Regulation of Filaggrin, Loricrin, and Involucrin by IL-4, IL-13, IL-17A, IL-22, AHR, and NRF2: Pathogenic Implications in Atopic Dermatitis


Abstract
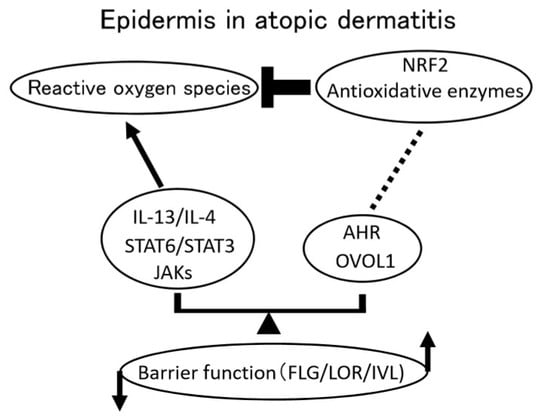
:
{kind=link}
{kind=link}
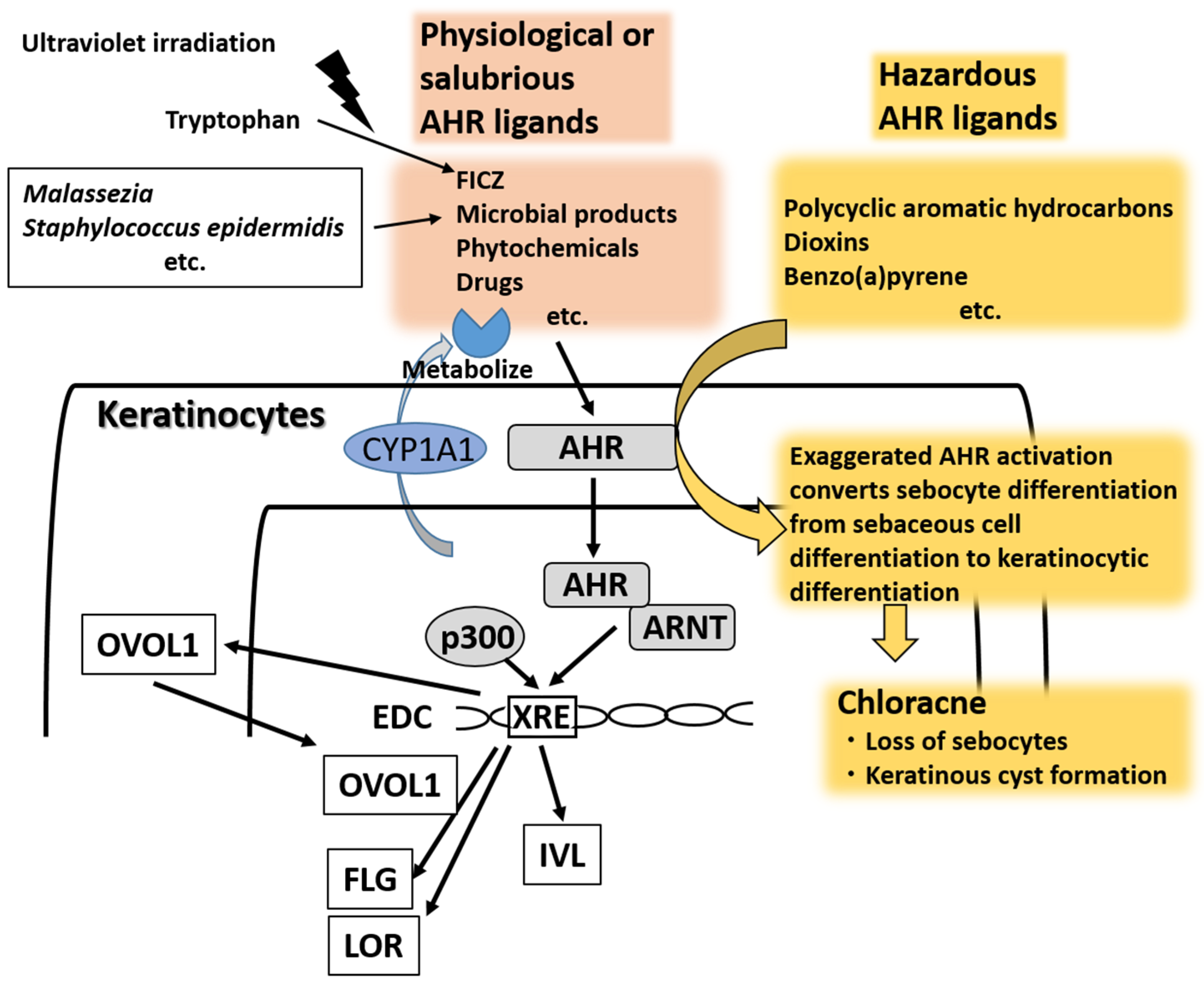{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Roles of IVL, LOR, FLG, and FLG2 in Epidermal Barrier Formation
3. Upregulation of IVL, LOR, and FLG by AHR Activation
4. Decreased Expression of IVL, LOR, and FLG in AD
5. Downregulation of IVL, LOR, and FLG by IL-4/IL-13
6. Medicinal Use of AHR/NRF2 Dual Activators for AD
7. Downregulation of IVL, LOR, and FLG by IL-22
8. Regulation of IVL, LOR, and FLG by IL-17A
9. Conclusions
Funding
Conflicts of Interest
References
- Ishitsuka, Y.; Roop, D.R. Loricrin: Past, present, and future. Int. J. Mol. Sci. 2020, 21, 2271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalinin, A.; Marekov, L.N.; Steinert, P.M. Assembly of the epidermal cornified cell envelope. J. Cell Sci. 2001, 114, 3069–3070. [Google Scholar] [PubMed]
- Karim, N.; Phinney, B.S.; Salemi, M.; Wu, P.W.; Naeem, M.; Rice, R.H. Human stratum corneum proteomics reveals cross-linking of a broad spectrum of proteins in cornified envelopes. Exp. Dermatol. 2019, 28, 618–622. [Google Scholar] [PubMed] [Green Version]
- Kennedy, L.H.; Sutter, C.H.; Leon Carrion, S.; Tran, Q.T.; Bodreddigari, S.; Kensicki, E.; Mohney, R.P.; Sutter, T.R. 2,3,7,8-Tetrachlorodibenzo-p-dioxin-mediated production of reactive oxygen species is an essential step in the mechanism of action to accelerate human keratinocyte differentiation. Toxicol. Sci. 2013, 132, 235–249. [Google Scholar]
- Kypriotou, M.; Huber, M.; Hohl, D. The human epidermal differentiation complex: Cornified envelope precursors, S100 proteins and the ‘fused genes’ family. Exp. Dermatol. 2012, 21, 643–649. [Google Scholar]
- Proksch, E.; Brandner, J.M.; Jensen, J.M. The skin: An indispensable barrier. Exp. Dermatol. 2008, 17, 1063–1072. [Google Scholar] [CrossRef]
- Ishida-Yamamoto, A.; Takahashi, H.; Presland, R.B.; Dale, B.A.; Iizuka, H. Translocation of profilaggrin N-terminal domain into keratinocyte nuclei with fragmented DNA in normal human skin and loricrin keratoderma. Lab. Investig. 1998, 78, 1245–1253. [Google Scholar]
- Pearton, D.J.; Dale, B.A.; Presland, R.B. Functional analysis of the profilaggrin N-terminal peptide: Identification of domains that regulate nuclear and cytoplasmic distribution. J. Investig. Dermatol. 2002, 119, 661–669. [Google Scholar] [CrossRef] [Green Version]
- Presland, R.B.; Kimball, J.R.; Kautsky, M.B.; Lewis, S.P.; Lo, C.Y.; Dale, B.A. Evidence for specific proteolytic cleavage of the N-terminal domain of human profilaggrin during epidermal differentiation. J. Investig. Dermatol. 1997, 108, 170–178. [Google Scholar] [CrossRef] [Green Version]
- Chiba, T.; Nakahara, T.; Kohda, F.; Ichiki, T.; Manabe, M.; Furue, M. Measurement of trihydroxy-linoleic acids in stratum corneum by tape-stripping: Possible biomarker of barrier function in atopic dermatitis. PLoS ONE 2019, 14, e0210013. [Google Scholar] [CrossRef]
- Mischke, D.; Korge, B.P.; Marenholz, I.; Volz, A.; Ziegler, A. Genes encoding structural proteins of epidermal cornification and S100 calcium-binding proteins form a gene complex (“epidermal differentiation complex”) on human chromosome 1q21. J. Investig. Dermatol. 1996, 106, 989–992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leśniak, W.; Graczyk-Jarzynka, A. The S100 proteins in epidermis: Topology and function. Biochim. Biophys. Acta 2015, 1850, 2563–2572. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, Z.A.; de Guzman Strong, C. Recent positive selection in genes of the mammalian epidermal differentiation complex locus. Front. Genet. 2017, 7, 227. [Google Scholar] [CrossRef] [Green Version]
- Furue, M.; Uchi, H.; Mitoma, C.; Hashimoto-Hachiya, A.; Tanaka, Y.; Ito, T.; Tsuji, G. Implications of tryptophan photoproduct FICZ in oxidative stress and terminal differentiation of keratinocytes. G. Ital. Dermatol. Venereol. 2019, 154, 37–41. [Google Scholar] [CrossRef] [PubMed]
- Kiyomatsu-Oda, M.; Uchi, H.; Morino-Koga, S.; Furue, M. Protective role of 6-formylindolo[3,2-b]carbazole (FICZ), an endogenous ligand for arylhydrocarbon receptor, in chronic mite-induced dermatitis. J. Dermatol. Sci. 2018, 90, 284–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sutter, C.H.; Bodreddigari, S.; Campion, C.; Wible, R.S.; Sutter, T.R. 2,3,7,8-Tetrachlorodibenzo-p-dioxin increases the expression of genes in the human epidermal differentiation complex and accelerates epidermal barrier formation. Toxicol. Sci. 2011, 124, 128–137. [Google Scholar] [CrossRef] [Green Version]
- Buommino, E.; Baroni, A.; Papulino, C.; Nocera, F.P.; Coretti, L.; Donnarumma, G.; De Filippis, A.; De Martino, L. Malassezia pachydermatis up-regulates AhR related CYP1A1 gene and epidermal barrier markers in human keratinocytes. Med. Mycol. 2018, 56, 987–993. [Google Scholar] [CrossRef] [Green Version]
- Furue, M.; Tsuji, G.; Mitoma, C.; Nakahara, T.; Chiba, T.; Morino-Koga, S.; Uchi, H. Gene regulation of filaggrin and other skin barrier proteins via aryl hydrocarbon receptor. J. Dermatol. Sci. 2015, 80, 83–88. [Google Scholar] [CrossRef]
- Yu, J.; Luo, Y.; Zhu, Z.; Zhou, Y.; Sun, L.; Gao, J.; Sun, J.; Wang, G.; Yao, X.; Li, W. A tryptophan metabolite of the skin microbiota attenuates inflammation in patients with atopic dermatitis through the aryl hydrocarbon receptor. J. Allergy Clin. Immunol. 2019, 143, 2108–2119. [Google Scholar] [CrossRef] [Green Version]
- Takei, K.; Mitoma, C.; Hashimoto-Hachiya, A.; Takahara, M.; Tsuji, G.; Nakahara, T.; Furue, M. Galactomyces fermentation filtrate prevents T helper 2-mediated reduction of filaggrin in an aryl hydrocarbon receptor-dependent manner. Clin. Exp. Dermatol. 2015, 40, 786–793. [Google Scholar] [CrossRef]
- Furue, M.; Hashimoto-Hachiya, A.; Tsuji, G. Antioxidative phytochemicals accelerate epidermal terminal differentiation via the AHR-OVOL1 pathway: Implications for atopic dermatitis. Acta Derm. Venereol. 2018, 98, 918–923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashimoto-Hachiya, A.; Tsuji, G.; Murai, M.; Yan, X.; Furue, M. Upregulation of FLG, LOR, and IVL expression by Rhodiola crenulata root extract via aryl hydrocarbon receptor: Differential involvement of OVOL. Int. J. Mol. Sci. 2018, 19, 1654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirano, A.; Goto, M.; Mitsui, T.; Hashimoto-Hachiya, A.; Tsuji, G.; Furue, M. Antioxidant Artemisia princeps extract enhances the expression of filaggrin and loricrin via the AHR/OVOL1 pathway. Int. J. Mol. Sci. 2017, 18, 1948. [Google Scholar] [CrossRef] [PubMed]
- Nakahara, T.; Mitoma, C.; Hashimoto-Hachiya, A.; Takahara, M.; Tsuji, G.; Uchi, H.; Yan, X.; Hachisuka, J.; Chiba, T.; Esaki, H.; et al. Antioxidant Opuntia ficus-indica extract activates AHR-NRF2 signaling and upregulates filaggrin and loricrin expression in human keratinocytes. J. Med. Food 2015, 18, 1143–1149. [Google Scholar] [CrossRef]
- Takei, K.; Mitoma, C.; Hashimoto-Hachiya, A.; Uchi, H.; Takahara, M.; Tsuji, G.; Kido-Nakahara, M.; Nakahara, T.; Furue, M. Antioxidant soybean tar Glyteer rescues T-helper-mediated downregulation of filaggrin expression via aryl hydrocarbon receptor. J. Dermatol. 2015, 42, 171–180. [Google Scholar] [CrossRef]
- Tsuji, G.; Hashimoto-Hachiya, A.; Kiyomatsu-Oda, M.; Takemura, M.; Ohno, F.; Ito, T.; Morino-Koga, S.; Mitoma, C.; Nakahara, T.; Uchi, H.; et al. Aryl hydrocarbon receptor activation restores filaggrin expression via OVOL1 in atopic dermatitis. Cell Death Dis. 2017, 8, e2931. [Google Scholar] [CrossRef] [Green Version]
- Furue, M.; Fuyuno, Y.; Mitoma, C.; Uchi, H.; Tsuji, G. Therapeutic agents with AHR inhibiting and NRF2 activating activity for managing chloracne. Antioxidants 2018, 7, 90. [Google Scholar] [CrossRef] [Green Version]
- Furue, M.; Tsuji, G. Chloracne and hyperpigmentation caused by exposure to hazardous aryl hydrocarbon receptor ligands. Int. J. Environ. Res. Public Health 2019, 16, 4864. [Google Scholar] [CrossRef] [Green Version]
- Ju, Q.; Fimmel, S.; Hinz, N.; Stahlmann, R.; Xia, L.; Zouboulis, C.C. 2,3,7,8-Tetrachlorodibenzo-p-dioxin alters sebaceous gland cell differentiation in vitro. Exp. Dermatol. 2011, 20, 320–325. [Google Scholar] [CrossRef]
- Suskind, R.R. Chloracne, “the hallmark of dioxin intoxication”. Scand. J. Work Environ. Health 1985, 11, 165–171. [Google Scholar]
- van den Bogaard, E.H.; Bergboer, J.G.; Vonk-Bergers, M.; van Vlijmen-Willems, I.M.; Hato, S.V.; van der Valk, P.G.; Schröder, J.M.; Joosten, I.; Zeeuwen, P.L.; Schalkwijk, J. Coal tar induces AHR-dependent skin barrier repair in atopic dermatitis. J. Clin. Investig. 2013, 123, 917–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Endo, H.; Sugioka, Y.; Nakagi, Y.; Saijo, Y.; Yoshida, T. A novel role of the NRF2 transcription factor in the regulation of arsenite-mediated keratin 16 gene expression in human keratinocytes. Environ. Health Perspect. 2008, 116, 873–879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.; Shin, J.M.; Jang, S.; Choi, D.K.; Seo, M.S.; Kim, H.R.; Sohn, K.C.; Im, M.; Seo, Y.J.; Lee, J.H.; et al. Role of nuclear factor E2-related factor 2 (Nrf2) in epidermal differentiation. Arch. Dermatol. Res. 2014, 306, 677–682. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, T.; Ishitsuka, Y.; Inoue, S.; Nakamura, Y.; Saito, A.; Okiyama, N.; Fujisawa, Y.; Furuta, J.; Watanabe, R.; Fujimoto, M. Nuclear factor erythroid 2-related factor 2 (Nrf2) regulates epidermal keratinization under psoriatic skin inflammation. Am. J. Pathol. 2020, 190, 577–585. [Google Scholar] [CrossRef]
- Schäfer, M.; Farwanah, H.; Willrodt, A.H.; Huebner, A.J.; Sandhoff, K.; Roop, D.; Hohl, D.; Bloch, W.; Werner, S. Nrf2 links epidermal barrier function with antioxidant defense. EMBO Mol. Med. 2012, 4, 364–379. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, M.; Willrodt, A.H.; Kurinna, S.; Link, A.S.; Farwanah, H.; Geusau, A.; Gruber, F.; Sorg, O.; Huebner, A.J.; Roop, D.R.; et al. Activation of Nrf2 in keratinocytes causes chloracne (MADISH)-like skin disease in mice. EMBO Mol. Med. 2014, 6, 442–457. [Google Scholar] [CrossRef]
- Schäfer, M.; Werner, S. Nrf2--A regulator of keratinocyte redox signaling. Free Radic. Biol. Med. 2015, 88, 243–252. [Google Scholar] [CrossRef]
- Yang, L.; Fan, X.; Cui, T.; Dang, E.; Wang, G. Nrf2 promotes keratinocyte proliferation in psoriasis through up-regulation of keratin 6, keratin 16, and keratin 17. J. Investig. Dermatol. 2017, 137, 2168–2176. [Google Scholar] [CrossRef] [Green Version]
- Vogel, C.F.A.; Van Winkle, L.S.; Esser, C.; Haarmann-Stemmann, T. The aryl hydrocarbon receptor as a target of environmental stressors—Implications for pollution mediated stress and inflammatory responses. Redox Biol. 2020, 34, 101530. [Google Scholar] [CrossRef]
- Fuyuno, Y.; Uchi, H.; Yasumatsu, M.; Morino-Koga, S.; Tanaka, Y.; Mitoma, C.; Furue, M. Perillaldehyde inhibits AHR signaling and activates NRF2 antioxidant pathway in human keratinocytes. Oxidative Med. Cell Longev. 2018, 2018, 9524657. [Google Scholar] [CrossRef]
- Takei, K.; Hashimoto-Hachiya, A.; Takahara, M.; Tsuji, G.; Nakahara, T.; Furue, M. Cynaropicrin attenuates UVB-induced oxidative stress via the AhR-Nrf2-Nqo1 pathway. Toxicol. Lett. 2015, 234, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Uchi, H.; Furue, M. Antioxidant cinnamaldehyde attenuates UVB-induced photoaging. J. Dermatol. Sci. 2019, 96, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Uchi, H.; Yasumatsu, M.; Morino-Koga, S.; Mitoma, C.; Furue, M. Inhibition of aryl hydrocarbon receptor signaling and induction of NRF2-mediated antioxidant activity by cinnamaldehyde in human keratinocytes. J. Dermatol. Sci. 2017, 85, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Guttman-Yassky, E.; Bissonnette, R.; Ungar, B.; Suárez-Fariñas, M.; Ardeleanu, M.; Esaki, H.; Suprun, M.; Estrada, Y.; Xu, H.; Peng, X.; et al. Dupilumab progressively improves systemic and cutaneous abnormalities in patients with atopic dermatitis. J. Allergy Clin. Immunol. 2019, 143, 155–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krueger, J.G.; Wharton, K.A., Jr.; Schlitt, T.; Suprun, M.; Torene, R.I.; Jiang, X.; Wang, C.Q.; Fuentes-Duculan, J.; Hartmann, N.; Peters, T.; et al. IL-17A inhibition by secukinumab induces early clinical, histopathologic, and molecular resolution of psoriasis. J. Allergy Clin. Immunol. 2019, 144, 750–763. [Google Scholar] [CrossRef] [Green Version]
- Guttman-Yassky, E.; Brunner, P.M.; Neumann, A.U.; Khattri, S.; Pavel, A.B.; Malik, K.; Singer, G.K.; Baum, D.; Gilleaudeau, P.; Sullivan-Whalen, M.; et al. Efficacy and safety of fezakinumab (an IL-22 monoclonal antibody) in adults with moderate-to-severe atopic dermatitis inadequately controlled by conventional treatments: A randomized, double-blind, phase 2a trial. J. Am. Acad. Dermatol. 2018, 78, 872–881. [Google Scholar] [CrossRef] [Green Version]
- Hayashida, S.; Uchi, H.; Takeuchi, S.; Esaki, H.; Moroi, Y.; Furue, M. Significant correlation of serum IL-22 levels with CCL17 levels in atopic dermatitis. J. Dermatol. Sci. 2011, 61, 78–79. [Google Scholar] [CrossRef]
- Cordoro, K.M.; Hitraya-Low, M.; Taravati, K.; Sandoval, P.M.; Kim, E.; Sugarman, J.; Pauli, M.L.; Liao, W.; Rosenblum, M.D. Skin-infiltrating, interleukin-22-producing T cells differentiate pediatric psoriasis from adult psoriasis. J. Am. Acad. Dermatol. 2017, 77, 417–424. [Google Scholar] [CrossRef]
- Gordon, K.B.; Blauvelt, A.; Papp, K.A.; Langley, R.G.; Luger, T.; Ohtsuki, M.; Reich, K.; Amato, D.; Ball, S.G.; Braun, D.K.; et al. Phase 3 trials of ixekizumab in moderate-to-severe plaque psoriasis. N. Engl. J. Med. 2016, 375, 345–356. [Google Scholar] [CrossRef]
- Boniface, K.; Bernard, F.X.; Garcia, M.; Gurney, A.L.; Lecron, J.C.; Morel, F. IL-22 inhibits epidermal differentiation and induces proinflammatory gene expression and migration of human keratinocytes. J. Immunol. 2005, 174, 3695–3702. [Google Scholar] [CrossRef] [Green Version]
- Nograles, K.E.; Zaba, L.C.; Guttman-Yassky, E.; Fuentes-Duculan, J.; Suárez-Fariñas, M.; Cardinale, I.; Khatcherian, A.; Gonzalez, J.; Pierson, K.C.; White, T.R.; et al. Th17 cytokines interleukin (IL)-17 and IL-22 modulate distinct inflammatory and keratinocyte-response pathways. Br. J. Dermatol. 2008, 159, 1092–1102. [Google Scholar] [CrossRef] [Green Version]
- Sa, S.M.; Valdez, P.A.; Wu, J.; Jung, K.; Zhong, F.; Hall, L.; Kasman, I.; Winer, J.; Modrusan, Z.; Danilenko, D.M.; et al. The effects of IL-20 subfamily cytokines on reconstituted human epidermis suggest potential roles in cutaneous innate defense and pathogenic adaptive immunity in psoriasis. J. Immunol. 2007, 178, 2229–2240. [Google Scholar] [CrossRef]
- Dale, B.A.; Presland, R.B.; Lewis, S.P.; Underwood, R.A.; Fleckman, P. Transient expression of epidermal filaggrin in cultured cells causes collapse of intermediate filament networks with alteration of cell shape and nuclear integrity. J. Investig. Dermatol. 1997, 108, 179–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, F.J.; Irvine, A.D.; Terron-Kwiatkowski, A.; Sandilands, A.; Campbell, L.E.; Zhao, Y.; Liao, H.; Evans, A.T.; Goudie, D.R.; Lewis-Jones, S.; et al. Loss-of-function mutations in the gene encoding filaggrin cause ichthyosis vulgaris. Nat. Genet. 2006, 38, 337–342. [Google Scholar] [PubMed]
- Nomura, T.; Sandilands, A.; Akiyama, M.; Liao, H.; Evans, A.T.; Sakai, K.; Ota, M.; Sugiura, H.; Yamamoto, K.; Sato, H.; et al. Unique mutations in the filaggrin gene in Japanese patients with ichthyosis vulgaris and atopic dermatitis. J. Allergy Clin. Immunol. 2007, 119, 434–440. [Google Scholar] [CrossRef] [PubMed]
- Palmer, C.N.; Irvine, A.D.; Terron-Kwiatkowski, A.; Zhao, Y.; Liao, H.; Lee, S.P.; Goudie, D.R.; Sandilands, A.; Campbell, L.E.; Smith, F.J.; et al. Common loss-of-function variants of the epidermal barrier protein filaggrin are a major predisposing factor for atopic dermatitis. Nat. Genet. 2006, 38, 441–446. [Google Scholar] [PubMed]
- Hanifin, J.M.; Rajka, G. Diagnostic features of atopic eczema. Acta Derm. Venereol. 1980, 92, 44–47. [Google Scholar]
- McLean, W.H. Filaggrin failure from ichthyosis vulgaris to atopic eczema and beyond. Br. J. Dermatol. 2016, 175, 4–7. [Google Scholar] [PubMed] [Green Version]
- Wu, Z.; Hansmann, B.; Meyer-Hoffert, U.; Gläser, R.; Schröder, J.M. Molecular identification and expression analysis of filaggrin-2, a member of the S100 fused-type protein family. PLoS ONE 2009, 4, e5227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasan, M.Z.; Kitamura, M.; Kawai, M.; Ohira, M.; Mori, K.; Shoju, S.; Takagi, K.; Tsukamoto, K.; Kawai, Y.; Inoue, A. Transcriptional profiling of lactic acid treated reconstructed human epidermis reveals pathways underlying stinging and itch. Toxicol. In Vitro 2019, 57, 164–173. [Google Scholar] [CrossRef] [PubMed]
- de Koning, H.D.; van den Bogaard, E.H.; Bergboer, J.G.; Kamsteeg, M.; van Vlijmen-Willems, I.M.; Hitomi, K.; Henry, J.; Simon, M.; Takashita, N.; Ishida-Yamamoto, A.; et al. Expression profile of cornified envelope structural proteins and keratinocyte differentiation-regulating proteins during skin barrier repair. Br. J. Dermatol. 2012, 166, 1245–1254. [Google Scholar] [CrossRef]
- Pendaries, V.; Le Lamer, M.; Cau, L.; Hansmann, B.; Malaisse, J.; Kezic, S.; Serre, G.; Simon, M. In a three-dimensional reconstructed human epidermis filaggrin-2 is essential for proper cornification. Cell Death Dis. 2015, 6, e1656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohamad, J.; Sarig, O.; Godsel, L.M.; Peled, A.; Malchin, N.; Bochner, R.; Vodo, D.; Rabinowitz, T.; Pavlovsky, M.; Taiber, S.; et al. Filaggrin 2 deficiency results in abnormal cell-cell adhesion in the cornified cell layers and causes peeling skin syndrome Type A. J. Investig. Dermatol. 2018, 138, 1736–1743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishida-Yamamoto, A.; Iizuka, H. Differences in involucrin immunolabeling within cornified cell envelopes in normal and psoriatic epidermis. J. Investig. Dermatol. 1995, 104, 391–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loertscher, J.A.; Sattler, C.A.; Allen-Hoffmann, B.L. 2,3,7,8-Tetrachlorodibenzo-p-dioxin alters the differentiation pattern of human keratinocytes in organotypic culture. Toxicol. Appl. Pharmacol. 2001, 175, 121–129. [Google Scholar] [CrossRef] [Green Version]
- Seguchi, T.; Cui, C.Y.; Kusuda, S.; Takahashi, M.; Aisu, K.; Tezuka, T. Decreased expression of filaggrin in atopic skin. Arch. Dermatol. Res. 1996, 288, 442–446. [Google Scholar] [CrossRef]
- Mitoma, C.; Mine, Y.; Utani, A.; Imafuku, S.; Muto, M.; Akimoto, T.; Kanekura, T.; Furue, M.; Uchi, H. Current skin symptoms of Yusho patients exposed to high levels of 2,3,4,7,8-pentachlorinated dibenzofuran and polychlorinated biphenyls in 1968. Chemosphere 2015, 137, 45–51. [Google Scholar] [CrossRef]
- Mitoma, C.; Uchi, H.; Tsukimori, K.; Todaka, T.; Kajiwara, J.; Shimose, T.; Akahane, M.; Imamura, T.; Furue, M. Current state of yusho and prospects for therapeutic strategies. Environ. Sci. Pollut. Res. Int. 2018, 25, 16472–16480. [Google Scholar] [CrossRef]
- Tsuji, G.; Takahara, M.; Uchi, H.; Takeuchi, S.; Mitoma, C.; Moroi, Y.; Furue, M. An environmental contaminant, benzo(a)pyrene, induces oxidative stress-mediated interleukin-8 production in human keratinocytes via the aryl hydrocarbon receptor signaling pathway. J. Dermatol. Sci. 2011, 62, 42–49. [Google Scholar] [CrossRef]
- Furue, M.; Takahara, M.; Nakahara, T.; Uchi, H. Role of AhR/ARNT system in skin homeostasis. Arch. Dermatol. Res. 2014, 306, 769–779. [Google Scholar]
- Kazlauskas, A.; Sundström, S.; Poellinger, L.; Pongratz, I. The hsp90 chaperone complex regulates intracellular localization of the dioxin receptor. Mol. Cell. Biol. 2001, 21, 2594–2607. [Google Scholar] [CrossRef] [Green Version]
- Lees, M.J.; Peet, D.J.; Whitelaw, M.L. Defining the role for XAP2 in stabilization of the dioxin receptor. J. Biol. Chem. 2003, 278, 35878–35888. [Google Scholar]
- Esser, C.; Rannug, A. The aryl hydrocarbon receptor in barrier organ physiology, immunology, and toxicology. Pharmacol. Rev. 2015, 67, 259–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mimura, J.; Fujii-Kuriyama, Y. Functional role of AhR in the expression of toxic effects by TCDD. Biochim. Biophys. Acta 2003, 1619, 263–268. [Google Scholar] [CrossRef]
- Loertscher, J.A.; Lin, T.M.; Peterson, R.E.; Allen-Hoffmann, B.L. In utero exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin causes accelerated terminal differentiation in fetal mouse skin. Toxicol. Sci. 2002, 68, 465–472. [Google Scholar] [PubMed] [Green Version]
- Muenyi, C.S.; Carrion, S.L.; Jones, L.A.; Kennedy, L.H.; Slominski, A.T.; Sutter, C.H.; Sutter, T.R. Effects of in utero exposure of C57BL/6J mice to 2,3,7,8-tetrachlorodibenzo-p-dioxin on epidermal permeability barrier development and function. Environ. Health Perspect. 2014, 122, 1052–1058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray, S.S.; Swanson, H.I. Alteration of keratinocyte differentiation and senescence by the tumor promoter dioxin. Toxicol. Appl. Pharmacol. 2003, 192, 131–145. [Google Scholar] [CrossRef]
- Sutter, C.H.; Yin, H.; Li, Y.; Mammen, J.S.; Bodreddigari, S.; Stevens, G.; Cole, J.A.; Sutter, T.R. EGF receptor signaling blocks aryl hydrocarbon receptor-mediated transcription and cell differentiation in human epidermal keratinocytes. Proc. Natl. Acad. Sci. USA 2009, 106, 4266–4271. [Google Scholar]
- Van den Bogaard, E.H.; Podolsky, M.A.; Smits, J.P.; Cui, X.; John, C.; Gowda, K.; Desai, D.; Amin, S.G.; Schalkwijk, J.; Perdew, G.H.; et al. Genetic and pharmacological analysis identifies a physiological role for the AHR in epidermal differentiation. J. Investig. Dermatol. 2015, 135, 1320–1328. [Google Scholar] [CrossRef] [Green Version]
- Kopf, P.G.; Walker, M.K. 2,3,7,8-Tetrachlorodibenzo-p-dioxin increases reactive oxygen species production in human endothelial cells via induction of cytochrome P4501A1. Toxicol. Appl. Pharmacol. 2010, 245, 91–99. [Google Scholar] [CrossRef] [Green Version]
- Bargo, P.R.; Walston, S.T.; Chu, M.; Seo, I.; Kollias, N. Non-invasive assessment of tryptophan fluorescence and confocal microscopy provide information on skin barrier repair dynamics beyond TEWL. Exp. Dermatol. 2013, 22, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Gillies, R.; Zonios, G.; Anderson, R.R.; Kollias, N. Fluorescence excitation spectroscopy provides information about human skin in vivo. J. Investig. Dermatol. 2000, 115, 704–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fritsche, E.; Schäfer, C.; Calles, C.; Bernsmann, T.; Bernshausen, T.; Wurm, M.; Hübenthal, U.; Cline, J.E.; Hajimiragha, H.; Schroeder, P.; et al. Lightening up the UV response by identification of the aryl hydrocarbon receptor as a cytoplasmatic target for ultraviolet B radiation. Proc. Natl. Acad. Sci. USA 2007, 104, 8851–8856. [Google Scholar] [CrossRef] [Green Version]
- Katiyar, S.K.; Matsui, M.S.; Mukhtar, H. Ultraviolet-B exposure of human skin induces cytochromes P450 1A1 and 1B1. J. Investig. Dermatol. 2000, 114, 328–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukhtar, H.; Del Tito, B.J., Jr.; Matgouranis, P.M.; Das, M.; Asokan, P.; Bickers, D.R. Additive effects of ultraviolet B and crude coal tar on cutaneous carcinogen metabolism: Possible relevance to the tumorigenicity of the Goeckerman regimen. J. Investig. Dermatol. 1986, 87, 348–353. [Google Scholar] [CrossRef] [Green Version]
- Wei, Y.D.; Rannug, U.; Rannug, A. UV-induced CYP1A1 gene expression in human cells is mediated by tryptophan. Chem. Biol. Interact. 1999, 118, 127–140. [Google Scholar] [CrossRef]
- Magiatis, P.; Pappas, P.; Gaitanis, G.; Mexia, N.; Melliou, E.; Galanou, M.; Vlachos, C.; Stathopoulou, K.; Skaltsounis, A.L.; Marselos, M.; et al. Malassezia yeasts produce a collection of exceptionally potent activators of the Ah (dioxin) receptor detected in diseased human skin. J. Investig. Dermatol. 2013, 133, 2023–2030. [Google Scholar] [CrossRef] [Green Version]
- Jin, U.H.; Karki, K.; Kim, S.B.; Safe, S. Inhibition of pancreatic cancer Panc1 cell migration by omeprazole is dependent on aryl hydrocarbon receptor activation of JNK. Biochem. Biophys. Res. Commun. 2018, 501, 751–757. [Google Scholar] [CrossRef]
- Tsuji, G.; Takahara, M.; Uchi, H.; Matsuda, T.; Chiba, T.; Takeuchi, S.; Yasukawa, F.; Moroi, Y.; Furue, M. Identification of ketoconazole as an AhR-Nrf2 activator in cultured human keratinocytes: The basis of its anti-inflammatory effect. J. Investig. Dermatol. 2012, 132, 59–68. [Google Scholar] [CrossRef] [Green Version]
- Tsuji, G.; Ito, T.; Chiba, T.; Mitoma, C.; Nakahara, T.; Uchi, H.; Furue, M. The role of the OVOL1-OVOL2 axis in normal and diseased human skin. J. Dermatol. Sci. 2018, 90, 227–231. [Google Scholar] [CrossRef] [Green Version]
- Furue, K.; Ito, T.; Tsuji, G.; Ulzii, D.; Vu, Y.H.; Kido-Nakahara, M.; Nakahara, T.; Furue, M. The IL-13-OVOL1-FLG axis in atopic dermatitis. Immunology 2019, 158, 281–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, T.; Tsuji, G.; Ohno, F.; Uchi, H.; Nakahara, T.; Hashimoto-Hachiya, A.; Yoshida, Y.; Yamamoto, O.; Oda, Y.; Furue, M. Activation of the OVOL1-OVOL2 axis in the hair bulb and in pilomatricoma. Am. J. Pathol. 2016, 186, 1036–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, T.; Tsuji, G.; Ohno, F.; Nakahara, T.; Uchi, H.; Furue, M. Potential role of the OVOL1-OVOL2 axis and c-Myc in the progression of cutaneous squamous cell carcinoma. Mod. Pathol. 2017, 30, 919–927. [Google Scholar] [CrossRef]
- Nair, M.; Teng, A.; Bilanchone, V.; Agrawal, A.; Li, B.; Dai, X. Ovol1 regulates the growth arrest of embryonic epidermal progenitor cells and represses c-myc transcription. J. Cell Biol. 2006, 173, 253–264. [Google Scholar] [CrossRef]
- Paternoster, L.; Standl, M.; Waage, J.; Baurecht, H.; Hotze, M.; Strachan, D.P.; Curtin, J.A.; Bønnelykke, K.; Tian, C.; Takahashi, A.; et al. Multi-ancestry genome-wide association study of 21,000 cases and 95,000 controls identifies new risk loci for atopic dermatitis. Nat. Genet. 2015, 47, 1449–1456. [Google Scholar]
- Wei, Y.D.; Bergander, L.; Rannug, U.; Rannug, A. Regulation of CYP1A1 transcription via the metabolism of the tryptophan-derived 6-formylindolo[3,2-b]carbazole. Arch. Biochem. Biophys. 2000, 383, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Knutson, J.C.; Poland, A. Response of murine epidermis to 2,3,7,8-tetrachlorodibenzo-p-dioxin: Interaction of the ah and hr loci. Cell 1982, 30, 225–234. [Google Scholar] [CrossRef]
- Furue, M.; Ulzii, D.; Vu, Y.H.; Tsuji, G.; Kido-Nakahara, M.; Nakahara, T. Pathogenesis of atopic dermatitis: Current paradigm. Iran. J. Immunol. 2019, 16, 97–107. [Google Scholar]
- Furue, M.; Chiba, T.; Tsuji, G.; Ulzii, D.; Kido-Nakahara, M.; Nakahara, T.; Kadono, T. Atopic dermatitis: Immune deviation, barrier dysfunction, IgE autoreactivity and new therapies. Allergol. Int. 2017, 66, 398–403. [Google Scholar] [CrossRef]
- Seo, E.; Yoon, J.; Jung, S.; Lee, J.; Lee, B.H.; Yu, J. Phenotypes of atopic dermatitis identified by cluster analysis in early childhood. J. Dermatol. 2019, 46, 117–123. [Google Scholar] [CrossRef]
- Arima, K.; Gupta, S.; Gadkari, A.; Hiragun, T.; Kono, T.; Katayama, I.; Demiya, S.; Eckert, L. Burden of atopic dermatitis in Japanese adults: Analysis of data from the 2013 National Health and Wellness Survey. J. Dermatol. 2018, 45, 390–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Igarashi, A.; Fujita, H.; Arima, K.; Inoue, T.; Dorey, J.; Fukushima, A.; Taguchi, Y. Health-care resource use and current treatment of adult atopic dermatitis patients in Japan: A retrospective claims database analysis. J. Dermatol. 2019, 46, 652–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, H.J.; Bae, J.Y.; Kim, J.E.; Na, C.H.; Park, G.H.; Bae, Y.I.; Shin, M.K.; Lee, Y.B.; Lee, U.H.; Jang, Y.H.; et al. Survey of disease awareness, treatment behavior and treatment satisfaction in patients with atopic dermatitis in Korea: A multicenter study. J. Dermatol. 2018, 45, 1172–1180. [Google Scholar] [CrossRef] [PubMed]
- Komura, Y.; Kogure, T.; Kawahara, K.; Yokozeki, H. Economic assessment of actual prescription of drugs for treatment of atopic dermatitis: Differences between dermatology and pediatrics in large-scale receipt data. J. Dermatol. 2018, 45, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, S.; Oba, J.; Esaki, H.; Furue, M. Non-corticosteroid adherence and itch severity influence perception of itch in atopic dermatitis. J. Dermatol. 2018, 45, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Elias, P.M. Primary role of barrier dysfunction in the pathogenesis of atopic dermatitis. Exp. Dermatol. 2018, 27, 847–851. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.E.; Leung, D.Y.M. Significance of skin barrier dysfunction in atopic dermatitis. Allergy Asthma Immunol. Res. 2008, 10, 207–215. [Google Scholar] [CrossRef] [Green Version]
- Rinaldi, A.O.; Morita, H.; Wawrzyniak, P.; Dreher, A.; Grant, S.; Svedenhag, P.; Akdis, C.A. Direct assessment of skin epithelial barrier by electrical impedance, spectroscopy. Allergy 2019, 74, 1934–1944. [Google Scholar] [CrossRef]
- Otsuka, A.; Doi, H.; Egawa, G.; Maekawa, A.; Fujita, T.; Nakamizo, S.; Nakashima, C.; Nakajima, S.; Watanabe, T.; Miyachi, Y.; et al. Possible new therapeutic strategy to regulate atopic dermatitis through upregulating filaggrin expression. J. Allergy Clin. Immunol. 2014, 133, 139–146. [Google Scholar] [CrossRef]
- Kim, B.E.; Leung, D.Y.M.; Boguniewicz, M.; Howell, M.D. Loricrin and involucrin expression is down-regulated by Th2 cytokines through STAT-6. Clin. Immunol. 2008, 126, 332–337. [Google Scholar] [CrossRef] [Green Version]
- Furue, M.; Iida, K.; Imaji, M.; Nakahara, T. Microbiome analysis of forehead skin in patients with atopic dermatitis and healthy subjects: Implication of Staphylococcus and Corynebacterium. J. Dermatol. 2018, 45, 876–877. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, K.; Moriwaki, M.; Miyake, R.; Hide, M. Staphylococcus aureus in atopic dermatitis: Strain-specific cell wall proteins and skin immunity. Allergol. Int. 2019, 68, 309–315. [Google Scholar] [CrossRef]
- Cascella, R.; Foti Cuzzola, V.; Lepre, T.; Galli, E.; Moschese, V.; Chini, L.; Mazzanti, C.; Fortugno, P.; Novelli, G.; Giardina, E. Full sequencing of the FLG gene in Italian patients with atopic eczema: Evidence of new mutations, but lack of an association. J. Investig. Dermatol. 2011, 131, 982–984. [Google Scholar] [CrossRef] [PubMed]
- Thawer-Esmail, F.; Jakasa, I.; Todd, G.; Wen, Y.; Brown, S.J.; Kroboth, K.; Campbell, L.E.; O’Regan, G.M.; McLean, W.H.; Irvine, A.D.; et al. South African amaXhosa patients with atopic dermatitis have decreased levels of filaggrin breakdown products but no loss-of-function mutations in filaggrin. J. Allergy Clin. Immunol. 2014, 133, 280–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasaki, T.; Furusyo, N.; Shiohama, A.; Takeuchi, S.; Nakahara, T.; Uchi, H.; Hirota, T.; Tamari, M.; Shimizu, N.; Ebihara, T.; et al. Filaggrin loss-of-function mutations are not a predisposing factor for atopic dermatitis in an Ishigaki Island under subtropical climate. J. Dermatol. Sci. 2014, 76, 10–15. [Google Scholar] [CrossRef]
- Howell, M.D.; Kim, B.E.; Gao, P.; Grant, A.V.; Boguniewicz, M.; DeBenedetto, A.; Schneider, L.; Beck, L.A.; Barnes, K.C.; Leung, D.Y. Cytokine modulation of atopic dermatitis filaggrin skin expression. J. Allergy Clin. Immunol. 2007, 120, 150–155. [Google Scholar] [CrossRef] [Green Version]
- Jurakic Toncic, R.; Kezic, S.; Jakasa, I.; Ljubojevic Hadzavdic, S.; Balic, A.; Petkovic, M.; Pavicic, B.; Zuzul, K.; Marinovic, B. Filaggrin loss-of-function mutations and levels of filaggrin degradation products in adult patients with atopic dermatitis in Croatia. J. Eur. Acad. Dermatol. Venereol. 2020. [Google Scholar] [CrossRef]
- Simpson, E.L.; Bieber, T.; Guttman-Yassky, E.; Beck, L.A.; Blauvelt, A.; Cork, M.J.; Silverberg, J.I.; Deleuran, M.; Kataoka, Y.; Lacour, J.P.; et al. Two phase 3 trials of dupilumab versus placebo in atopic dermatitis. N. Engl. J. Med. 2016, 375, 2335–2348. [Google Scholar] [CrossRef]
- Tsoi, L.C.; Rodriguez, E.; Degenhardt, F.; Baurecht, H.; Wehkamp, U.; Volks, N.; Szymczak, S.; Swindell, W.R.; Sarkar, M.K.; Raja, K.; et al. Atopic dermatitis is an IL-13-dominant disease with greater molecular heterogeneity compared to psoriasis. J. Investig. Dermatol. 2019, 139, 1480–1489. [Google Scholar] [CrossRef] [Green Version]
- Wollenberg, A.; Howell, M.D.; Guttman-Yassky, E.; Silverberg, J.I.; Kell, C.; Ranade, K.; Moate, R.; van der Merwe, R. Treatment of atopic dermatitis with tralokinumab, an anti-IL-13 mAb. J. Allergy Clin. Immunol. 2019, 143, 135–141. [Google Scholar] [CrossRef] [Green Version]
- Jensen, J.M.; Fölster-Holst, R.; Baranowsky, A.; Schunck, M.; Winoto-Morbach, S.; Neumann, C.; Schütze, S.; Proksch, E. Impaired sphingomyelinase activity and epidermal differentiation in atopic dermatitis. J. Investig. Dermatol. 2004, 122, 1423–1431. [Google Scholar] [CrossRef] [Green Version]
- Pellerin, L.; Henry, J.; Hsu, C.Y.; Balica, S.; Jean-Decoster, C.; Méchin, M.C.; Hansmann, B.; Rodriguez, E.; Weindinger, S.; Schmitt, A.M.; et al. Defects of filaggrin-like proteins in both lesional and nonlesional atopic skin. J. Allergy Clin. Immunol. 2013, 131, 1094–1102. [Google Scholar] [CrossRef]
- Broccardo, C.J.; Mahaffey, S.; Schwarz, J.; Wruck, L.; David, G.; Schlievert, P.M.; Reisdorph, N.A.; Leung, D.Y. Comparative proteomic profiling of patients with atopic dermatitis based on history of eczema herpeticum infection and Staphylococcus aureus colonization. J. Allergy Clin. Immunol. 2011, 127, 186–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trzeciak, M.; Sakowicz-Burkiewicz, M.; Wesserling, M.; Dobaczewska, D.; Gleń, J.; Nowicki, R.; Pawelczyk, T. Expression of cornified envelope proteins in skin and its relationship with atopic dermatitis phenotype. Acta Derm. Venereol. 2017, 97, 36–41. [Google Scholar] [CrossRef] [Green Version]
- Guttman-Yassky, E.; Ungar, B.; Malik, K.; Dickstein, D.; Suprun, M.; Estrada, Y.D.; Xu, H.; Peng, X.; Oliva, M.; Todd, D.; et al. Molecular signatures order the potency of topically applied anti-inflammatory drugs in patients with atopic dermatitis. J. Allergy Clin. Immunol. 2017, 140, 1032–1042. [Google Scholar] [CrossRef] [Green Version]
- Meisser, S.S.; Altunbulakli, C.; Bandier, J.; Opstrup, M.S.; Castro-Giner, F.; Akdis, M.; Bonefeld, C.M.; Johansen, J.D.; Akdis, C.A. Skin barrier damage after exposure to paraphenylenediamine. J. Allergy Clin. Immunol. 2020, 145, 619–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amano, W.; Nakajima, S.; Kunugi, H.; Numata, Y.; Kitoh, A.; Egawa, G.; Dainichi, T.; Honda, T.; Otsuka, A.; Kimoto, Y.; et al. The Janus kinase inhibitor JTE-052 improves skin barrier function through suppressing signal transducer and activator of transcription 3 signaling. J. Allergy Clin. Immunol. 2015, 136, 667–677. [Google Scholar] [CrossRef] [Green Version]
- Clarysse, K.; Pfaff, C.M.; Marquardt, Y.; Huth, L.; Kortekaas Krohn, I.; Kluwig, D.; Lüscher, B.; Gutermuth, J.; Baron, J. JAK1/3 inhibition preserves epidermal morphology in full-thickness 3D skin models of atopic dermatitis and psoriasis. J. Eur. Acad. Dermatol. Venereol. 2019, 33, 367–375. [Google Scholar] [CrossRef] [PubMed]
- Ranasinghe, C.; Trivedi, S.; Wijesundara, D.K.; Jackson, R.J. IL-4 and IL-13 receptors: Roles in immunity and powerful vaccine adjuvants. Cytokine Growth Factor Rev. 2014, 25, 437–442. [Google Scholar] [CrossRef]
- Murata, T.; Taguchi, J.; Puri, R.K.; Mohri, H. Sharing of receptor subunits and signal transduction pathway between the IL-4 and IL-13 receptor system. Int. J. Hematol. 1999, 69, 13–20. [Google Scholar]
- Junttila, I.S. Tuning the cytokine responses: An update on interleukin (IL)-4 and IL-13 receptor complexes. Front. Immunol. 2018, 9, 888. [Google Scholar] [CrossRef] [PubMed]
- Furue, M.; Ulzii, D.; Nakahara, T.; Tsuji, G.; Furue, K.; Hashimoto-Hachiya, A.; Kido-Nakahara, M. Implications of IL-13Rα2 in atopic skin inflammation. Allergol. Int. 2020. [Google Scholar] [CrossRef]
- Ulzii, D.; Kido-Nakahara, M.; Nakahara, T.; Tsuji, G.; Furue, K.; Hashimoto-Hachiya, A.; Furue, M. Scratching counteracts IL-13 signaling by upregulating the decoy receptor IL-13Rα2 in keratinocytes. Int. J. Mol. Sci. 2019, 20, 3324. [Google Scholar] [CrossRef] [Green Version]
- Harada, N.; Higuchi, K.; Wakao, H.; Hamasaki, N.; Izuhara, K. Identification of the critical portions of the human IL-4 receptor alpha chain for activation of STAT6. Biochem. Biophys. Res. Commun. 1998, 246, 675–680. [Google Scholar] [CrossRef] [PubMed]
- Izuhara, K.; Heike, T.; Otsuka, T.; Yamaoka, K.; Mayumi, M.; Imamura, T.; Niho, Y.; Harada, N. Signal transduction pathway of interleukin-4 and interleukin-13 in human B cells derived from X-linked severe combined immunodeficiency patients. J. Biol. Chem. 1996, 271, 619–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murata, T.; Noguchi, P.D.; Puri, R.K. Receptors for interleukin (IL)-4 do not associate with the common gamma chain, and IL-4 induces the phosphorylation of JAK2 tyrosine kinase in human colon carcinoma cells. J. Biol. Chem. 1995, 270, 30829–30836. [Google Scholar] [CrossRef] [Green Version]
- Murata, T.; Husain, S.R.; Mohri, H.; Puri, R.K. Two different IL-13 receptor chains are expressed in normal human skin fibroblasts, and IL-4 and IL-13 mediate signal transduction through a common pathway. Int. Immunol. 1998, 10, 1103–1110. [Google Scholar] [CrossRef]
- Jin, S.H.; Choi, D.; Chun, Y.J.; Noh, M. Keratinocyte-derived IL-24 plays a role in the positive feedback regulation of epidermal inflammation in response to environmental and endogenous toxic stressors. Toxicol. Appl. Pharmacol. 2014, 280, 199–206. [Google Scholar] [CrossRef]
- Zhang, W.; Sakai, T.; Matsuda-Hirose, H.; Goto, M.; Yamate, T.; Hatano, Y. Cutaneous permeability barrier function in signal transducer and activator of transcription 6-deficient mice is superior to that in wild-type mice. J. Dermatol. Sci. 2018, 92, 54–61. [Google Scholar] [CrossRef] [Green Version]
- Olsan, E.E.; West, J.D.; Torres, J.A.; Doerr, N.; Weimbs, T. Identification of targets of IL-13 and STAT6 signaling in polycystic kidney disease. Am. J. Physiol. Renal. Physiol. 2018, 315, F86–F96. [Google Scholar] [CrossRef]
- Zissler, U.M.; Chaker, A.M.; Effner, R.; Ulrich, M.; Guerth, F.; Piontek, G.; Dietz, K.; Regn, M.; Knapp, B.; Theis, F.J.; et al. Interleukin-4 and interferon-γ orchestrate an epithelial polarization in the airways. Mucosal Immunol. 2016, 9, 917–926. [Google Scholar] [CrossRef] [PubMed]
- Masuoka, M.; Shiraishi, H.; Ohta, S.; Suzuki, S.; Arima, K.; Aoki, S.; Toda, S.; Inagaki, N.; Kurihara, Y.; Hayashida, S.; et al. Periostin promotes chronic allergic inflammation in response to Th2 cytokines. J. Clin. Investig. 2012, 122, 2590–2600. [Google Scholar] [CrossRef] [Green Version]
- Mitamura, Y.; Murai, M.; Mitoma, C.; Furue, M. NRF2 activation inhibits both TGF-β1- and IL-13-mediated periostin expression in fibroblasts: Benefit of cinnamaldehyde for antifibrotic treatment. Oxidative Med. Cell Longev. 2018, 2018, 2475047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitamura, Y.; Nunomura, S.; Nanri, Y.; Ogawa, M.; Yoshihara, T.; Masuoka, M.; Tsuji, G.; Nakahara, T.; Hashimoto-Hachiya, A.; Conway, S.J.; et al. The IL-13/periostin/IL-24 pathway causes epidermal barrier dysfunction in allergic skin inflammation. Allergy 2018, 73, 1881–1891. [Google Scholar] [CrossRef] [PubMed]
- Mitamura, Y.; Nunomura, S.; Furue, M.; Izuhara, K. IL-24: A new player in the pathogenesis of pro-inflammatory and allergic skin diseases. Allergol. Int. 2020. [Google Scholar] [CrossRef]
- Niess, J.H.; Hruz, P.; Kaymak, T. The interleukin-20 cytokines in intestinal diseases. Front. Immunol. 2018, 9, 1373. [Google Scholar] [CrossRef]
- Furue, M.; Uchi, H.; Mitoma, C.; Hashimoto-Hachiya, A.; Chiba, T.; Ito, T.; Nakahara, T.; Tsuji, G. Antioxidants for healthy skin: The emerging role of aryl hydrocarbon receptors and nuclear factor-erythroid 2-related factor-2. Nutrients 2017, 9, 223. [Google Scholar] [CrossRef]
- Yasukawa, S.; Miyazaki, Y.; Yoshii, C.; Nakaya, M.; Ozaki, N.; Toda, S.; Kuroda, E.; Ishibashi, K.; Yasuda, T.; Natsuaki, Y.; et al. An ITAM-Syk-CARD9 signalling axis triggers contact hypersensitivity by stimulating IL-1 production in dendritic cells. Nat. Commun. 2014, 5, 3755. [Google Scholar] [CrossRef] [Green Version]
- Eto, H.; Tsuji, G.; Chiba, T.; Furue, M.; Hyodo, F. Non-invasive evaluation of atopic dermatitis based on redox status using in vivo dynamic nuclear polarization magnetic resonance imaging. Free Radic. Biol. Med. 2017, 103, 209–215. [Google Scholar] [CrossRef]
- Bissonnette, R.; Poulin, Y.; Zhou, Y.; Tan, J.; Hong, H.C.; Webster, J.; Ip, W.; Tang, L.; Lyle, M. Efficacy and safety of topical WBI-1001 in patients with mild to severe atopic dermatitis: Results from a 12-week, multicentre, randomized, placebo-controlled double-blind trial. Br. J. Dermatol. 2012, 166, 853–860. [Google Scholar] [CrossRef]
- Peppers, J.; Paller, A.S.; Maeda-Chubachi, T.; Wu, S.; Robbins, K.; Gallagher, K.; Kraus, J.E. A phase 2, randomized dose-finding study of tapinarof (GSK2894512 cream) for the treatment of atopic dermatitis. J. Am. Acad. Dermatol. 2019, 80, 89–98. [Google Scholar] [CrossRef]
- Smith, S.H.; Jayawickreme, C.; Rickard, D.J.; Nicodeme, E.; Bui, T.; Simmons, C.; Coquery, C.M.; Neil, J.; Pryor, W.M.; Mayhew, D.; et al. Tapinarof is a natural AhR agonist that resolves skin inflammation in mice and humans. J. Investig. Dermatol. 2017, 137, 2110–2119. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, K.; Ito, K.; Namikawa, S. Anti-inflammatory activity of the dry distillation tar of delipidated soybean (Glyteer) (3). Effects on type I-type IV allergic reaction. Nihon Yakurigaku Zasshi 1990, 95, 149–157. (In Japanese) [Google Scholar] [CrossRef] [PubMed]
- Richardson, W.H.; Schmidt, T.M.; Nealson, K.H. Identification of an anthraquinone pigment and a hydroxystilbene antibiotic from Xenorhabdus luminescens. Appl. Environ. Microbiol. 1988, 54, 1602–1605. [Google Scholar] [CrossRef] [Green Version]
- Zang, Y.N.; Jiang, D.L.; Cai, L.; Chen, X.; Wang, Q.; Xie, Z.W.; Liu, Y.; Zhang, C.Y.; Jing, S.; Chen, G.H.; et al. Use of a dose-response model to guide future clinical trial of Benvitimod cream to treat mild and moderate psoriasis. Int. J. Clin. Pharmacol. Ther. 2016, 54, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Bissonnette, R.; Vasist, L.S.; Bullman, J.N.; Collingwood, T.; Chen, G.; Maeda-Chubachi, T. Systemic pharmacokinetics, safety, and preliminary efficacy of topical AhR agonist Tapinarof: Results of a phase 1 study. Clin. Pharmacol. Drug Dev. 2018, 7, 524–531. [Google Scholar] [CrossRef] [PubMed]
- Hirakawa, S.; Saito, R.; Ohara, H.; Okuyama, R.; Aiba, S. Dual oxidase 1 induced by Th2 cytokines promotes STAT6 phosphorylation via oxidative inactivation of protein tyrosine phosphatase 1B in human epidermal keratinocytes. J. Immunol. 2011, 186, 4762–4770. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Malumbres, R.; Shields, B.; Jiang, X.; Sarosiek, K.A.; Natkunam, Y.; Tiganis, T.; Lossos, I.S. PTP1B is a negative regulator of interleukin 4-induced STAT6 signaling. Blood 2008, 112, 4098–4108. [Google Scholar]
- Sharma, P.; Chakraborty, R.; Wang, L.; Min, B.; Tremblay, M.L.; Kawahara, T.; Lambeth, J.D.; Haque, S.J. Redox regulation of interleukin-4 signaling. Immunity 2008, 29, 551–564. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, Y.; Ito, T.; Tsuji, G.; Furue, M. Baicalein inhibits benzo[a]pyrene-induced toxic response by downregulating Src phosphorylation and by upregulating NRF2-HMOX1 system. Antioxidants 2020, 9, 507. [Google Scholar] [CrossRef]
- Hashimoto-Hachiya, A.; Tsuji, G.; Furue, M. Antioxidants cinnamaldehyde and Galactomyces fermentation filtrate downregulate senescence marker CDKN2A/p16INK4A via NRF2 activation in keratinocytes. J. Dermatol. Sci. 2019, 96, 53–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Min, J.; Mao, X.; Wang, X.; Yang, Y.; Chen, Y. Edaravone ameliorates experimental autoimmune thyroiditis in rats through HO-1-dependent STAT3/PI3K/Akt pathway. Am. J. Transl. Res. 2018, 10, 2037–2046. [Google Scholar] [PubMed]
- Park, B.; Lim, J.W.; Kim, H. Lycopene treatment inhibits activation of Jak1/Stat3 and Wnt/β-catenin signaling and attenuates hyperproliferation in gastric epithelial cells. Nutr. Res. 2019, 70, 70–81. [Google Scholar] [CrossRef] [PubMed]
- Dao, T.T.P.; Song, K.; Kim, J.Y.; Kim, Y.S. Igalan from Inula helenium (L.) suppresses the atopic dermatitis-like response in stimulated HaCaT keratinocytes via JAK/STAT3 signaling. Inflamm. Res. 2020, 69, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Song, K.; Hiebert, P.; Werner, S.; Kim, T.G.; Kim, Y.S. Tussilagonone ameliorates psoriatic features in keratinocytes and imiquimod-induced psoriasis-like lesions in mice via NRF2 activation. J. Investig. Dermatol. 2020, 140, 1223–1232. [Google Scholar] [CrossRef]
- Takemura, M.; Nakahara, T.; Hashimoto-Hachiya, A.; Furue, M.; Tsuji, G. Glyteer, soybean tar, impairs IL-4/Stat6 signaling in murine bone marrow-derived dendritic cells: The basis of its therapeutic effect on atopic dermatitis. Int. J. Mol. Sci. 2018, 19, 1169. [Google Scholar] [CrossRef] [Green Version]
- Miake, S.; Tsuji, G.; Takemura, M.; Hashimoto-Hachiya, A.; Vu, Y.H.; Furue, M.; Nakahara, T. IL-4 augments IL-31/IL-31 receptor alpha interaction leading to enhanced Ccl17 and Ccl22 production in dendritic cells: Implications for atopic dermatitis. Int. J. Mol. Sci. 2019, 20, 4053. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, G.; Kanaji, S.; Hirano, A.; Arima, K.; Shinagawa, A.; Goda, C.; Yasunaga, S.; Ikizawa, K.; Yanagihara, Y.; Kubo, M.; et al. Induction and activation of the aryl hydrocarbon receptor by IL-4 in B cells. Int. Immunol. 2005, 17, 797–805. [Google Scholar] [CrossRef]
- Furue, M.; Hashimoto-Hachiya, A.; Tsuji, G. Aryl hydrocarbon receptor in atopic dermatitis and psoriasis. Int. J. Mol. Sci. 2019, 20, 5424. [Google Scholar] [CrossRef] [Green Version]
- Tsuji, G.; Hashimoto-Hachiya, A.; Yen, V.H.; Miake, S.; Takemura, M.; Mitamura, Y.; Ito, T.; Murata, M.; Furue, M.; Nakahara, T. Aryl hydrocarbon receptor activation downregulates IL-33 expression in keratinocytes via Ovo-like 1. J. Clin. Med. 2020, 9, 891. [Google Scholar] [CrossRef] [Green Version]
- Czarnowicki, T.; Esaki, H.; Gonzalez, J.; Malajian, D.; Shemer, A.; Noda, S.; Talasila, S.; Berry, A.; Gray, J.; Becker, L.; et al. Early pediatric atopic dermatitis shows only a cutaneous lymphocyte antigen (CLA)(+) TH2/TH1 cell imbalance, whereas adults acquire CLA(+) TH22/TC22 cell subsets. J. Allergy Clin. Immunol. 2015, 136, 941–951. [Google Scholar] [CrossRef] [Green Version]
- Esaki, H.; Brunner, P.M.; Renert-Yuval, Y.; Czarnowicki, T.; Huynh, T.; Tran, G.; Lyon, S.; Rodriguez, G.; Immaneni, S.; Johnson, D.B.; et al. Early-onset pediatric atopic dermatitis is T(H)2 but also T(H)17 polarized in skin. J. Allergy Clin. Immunol. 2016, 138, 1639–1651. [Google Scholar] [CrossRef] [Green Version]
- Gittler, J.K.; Shemer, A.; Suárez-Fariñas, M.; Fuentes-Duculan, J.; Gulewicz, K.J.; Wang, C.Q.; Mitsui, H.; Cardinale, I.; de Guzman Strong, C.; Krueger, J.G.; et al. Progressive activation of T(H)2/T(H)22 cytokines and selective epidermal proteins characterizes acute and chronic atopic dermatitis. J. Allergy Clin. Immunol. 2012, 130, 1344–1354. [Google Scholar] [CrossRef] [Green Version]
- Eyerich, K.; Dimartino, V.; Cavani, A. IL-17 and IL-22 in immunity: Driving protection and pathology. Eur. J. Immunol. 2017, 47, 607–614. [Google Scholar] [CrossRef] [Green Version]
- Nograles, K.E.; Zaba, L.C.; Shemer, A.; Fuentes-Duculan, J.; Cardinale, I.; Kikuchi, T.; Ramon, M.; Bergman, R.; Krueger, J.G.; Guttman-Yassky, E. IL-22-producing “T22” T cells account for upregulated IL-22 in atopic dermatitis despite reduced IL-17-producing TH17 T cells. J. Allergy Clin. Immunol. 2009, 123, 1244–1252. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.S.; Cella, M.; McDonald, K.G.; Garlanda, C.; Kennedy, G.D.; Nukaya, M.; Mantovani, A.; Kopan, R.; Bradfield, C.A.; Newberry, R.D.; et al. AHR drives the development of gut ILC22 cells and postnatal lymphoid tissues via pathways dependent on and independent of Notch. Nat. Immunol. 2011, 13, 144–151. [Google Scholar] [CrossRef]
- Nikamo, P.; Cheuk, S.; Lysell, J.; Enerbäck, C.; Bergh, K.; Xu Landén, N.; Eidsmo, L.; Ståhle, M. Genetic variants of the IL22 promoter associate to onset of psoriasis before puberty and increased IL-22 production in T cells. J. Investig. Dermatol. 2014, 134, 1535–1541. [Google Scholar] [CrossRef] [Green Version]
- Schiering, C.; Vonk, A.; Das, S.; Stockinger, B.; Wincent, E. Cytochrome P4501-inhibiting chemicals amplify aryl hydrocarbon receptor activation and IL-22 production in T helper 17 cells. Biochem. Pharmacol. 2018, 151, 47–58. [Google Scholar] [CrossRef]
- Trifari, S.; Spits, H. IL-22-producing CD4+ T cells: Middle-men between the immune system and its environment. Eur. J. Immunol. 2010, 40, 2369–2371. [Google Scholar] [CrossRef]
- Brunner, P.M.; Pavel, A.B.; Khattri, S.; Leonard, A.; Malik, K.; Rose, S.; Jim On, S.; Vekaria, A.S.; Traidl-Hoffmann, C.; Singer, G.K.; et al. Baseline IL-22 expression in patients with atopic dermatitis stratifies tissue responses to fezakinumab. J. Allergy Clin. Immunol. 2019, 143, 142–154. [Google Scholar] [CrossRef]
- Jones, B.C.; Logsdon, N.J.; Walter, M.R. Structure of IL-22 bound to its high-affinity IL-22R1 chain. Structure 2008, 16, 1333–1344. [Google Scholar] [CrossRef] [Green Version]
- Wolk, K.; Kunz, S.; Witte, E.; Friedrich, M.; Asadullah, K.; Sabat, R. IL-22 increases the innate immunity of tissues. Immunity 2004, 21, 241–254. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Zheng, S.G. Interleukin-22: A likely target for treatment of autoimmune diseases. Autoimmun. Rev. 2014, 13, 615–620. [Google Scholar] [CrossRef] [Green Version]
- Lejeune, D.; Dumoutier, L.; Constantinescu, S.; Kruijer, W.; Schuringa, J.J.; Renauld, J.C. Interleukin-22 (IL-22) activates the JAK/STAT, ERK, JNK, and p38 MAP kinase pathways in a rat hepatoma cell line. Pathways that are shared with and distinct from IL-10. J. Biol. Chem. 2002, 277, 33676–33682. [Google Scholar] [CrossRef] [Green Version]
- Avitabile, S.; Odorisio, T.; Madonna, S.; Eyerich, S.; Guerra, L.; Eyerich, K.; Zambruno, G.; Cavani, A.; Cianfarani, F. Interleukin-22 promotes wound repair in diabetes by improving keratinocyte pro-healing functions. J. Investig. Dermatol. 2015, 135, 2862–2870. [Google Scholar] [CrossRef] [Green Version]
- Gutowska-Owsiak, D.; Schaupp, A.L.; Salimi, M.; Taylor, S.; Ogg, G.S. Interleukin-22 downregulates filaggrin expression and affects expression of profilaggrin processing enzymes. Br. J. Dermatol. 2011, 165, 492–498. [Google Scholar] [CrossRef] [PubMed]
- Tohyama, M.; Hanakawa, Y.; Shirakata, Y.; Dai, X.; Yang, L.; Hirakawa, S.; Tokumaru, S.; Okazaki, H.; Sayama, K.; Hashimoto, K. IL-17 and IL-22 mediate IL-20 subfamily cytokine production in cultured keratinocytes via increased IL-22 receptor expression. Eur. J. Immunol. 2009, 39, 2779–2788. [Google Scholar] [CrossRef]
- Noh, M.; Yeo, H.; Ko, J.; Kim, H.K.; Lee, C.H. MAP17 is associated with the T-helper cell cytokine-induced down-regulation of filaggrin transcription in human keratinocytes. Exp. Dermatol. 2010, 19, 355–362. [Google Scholar] [CrossRef]
- Jang, M.; Kim, H.; Kim, Y.; Choi, J.; Jeon, J.; Hwang, Y.; Kang, J.S.; Lee, W.J. The crucial role of IL-22 and its receptor in thymus and activation regulated chemokine production and T-cell migration by house dust mite extract. Exp. Dermatol. 2016, 25, 598–603. [Google Scholar] [CrossRef]
- Gläser, R.; Meyer-Hoffert, U.; Harder, J.; Cordes, J.; Wittersheim, M.; Kobliakova, J.; Fölster-Holst, R.; Proksch, E.; Schröder, J.M.; Schwarz, T. The antimicrobial protein psoriasin (S100A7) is upregulated in atopic dermatitis and after experimental skin barrier disruption. J. Investig. Dermatol. 2009, 129, 641–649. [Google Scholar]
- Bansal, G.; Das, D.; Hsieh, C.Y.; Wang, Y.H.; Gilmore, B.A.; Wong, C.M.; Suzuki, Y.J. IL-22 activates oxidant signaling in pulmonary vascular smooth muscle cells. Cell. Signal. 2013, 25, 2727–2733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, K.A.; Suh, J.W.; Lee, K.H.; Kang, J.L.; Woo, S.Y. IL-17 and IL-22 enhance skin inflammation by stimulating the secretion of IL-1β by keratinocytes via the ROS-NLRP3-caspase-1 pathway. Int. Immunol. 2012, 24, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Sun, L.; Li, W.; Wang, Y.; Li, X.; Liu, Y. Overexpression of macrophage stimulating 1 enhances the anti-tumor effects of IL-24 in esophageal cancer via inhibiting ERK-Mfn2 signaling-dependent mitophagy. Biomed. Pharmacother. 2019, 114, 108844. [Google Scholar] [CrossRef] [PubMed]
- Palombo, R.; Savini, I.; Avigliano, L.; Madonna, S.; Cavani, A.; Albanesi, C.; Mauriello, A.; Melino, G.; Terrinoni, A. Luteolin-7-glucoside inhibits IL-22/STAT3 pathway, reducing proliferation, acanthosis, and inflammation in keratinocytes and in mouse psoriatic model. Cell Death Dis. 2016, 7, e2344. [Google Scholar] [CrossRef]
- Furue, K.; Ito, T.; Furue, M. Differential efficacy of biologic treatments targeting the TNF-α/IL-23/IL-17 axis in psoriasis and psoriatic arthritis. Cytokine 2018, 111, 182–188. [Google Scholar] [CrossRef]
- Furue, K.; Ito, T.; Tsuji, G.; Kadono, T.; Furue, M. Psoriasis and the TNF/IL23/IL17 axis. G. Ital. Dermatol. Venereol. 2019, 154, 418–424. [Google Scholar] [CrossRef]
- Kamata, M.; Tada, Y. Safety of biologics in psoriasis. J. Dermatol. 2018, 45, 279–286. [Google Scholar] [CrossRef]
- Langley, R.G.; Elewski, B.E.; Lebwohl, M.; Reich, K.; Griffiths, C.E.; Papp, K.; Puig, L.; Nakagawa, H.; Spelman, L.; Sigurgeirsson, B.; et al. Secukinumab in plaque psoriasis--results of two phase 3 trials. N. Engl. J. Med. 2014, 371, 326–338. [Google Scholar] [CrossRef] [Green Version]
- Gordon, K.B.; Armstrong, A.W.; Foley, P.; Song, M.; Shen, Y.K.; Li, S.; Muñoz-Elías, E.J.; Branigan, P.; Liu, X.; Reich, K. Guselkumab efficacy after withdrawal is associated with suppression of serum IL-23-regulated IL-17 and IL-22 in psoriasis: VOYAGE 2 Study. J. Investig. Dermatol. 2019, 139, 2437–2446. [Google Scholar] [CrossRef]
- Lebwohl, M.; Strober, B.; Menter, A.; Gordon, K.; Weglowska, J.; Puig, L.; Papp, K.; Spelman, L.; Toth, D.; Kerdel, F.; et al. Phase 3 studies comparing brodalumab with ustekinumab in psoriasis. N. Engl. J. Med. 2015, 373, 1318–1328. [Google Scholar] [CrossRef]
- Momose, M.; Asahina, A.; Umezawa, Y.; Nakagawa, H. Long-term clinical efficacy and safety of secukinumab for Japanese patients with psoriasis: A single-center experience. J. Dermatol. 2018, 45, 318–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogawa, E.; Sato, Y.; Minagawa, A.; Okuyama, R. Pathogenesis of psoriasis and development of treatment. J. Dermatol. 2018, 45, 264–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komatsu-Fujii, T.; Honda, T.; Otsuka, A.; Kabashima, K. Inverse responses of the skin and nail lesions of psoriatic arthritis to an anti-interleukin-17A antibody and an anti-tumor necrosis factor-α antibody. J. Dermatol. 2019, 46, e440–e441. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.G.; Huang, Y.H.; Lee, J.H.; Lee, S.C.; Kim, T.G.; Aw, D.C.; Bao, W.; Dee, C.M.A.; Guana, A.; Tsai, T.F. Secukinumab demonstrates superior efficacy and a faster response in clearing skin in Asian subjects with moderate to severe plaque psoriasis compared with ustekinumab: Subgroup analysis from the CLEAR study. J. Dermatol. 2019, 46, 752–758. [Google Scholar] [CrossRef]
- Okubo, Y.; Ohtsuki, M.; Morita, A.; Yamaguchi, M.; Shima, T.; Tani, Y.; Nakagawa, H. Long-term efficacy and safety of secukinumab in Japanese patients with moderate to severe plaque psoriasis: 3-year results of a double-blind extension study. J. Dermatol. 2019, 46, 186–192. [Google Scholar] [CrossRef]
- Assefa, G.T.; Kaneko, S.; Oguro, H.; Morita, E. Treatment of psoriasis and psoriatic arthritis with secukinumab after unsatisfactory response to ustekinumab in multiple sclerosis patient. J. Dermatol. 2019, 46, e112–e113. [Google Scholar] [CrossRef]
- Chiricozzi, A.; Guttman-Yassky, E.; Suárez-Fariñas, M.; Nograles, K.E.; Tian, S.; Cardinale, I.; Chimenti, S.; Krueger, J.G. Integrative responses to IL-17 and TNF-α in human keratinocytes account for key inflammatory pathogenic circuits in psoriasis. J. Investig. Dermatol. 2011, 131, 677–687. [Google Scholar] [CrossRef]
- Krueger, J.G.; Brunner, P.M. Interleukin-17 alters the biology of many cell types involved in the genesis of psoriasis, systemic inflammation and associated comorbidities. Exp. Dermatol. 2018, 27, 115–123. [Google Scholar] [CrossRef] [Green Version]
- Zenobia, C.; Hajishengallis, G. Basic biology and role of interleukin-17 in immunity and inflammation. Periodontology 2000 2015, 69, 142–159. [Google Scholar] [CrossRef]
- Brembilla, N.C.; Senra, L.; Boehncke, W.H. The IL-17 family of cytokines in psoriasis: IL-17A and beyond. Front. Immunol. 2018, 9, 1682. [Google Scholar] [CrossRef] [Green Version]
- McGeachy, M.J.; Cua, D.J.; Gaffen, S.L. The IL-17 family of cytokines in health and disease. Immunity 2019, 50, 892–906. [Google Scholar] [CrossRef] [PubMed]
- Okada, S.; Puel, A.; Casanova, J.L.; Kobayashi, M. Chronic mucocutaneous candidiasis disease associated with inborn errors of IL-17 immunity. Clin. Transl. Immunol. 2016, 5, e114. [Google Scholar] [CrossRef]
- Su, Y.; Huang, J.; Zhao, X.; Lu, H.; Wang, W.; Yang, X.O.; Shi, Y.; Wang, X.; Lai, Y.; Dong, C. Interleukin-17 receptor D constitutes an alternative receptor for interleukin-17A important in psoriasis-like skin inflammation. Sci. Immunol. 2019, 4, eaau9657. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.Y.; Jia, K.; Zhang, Y. IL-17 promotes keratinocyte proliferation via the downregulation of C/EBPα. Exp. Ther. Med. 2016, 11, 631–636. [Google Scholar] [CrossRef]
- Chiricozzi, A.; Nograles, K.E.; Johnson-Huang, L.M.; Fuentes-Duculan, J.; Cardinale, I.; Bonifacio, K.M.; Gulati, N.; Mitsui, H.; Guttman-Yassky, E.; Suárez-Fariñas, M.; et al. IL-17 induces an expanded range of downstream genes in reconstituted human epidermis model. PLoS ONE 2014, 9, e90284. [Google Scholar] [CrossRef] [Green Version]
- Sun, D.P.; Yeh, C.H.; So, E.; Wang, L.Y.; Wei, T.S.; Chang, M.S.; Hsing, C.H. Interleukin (IL)-19 promoted skin wound healing by increasing fibroblast keratinocyte growth factor expression. Cytokine 2013, 62, 360–368. [Google Scholar] [CrossRef] [PubMed]
- Witte, E.; Kokolakis, G.; Witte, K.; Philipp, S.; Doecke, W.D.; Babel, N.; Wittig, B.M.; Warszawska, K.; Kurek, A.; Erdmann-Keding, M.; et al. IL-19 is a component of the pathogenetic IL-23/IL-17 cascade in psoriasis. J. Investig. Dermatol. 2014, 134, 2757–2767. [Google Scholar] [CrossRef] [Green Version]
- Lai, X.; Li, X.; Chang, L.; Chen, X.; Huang, Z.; Bao, H.; Huang, J.; Yang, L.; Wu, X.; Wang, Z.; et al. IL-19 up-regulates mucin 5AC production in patients with chronic rhinosinusitis via STAT3 pathway. Front. Immunol. 2019, 10, 1682. [Google Scholar] [CrossRef] [Green Version]
- House, J.S.; Zhu, S.; Ranjan, R.; Linder, K.; Smart, R.C. C/EBPalpha and C/EBPbeta are required for sebocyte differentiation and stratified squamous differentiation in adult mouse skin. PLoS ONE 2010, 5, e9837. [Google Scholar] [CrossRef] [Green Version]
- Crish, J.F.; Gopalakrishnan, R.; Bone, F.; Gilliam, A.C.; Eckert, R.L. The distal and proximal regulatory regions of the involucrin gene promoter have distinct functions and are required for in vivo involucrin expression. J. Investig. Dermatol. 2006, 126, 305–314. [Google Scholar] [CrossRef] [Green Version]
- Gutowska-Owsiak, D.; Schaupp, A.L.; Salimi, M.; Selvakumar, T.A.; McPherson, T.; Taylor, S.; Ogg, G.S. IL-17 downregulates filaggrin and affects keratinocyte expression of genes associated with cellular adhesion. Exp. Dermatol. 2012, 21, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Min, H.J.; Song, H.; Choi, S.Y.; Kim, T.H.; Cho, H.J.; Yoon, J.H.; Kim, C.H. Th2 cytokines differentially regulate psoriasin expression in human nasal epithelia. Am. J. Rhinol. Allergy 2014, 28, 449–453. [Google Scholar] [CrossRef] [PubMed]
- Bissonnette, R.; Bolduc, C.; Maari, C.; Nigen, S.; Webster, J.M.; Tang, L.; Lyle, M. Efficacy and safety of topical WBI-1001 in patients with mild to moderate psoriasis: Results from a randomized double-blind placebo-controlled, phase II trial. J. Eur. Acad. Dermatol. Venereol. 2012, 26, 1516–1521. [Google Scholar] [CrossRef] [PubMed]
- Robbins, K.; Bissonnette, R.; Maeda-Chubachi, T.; Ye, L.; Peppers, J.; Gallagher, K.; Kraus, J.E. Phase 2, randomized dose-finding study of tapinarof (GSK2894512 cream) for the treatment of plaque psoriasis. J. Am. Acad. Dermatol. 2019, 80, 714–721. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, H.; Zhu, X.; Feng, D.; Gong, J.; Han, T. Interleukin-24 induces neuroblastoma SH-SY5Y cell differentiation, growth inhibition, and apoptosis by promoting ROS production. J. Interferon Cytokine Res. 2013, 33, 709–714. [Google Scholar] [CrossRef]






© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Furue, M. Regulation of Filaggrin, Loricrin, and Involucrin by IL-4, IL-13, IL-17A, IL-22, AHR, and NRF2: Pathogenic Implications in Atopic Dermatitis. Int. J. Mol. Sci. 2020, 21, 5382. https://doi.org/10.3390/ijms21155382
Furue M. Regulation of Filaggrin, Loricrin, and Involucrin by IL-4, IL-13, IL-17A, IL-22, AHR, and NRF2: Pathogenic Implications in Atopic Dermatitis. International Journal of Molecular Sciences. 2020; 21(15):5382. https://doi.org/10.3390/ijms21155382
Chicago/Turabian StyleFurue, Masutaka. 2020. "Regulation of Filaggrin, Loricrin, and Involucrin by IL-4, IL-13, IL-17A, IL-22, AHR, and NRF2: Pathogenic Implications in Atopic Dermatitis" International Journal of Molecular Sciences 21, no. 15: 5382. https://doi.org/10.3390/ijms21155382