Rep. 2015, 35, e00206. [Google Scholar] [CrossRef]
- Nami, F.; Basiri, M.; Satarian, L.; Curtiss, C.; Baharvand, H.; Verfaillie, C. Strategies for In Vivo Genome Editing in Nondividing Cells. Trends Biotechnol. 2018, 36, 770–786. [Google Scholar] [CrossRef]
- Papapetrou, E.P.; Schambach, A. Gene Insertion into Genomic Safe Harbors for Human Gene Therapy. Mol. Ther. 2016, 24, 678–684. [Google Scholar] [CrossRef] [Green Version]
- Ou, L.; Przybilla, M.J.; Tăbăran, A.F.; Overn, P.; O’Sullivan, M.G.; Jiang, X.; Sidhu, R.; Kell, P.J.; Ory, D.S.; Whitley, C.B. A novel gene editing system to treat both Tay-Sachs and Sandhoff diseases. Gene Ther. 2020, 27, 226–236. [Google Scholar] [CrossRef]
- Cohen, J. Prime Editing Promises to be a Cut above CRISPR. Science 2019, 366, 406. [Google Scholar] [CrossRef]
- Santos, R.; Amaral, O. Advances in Sphingolipidoses: CRISPR-Cas9 Editing as an Option for Modelling and Therapy. Int. J. Mol. Sci. 2019, 20, 5897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ledford, H. Super-Precise New CRISPR Tool could Tackle a Plethora of Genetic Diseases. Nature 2019, 574, 464–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawamoto, K.; Stapleton, M.; Almeciga-Diaz, C.J.; Espejo-Mojica, A.J.; Losada, J.C.; Suarez, D.A.; Tomatsu, S. Therapeutic Options for Mucopolysaccharidoses: Current and Emerging Treatments. Drugs 2019, 79, 1103–1134. [Google Scholar] [CrossRef] [PubMed]
- Hassan, S.; Sidransky, E.; Tayebi, N. The Role of Epigenetics in Lysosomal Storage Disorders: Uncharted Territory. Mol. Genet. Metab. 2017, 122, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Rutten, M.G.S.; Rots, M.G.; Oosterveer, M.H. Exploiting Epigenetics for the Treatment of Inborn Errors of Metabolism. J. Inherit. Metab. Dis. 2020, 43, 63–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease | Affected Gene | Affected Protein | Accumulated Substrate * | Common Findings | Ref | ||
|---|---|---|---|---|---|---|---|
| Onset | Symptoms | Neuroimaging | |||||
| TSD | HEXA | HexA | GM2 Ganglioside | Infantile Acute | Seizures, axial hypotonia, cherry-red spot, regression in developmental milestones, exaggerated startle response | Bilateral thalamic involvement, brain atrophy, hypomyelination | [50,54,55] |
| SD | HEXB | HexA, HexB | GM2 Ganglioside, Globoside | Juvenile Subacute | Ataxia, myoclonus, motor regression, psychotic episodes, intellectual disability, progressive clumsiness | Cerebellar atrophy | [51,56] |
| AB Variant | GM2A | GM2AP | GM2 Ganglioside | Adult Chronic | Dysphagia, muscle atrophy, cerebellar ataxia, dysarthric speech, manic depression, muscle weakness, psychotic episodes | Severe cerebellar atrophy, hypodensity of the thalamus | [4,6,57] |
| Therapy | NCT Number | Intervention | Status | Phase | Country |
|---|---|---|---|---|---|
| Pharmacological Chaperone | NCT00679744 | Pyrimethamine | Withdrawn | Phase 1 | USA |
| NCT01102686 | Pyrimethamine | Completed | Phase 1/2 | Canada | |
| Substrate Reduction Therapy | NCT00418847 | Miglustat | Completed | Phase 2 | Canada |
| NCT03822013 | Miglustat | Recruiting | Phase 3 | Iran | |
| NCT00672022 | Miglustat | Completed | Phase 3 | USA | |
| NCT04221451 | Venglustat | Recruiting | Phase 3 | USA | |
| NCT02030015 | Miglustat and ketogenic diet | Recruiting | Phase 4 | USA | |
| HSCT | NCT01372228 | Enriched hematopoietic stem cell infusion | Active, not recruiting | Phase 1/2 | USA |
| NCT00176904 | Chemotherapy and hematopoietic cell transplantation | Completed | Phase 1/2 | USA | |
| NCT01626092 | Chemotherapy, total body irradiation with marrow boosting, and hematopoietic stem cell transplantation | Completed | Phase 1/2 | USA | |
| NCT00383448 | Chemotherapy, total body irradiation, and hematopoietic stem cell transplantation | Completed | Phase 2 | USA | |
| Umbilical Cord Blood Transplantation (UBC) | NCT02254863 | UBC-derived oligodendrocyte-like cells | Recruiting | Phase 1 | USA |
| NCT01003912 | Fetal UCB transplantation | Withdrawn | Phase 1 | USA | |
| NCT00654433 | UBC cells expressing high levels of the intracellular enzyme aldehyde dehydrogenase | Terminated | Phase 3 | USA | |
| Cerebellar Ataxia Treatment | NCT03759665 | N-Acetyl-L-Leucine | Recruiting | Phase 2 | USA |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leal, A.F.; Benincore-Flórez, E.; Solano-Galarza, D.; Garzón Jaramillo, R.G.; Echeverri-Peña, O.Y.; Suarez, D.A.; Alméciga-Díaz, C.J.; Espejo-Mojica, A.J. GM2 Gangliosidoses: Clinical Features, Pathophysiological Aspects, and Current Therapies. Int. J. Mol. Sci. 2020, 21, 6213. https://doi.org/10.3390/ijms21176213
Leal AF, Benincore-Flórez E, Solano-Galarza D, Garzón Jaramillo RG, Echeverri-Peña OY, Suarez DA, Alméciga-Díaz CJ, Espejo-Mojica AJ. GM2 Gangliosidoses: Clinical Features, Pathophysiological Aspects, and Current Therapies. International Journal of Molecular Sciences. 2020; 21(17):6213. https://doi.org/10.3390/ijms21176213
Chicago/Turabian StyleLeal, Andrés Felipe, Eliana Benincore-Flórez, Daniela Solano-Galarza, Rafael Guillermo Garzón Jaramillo, Olga Yaneth Echeverri-Peña, Diego A. Suarez, Carlos Javier Alméciga-Díaz, and Angela Johana Espejo-Mojica. 2020. "GM2 Gangliosidoses: Clinical Features, Pathophysiological Aspects, and Current Therapies" International Journal of Molecular Sciences 21, no. 17: 6213. https://doi.org/10.3390/ijms21176213