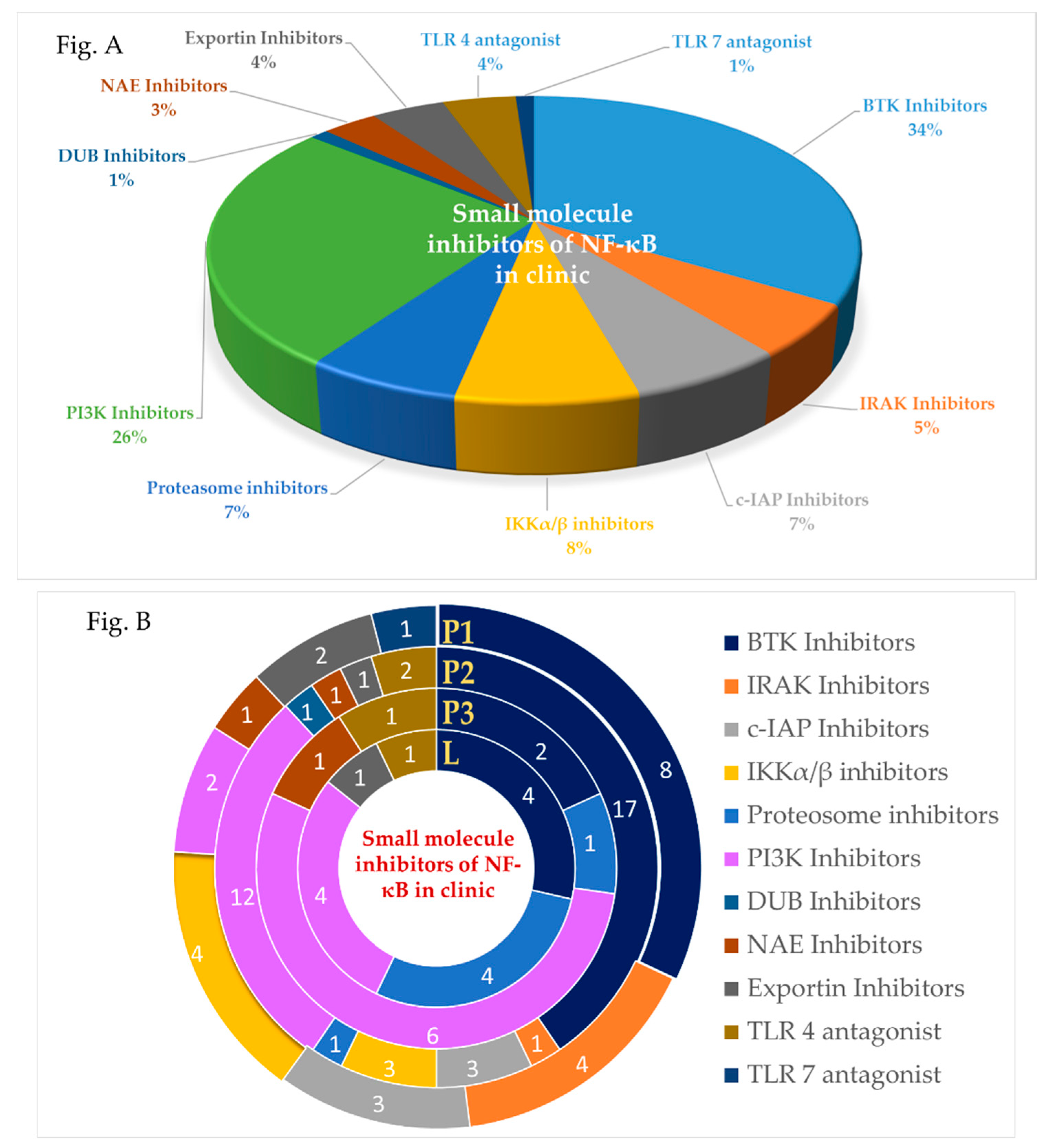
Small Molecule NF-κB Pathway Inhibitors in Clinic

Abstract
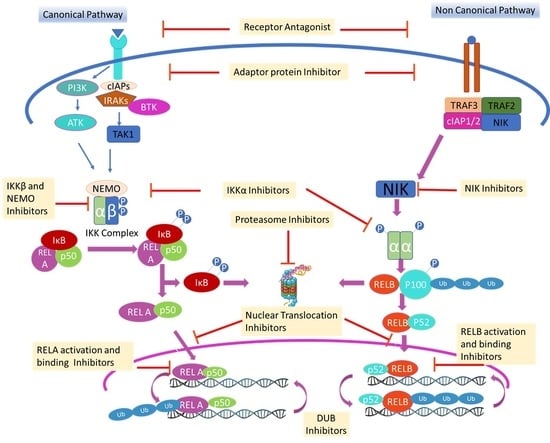
:
1. Introduction
2. Molecules that Inhibit Upstream IKK Complex in NF-κB Pathway
2.1. Inhibition of Cell Membrane Receptor Targets

{kind=link}
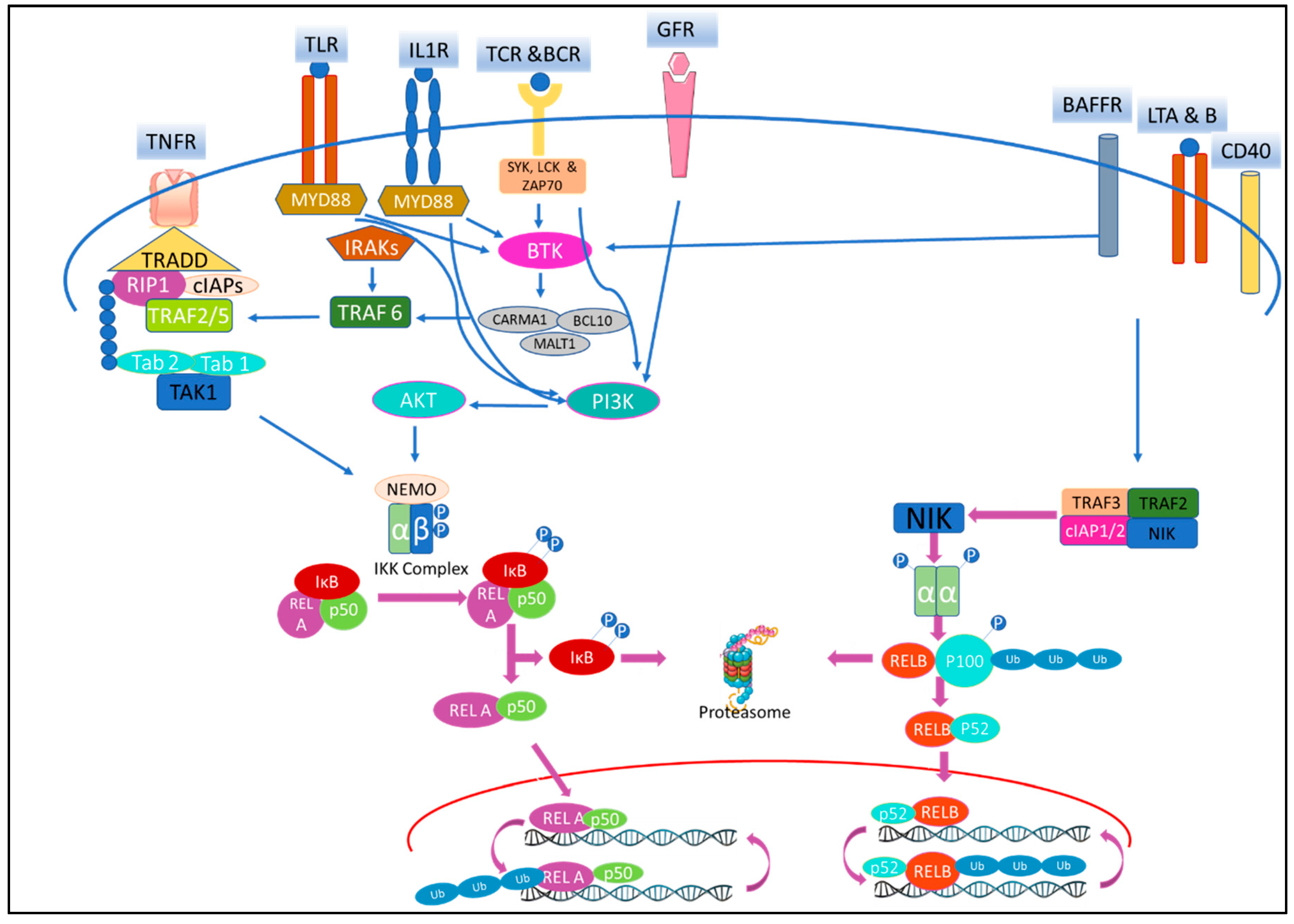{kind=link}
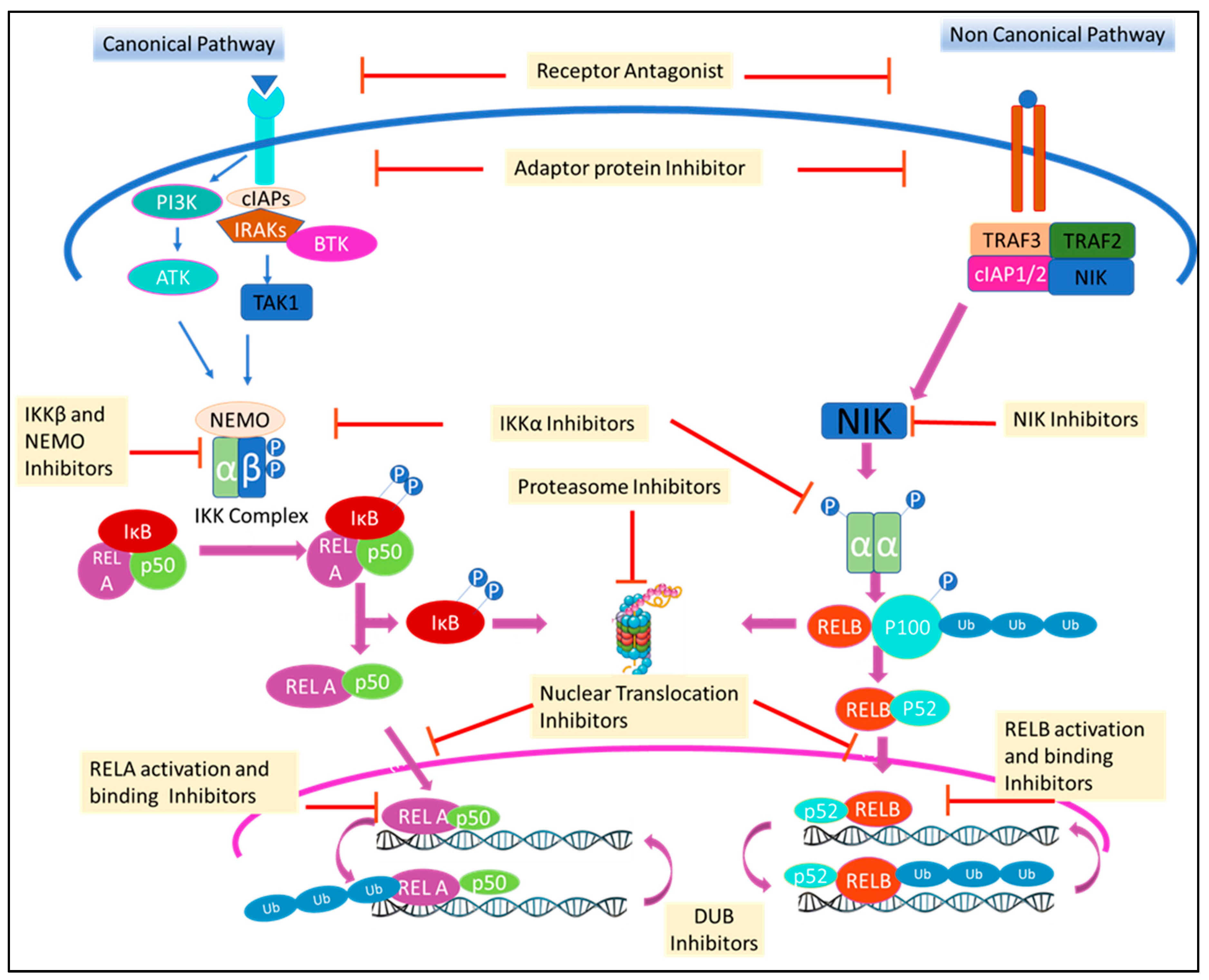{kind=link}
{kind=link}
{kind=link}
| Drug | Originator/Developer | Stage | Indication | Trail No. | Other Information |
|---|---|---|---|---|---|
| Toll like Receptor 4 (TLR4) antagonist | |||||
| Ibudilast (MN-166) | MediciNova Inc | Marketed in Japan | Allergic conjunctivitis, Asthma, Cerebrovascular disorders | NCT01860807; NCT04057898 |
|
| Phase3 | Amyotrophic Lateral Sclerosis | NCT04057898 | |||
| Phase 1/2 | Glioblastoma | NCT03782415 | |||
| MediciNova Inc/NIH | Phase 2 | MS | NCT01982942 | ||
| JKB-122 | Jenken Biosciences/TaiwanJ Pharmaceuticals | Phase 3 | Autoimmune Hepatitis | NCT04371718 |
|
| Phase 2 | Autoimmune Hepatitis | NCT02556372 | |||
| Phase 2 | Chronic hepatitis C | NCT02293941 | |||
| Phase 2 | NASH | NCT04255069 | |||
| VB-201 (TLR2/4 antagonist) | VBL Therapeutics | Phase 2 | Psoriasis | NCT01001468; NCT01837420 |
|
| Phase 2 | Ulcerative colitis | NCT01839214 | |||
| JKB-121 | Manal Abdelmalek, Duke University/TaiwanJ Pharmaceuticals | Phase 2 | NASH | NCT02442687 |
|
| Toll like Receptor 7 (TLR7) antagonist | |||||
| CPG-52364 | Pfizer | Phase 1 | Healthy Volunteers (For SLE) | NCT00547014 | Discontinued for SLE in Jan2020. |
2.2. Inhibition of Receptor Adaptor Protein
2.2.1. Bruton’s Tyrosine Kinase (BTK) Inhibitors
| Drug | Originator/Developer | Stage | Indication | Trail No. | Other Information |
|---|---|---|---|---|---|
| Bruton’s tyrosine kinase (BTK) inhibitors | |||||
| Zanubrutinib | BeiGene | Launched at 2019 | MCL | NCT04002297 |
|
| Preregistration | CLL | NCT03734016 | |||
| Phase 3 | BCL, RRWM | NCT03332173 | |||
| Phase 2 | DLBCL, Follicular lymphoma, Lymphoma, CLL, Lymphosarcoma, FL, MZL | NCT03145064; NCT04282018 | |||
| Acalabrutinib | AstraZeneca/Parexel | Phase 3 | CLL | NCT04008706 |
|
| Acerta Pharma/AstraZeneca, Biologics Inc, Merck | Launched 2017 | MCL | NCT02972840 | ||
| Acerta Pharma | Phase 3 | CLL | NCT02970318 | ||
| Acerta Pharma/AstraZeneca | Phase 2 | COVID-19 | NCT04346199, NCT04380688 | ||
| Phase 2 | Chronic GVHD | NCT04198922 | |||
| Acerta Pharma | Phase 2 | WM | NCT02180724 | ||
| Phase 2 | Metastatic Pancreatic Cancer | NCT02570711 | |||
| Phase 2 | RA | NCT02387762 | |||
| Acerta Pharma BV Merck Sharp & Dohme Corp. | Phase 2 | Ovarian Cancer | NCT02537444 | ||
| Phase 2 | NSCLC | NCT02448303 | |||
| Phase 2 | Squamous Cell Carcinoma of the Head and Neck | NCT02454179 | |||
| Acerta Pharma/Swedish Medical centre | Phase 2 | DLBCL | NCT03736616 | ||
| Ibrutinib | Janssen/Pharmacyclics; M.D. Anderson Cancer Center | Launched 2013 | CLL | NCT02801578 |
|
| Launched 2013 | WM | NCT02165397 | |||
| Launched 2013 | MCL | NCT01646021 | |||
| Phase 2 | GVHD | NCT02195869 | |||
| Janssen/Pharmacyclics | Phase 3 | Metastatic Pancreatic Adenocarcinoma | NCT02436668 | ||
| Phase 3 | DLBCL | NCT01855750 | |||
| Phase 3 | RRCLL, SLL | NCT01578707 | |||
| Dasatinib | BMS/Dana-Farber Cancer Institute | Launched 2006 | CML, acute cell lymphoblastic leukemia-lymphoma | NCT00123487, NCT03020030 |
|
| BMS | Phase 3 | Prostatic Neoplasms | NCT00744497 | ||
| BMS/NCI | Phase 2 | DLBCL | NCT00608361 | ||
| Phase 2 | Rhabdomyosarcoma | NCT0304170 | |||
| BMS/OSI pharma/M.D. Anderson Cancer Center | Phase 2 | NSCLC | NCT00826449 | ||
| BMS/Massachusetts General Hospital | Phase 2 | CLL | NCT00438854 | ||
| Phase 2 | Cholangiocarcinoma | NCT02428855 | |||
| BMS | Phase 2 | Breast Cancer | NCT00767520 | ||
| BMS/Jorge J. Castillo, MD | Phase 2 | WM | NCT04115059 | ||
| Tirabrutinib | Ono Pharmaceutical/Gilead Sciences | Registered | Lymphoma | NCT03162536 |
|
| Ono Pharmaceutical/Gilead Sciences | Preregistration | WM | NCT03740529 | ||
| Rilzabrutinib | Principia Biopharma | Phase 3 | Pemphigus vulgaris | NCT02704429 | Orphan drug status for Pemphigus vulgaris and Idiopathic thrombocytopenic purpura. |
| Evobrutinib | Merck Serono/EMD Serono | Phase 3 | MS | NCT04032158 | TEAE: Nasopharyngitis and increases in levels of alanine aminotransferase, aspartate aminotransferase and lipase [65]. |
| Merck Serono/EMD Serono | Phase 2 | RA | NCT02784106 | ||
| Orelabrutinib | InnoCare Pharma/Beijing | Phase 2 | SLE |
| |
| MZL | |||||
| ABBV-105 | AbbVie | Phase 2 | SLE | NCT03978520 | Focus is on ABBV-599 which is the ABBV-105/upadacitinib-combination |
| RA | NCT03682705 | ||||
| ABBV-599 | AbbVie | Phase 2 | RA, SLE | NCT03682705, NCT03978520 | |
| SAR-442168 | Principia Biopharma/Sanofi | Phase 2 | Relapsing MS | NCT03996291 | Common TEAE: Headache, upper respiratory tract infection and nasopharyngitis [67]. |
| Branebrutinib | BMS | Phase 2 | RA, SLE, Sjogren’s syndrome | NCT04186871 | In phase 1, no serious TEAE observed [68]. |
| TAS-5315 | Taiho Pharmaceutical | Phase 2 | RA | NCT03605251 | Observed decrease in platelet aggregation and prolonged bleeding time in phase 1 [69]. |
| Remibrutinib | Novartis | Phase 2 | Asthma, Sjogren’s syndrome, Urticaria | NCT03944707, NCT04035668, NCT04109313 | Phase 2 trial in Urticaria suspended due to COVID-19. |
| BMS-986142 | BMS | Phase 2 | RA | NCT02638948 |
|
| Sjogren’s syndrome | NCT02843659 | ||||
| Fenebrutinib | Genentech | Phase 2 | Urticaria | NCT03693625 | Higher doses may increase liver enzymes [71]. |
| Poseltinib | Hanmi Pharmaceutical/Eli Lilly | Phase 2 | RA | NCT02628028 | Phase 2 discontinued as study failed to demonstrate its target effectiveness in the interim results [72]. |
| Spebrutinib | Celegene | Phase 2 | RA | NCT01975610 | Celgene acquired by Bristol-Myers Squibb. |
| Phase 1 | CLL | NCT01732861 | |||
| DTRMWXHS-12 | Zhejiang DTRM Biopharma | Phase 2 | DLBCL, R and RCLL, Follicular Lymphoma | NCT04305444 | No development reported for B-cell lymphoma. |
| Phase 1 | MCL | NCT03836768 | |||
| Phase 1 | CLL, BCL | NCT02891590 | Evaluate the safety, tolerability and PK profile. | ||
| CT-1530 | Centaurus Biopharma Co., Ltd. | Phase 1/2 | B Cell-NHL, CLL, WM | NCT02981745 | Discontinued for all indications |
| REDX08608 | Redx Pharma/Loxo oncology | Phase 1/2 | CLL/SLL or NHL | NCT03740529 | In basal cell cancer, no development reported. |
| M-7583 | EMD Serono | Phase 1/2 | MCL, DLBCL, Relapsed/Refractory B Cell Malignancies | NCT02825836 | TEAE: Neutropenia, febrile neutropenia and pneumonia [73]. |
| ARQ-531 | ArQule/Merck | Phase 1/2 | Hematological malignancies | NCT03162536 | Well-tolerated through 65 mg QD [74]. |
| Vecabrutinib | Biogen Idec, Sunesis Pharmaceuticals | Phase 1/2 | Hematological malignancies | NCT03037645 | TEAE: Anemia, headache and night sweats. |
| TAK-020 | Takeda | Phase 1 | RA | NCT02413255 | TEAE; Abdominal distension, upper abdominal pain, nausea, and headache |
| BIIB068 | Biogen | Phase 1 | SLE | NCT02829541 | No update beyond phase 1 |
| AC-0058TA | ACEA Biosciences | Phase 1 | SLE | NCT03878303 | Phase 1 for Autoimmune disorders completed in 2017 and no further progress reported. |
| SN-1011 | Sinomab | Phase 1 | Autoimmune disorder | NCT04041544 | |
| BIIB-091 | Biogen | Phase 1 | Healthy Volunteer (MS) | NCT03943056 | Completes phase 1 trial for Multiple sclerosis. |
| TG-1701 | Eternity Bioscience/TG Therapeutics | Phase 1 | Healthy Volunteer (NHL, CLL) | NCT04291846 | Encouraging safety profile. |
| CG-806 | CrystalGenomics, Aptose Biosciences | Phase 1 | CLL, SLL, NHL | NCT03893682 | No drug-related dose-limiting toxicities. |
| Interleukin-1 receptor-associated kinase (IRAK) inhibitors | |||||
| PF-06650833 | Pfizer | Phase 2 | RA | NCT02996500 | TEAE: Infections and infestations [75]. |
| CA-4948 | Curis Pharmaceuticals | Phase 1 | AML, MDS | NCT04278768 | Adverse events: amylase/lipase increased neutrophil count decreased, rash and rhabdomyolysis [76]. |
| Hematological malignancies | NCT03328078 | ||||
| R-835 | Rigel Pharmaceuticals | Phase 1 | Autoimmune and Inflammatory Diseases | Rigel initiates phase 1 clinical trial [77]. | |
| BAY-1834845 | Bayer | Phase 1 | Pelvic Inflammatory Disease | NCT03054402 | No update beyond phase 1 for Pelvic Inflammatory Disease |
| Inflammation | NCT03244462 | ||||
| Psoriasis | NCT03493269 | ||||
| BAY-1830839 | Bayer | Phase 1 | RA | NCT03540615 | |
| Health volunteers (For RA) | NCT03965728 | ||||
| Cellular Inhibitor of Apoptosis Proteins (c-IAP) inhibitors | |||||
| Birinapant (TL32711) | Jonsson Comprehensive Cancer Center, NCI | Phase 2 | High Grade Ovarian, Fallopian Tube, Primary Peritoneal Cancer | NCT02756130 | Birinapant and pembrolizumab combination had futile outcome in patients with MSS colorectal cancer [78]. |
| TetraLogic Pharmaceuticals | Phase 1 | Hepatitis B | NCT02288208 | ||
| APG-1387 (SM-1387) | Ascentage Pharma | Phase 2 | Advanced Solid Tumors | NCT04284488 |
|
| Phase 2 | Myelofibrosis | NCT04354727 | |||
| Phase 1 | Advanced Solid Tumors or Hematologic Malignancies | NCT03386526 | |||
| Ascentage Pharma, HealthQuest Pharma | Phase 1 | Chronic Hepatitis B | NCT03585322 | ||
| LCL-161 | Mayo Clinic | Phase 2 | RR Plasma Cell Myeloma | NCT01955434 |
|
| MD Anderson Cancer Center, NCI, Novartis | Phase 2 | Myelofibrosis | NCT02098161 | ||
| Novartis Pharmaceuticals | Phase 2 | Breast Cancer | NCT01617668 | ||
| Phase 2 | Small Cell Lung Cancer Ovarian Cancer | NCT02649673 | |||
| US Oncology Research Novartis, Delta Clinical Research, LLC | Phase 1 | Metastatic Pancreatic Cancer | NCT01934634 | ||
| Novartis Pharmaceuticals | Phase 1 | Neoplasms | NCT01968915 | ||
| Phase 1 | Solid Tumors | NCT01240655 | |||
| Phase 1 | Advanced Solid Tumors | NCT01098838 | |||
| Phase 1 | MM | NCT03111992 | |||
| ASTX660 | Astex Pharmaceuticals | Phase 1 | AML | NCT04155580 | Common TEAE: Anemia, increased lipase and lymphopenia [81]. |
| Debio 1143 (AT-406) | Debiopharm | Phase 1 | Advanced Solid Tumors and Lymphomas | NCT01078649 | TEAE: Mucositis, dysphagia and anemia [82]. |
| CUDC-427 | Curis Pharmaceuticals | Phase 1 | Lymphoma | NCT01908413 | Few patients discontinued and TEAE includes pruritus and fatigue [83]. |
2.2.2. Interleukin-1 Receptor-associated Kinase (IRAK)
2.2.3. Phosphatidyl Inositol-3 Kinases (PI3K)/AKT
2.2.4. Cellular Inhibitor of Apoptosis Proteins (c-IAP)
3. Inhibition on IKK Complex in NF-κB Pathway
3.1. IKKα and IKKβ Inhibitors
3.2. Inhibitor of Nuclear Factor Kappa-B Kinase Subunit Gamma (IKKγ) Inhibitors
3.3. IKKε and Tank Binding Kinase 1 (TBK1) Inhibitors
| Drug | Originator/ Developer | Stage | Indication | Trail No. | Other Information |
|---|---|---|---|---|---|
| IKK α/β Inhibitors | |||||
| CHS-828 | Leo Pharma | Discontinued at phase 2 | Solid tumor | NCT00003979 |
|
| IMD-1041 (Pro-drug of IMD-0354) | Institute of Medicinal Molecular Design | Phase 2 | COPD | NCT00883584 |
|
| NA | Age-related macular degeneration, DM, Glaucoma, PF | NA | |||
| SAR-113945 | Sanofi | Discontinued at Phase 2 | OA | NCT01598415 |
|
| Discontinued at Phase 1 | OA | NCT01113333, NCT01463488, NCT01511549 | |||
| MLN-0415 | Millennium Pharmaceuticals | Discontinued at Phase 1 | Arthritis, Inflammation, MS | NA | Unfavorable safety profile in Phase 1. |
| VGX-1027 | VGX Pharmaceuticals | Phase 1 | Healthy subjects (RA) | NCT00627120 | No development reported for RA or other diseases. |
| Phase 1 | Healthy subjects (RA) | NCT00760396 | |||
| Teglarinad Chloride (EB-1627; GMX1777) | Leo Pharma | Phase 1 | Malignant melanoma | NCT00724841, | Multi dose study conducted with combination of Temozolomide. |
| Phase 1 | Lymphoma, Solid tumors | NCT00457574 | Single therapy was performed. | ||
| AS-602868 | Merck | Discontinued at Phase 1 | Hematological malignancies | NA | Also, inhibit FLT3. |
3.4. NF-κB Inducing Kinase (NIK) Inhibitors
4. Molecules that Inhibit Ubiquitin-Proteasome System (UPS)
4.1. Proteasome Inhibitors
| Drug | Originator/Developer | Stage | Indication | Trail No. | Purpose/Other Information |
|---|---|---|---|---|---|
| Proteasome Inhibitors | |||||
| Disulfiram | National Institute for Health and Welfare, Finland | Launched 1951 | Alcohol dependence | NCT00435435 |
|
| Sahlgrenska University Hospital, Sweden | Phase 3 | Recurrent Glioblastoma | NCT02678975 | ||
| Phase 3 | Lung Cancer | NCT00312819 | |||
| University of Utah | Phase 2 | refractory disseminated malignant melanoma | NCT02101008 | ||
| National Cancer Institute (NCI), Slovakia | Phase 2 | HER2 negative breast cancer | NCT04265274 | ||
| NCI, Slovakia | Phase 2 | Germ Cell Tumor | NCT03950830 | ||
| Mayo Clinic; NCI | Phase 1 | Metastatic Pancreatic Cancer | NCT02671890 | ||
| Bortezomib | Millennium/Takeda Pharmaceuticals | Launched 2003 | MCL, MM, WM | NCT00722137, NCT00257114, NCT02844322 | |
| The Rogosin Institute | Phase 4 | Chronic Kidney Disease and IgA Nephropathy | NCT01103778 | ||
| Melanoma Institute Australia | Phase 4 | Melanoma | NCT02645149 | ||
| Janssen Research & Development, LLC | Phase 3 | Amyloidosis | NCT03201965 | ||
| University Hospital Southampton NHS Foundation Trust; Janssen-Cilag Ltd. | Phase 3 | DLBCL | NCT01324596 | ||
| Millennium/Takeda Pharmaceuticals | Phase 3 | Relapsed or Refractory B-cell NHL | NCT00312845 | ||
| European Organisation for Research and Treatment of Cancer (EORTC) | Phase 3 | refractory or recurrent cutaneous TCL | NCT01386398 | ||
| Zhengang Yuan, Eastern Hepatobiliary Surgery Hospital, China | Phase 3 | Intrahepatic Cholangiocarcinoma | NCT03345303 | ||
| University Hospital Heidelberg | Phase 2 | AML | NCT04173585 | ||
| Millennium Pharmaceuticals | Phase 2 | NSCLC | NCT01833143 | ||
| Tianjin Medical University General Hospital | Phase 2 | Neuromyelitis Optica Spectrum Disorder | NCT02893111 | ||
| Northwestern University National Heart, Lung, and Blood Institute (NHLBI) | Phase 2 | PF | NCT02370693 | ||
| Sidney Kimmel Cancer Center at Thomas Jefferson University | Phase 2 | GVHD | NCT00408928 | ||
| Ixazomib | Millennium/Takeda Pharmaceuticals | Launched at 2015 | MM | NCT03173092 |
|
| Millennium/Takeda Pharmaceuticals | Phase 3 | Relapsed or Refractory Systemic Light Chain Amyloidosis | NCT01659658 | ||
| Takeda Pharmaceuticals | Phase 2 | Immune Thrombocytopenia and Autoimmune Hemolytic Anemia | NCT03965624 | ||
| Millennium/Takeda Pharmaceuticals | Phase 2 | Myeloid and Lymphoid Hematologic Malignancy | NCT03082677 | ||
| Takeda Pharmaceuticals | Phase 2 | MCL | NCT03616782 | ||
| Millennium Pharmaceuticals | Phase 2 | Kidney Diseases andEnd stage Renal Disease | NCT03213158 | ||
| Millennium Pharmaceuticals/NCI | Phase 2 | B-cell NHL | NCT02339922 | ||
| Carfilzomib (Kyprolis) | Proteolix/Onyx Pharmaceuticals, AbbVie, Genentech & others, Amgen | Launched at 2012 | MM | NCT03934684 |
|
| Amgen, Janssen, LP | Phase 3 | RRMM | NCT02412878 | ||
| Onyx Therapeutics, Inc | Phase 2 | MCL | NCT02042950 | ||
| SCRI Development Innovations, LLC, Amgen | Phase 2 | Neuroendocrine Cancer | NCT02318784 | ||
| Onyx Therapeutics, Inc. | Phase 2 | Refractory Renal Cell Carcinoma | NCT01775930 | ||
| Amgen | Phase 2 | Metastatic Castration-resistant Prostate Cancer | NCT02047253 | ||
| Fred Hutchinson Cancer Research Center, NCI | Phase 2 | Chronic GVHD | NCT02491359 | ||
| M.D. Anderson Cancer Center; Amgen | Phase 1 | MCL, TCL, DLBCL | NCT01926665 | ||
| Marizomib | Nereus Pharmaceuticals/Celgene | Phase 3 | Glioblastoma | NCT03345095 |
|
| Celgene | Phase 2 | MM | NCT00461045 | ||
| NCI | Phase 2 | Anaplastic Ependymoma | NCT03727841 | ||
| Celgene | Phase 1 | NSCLC, Pancreatic Cancer, Melanoma, Lymphoma, MM | NCT00667082 | ||
| Oprozomib | Onyx Pharmaceuticals | Phase 1/2 | R and/or R MM | NCT01832727 |
|
| Amgen | Phase 1/2 | Advanced HCC | NCT02227914 | ||
| Amgen | Phase 1/2 | MM, WM | NCT01416428 | ||
| Amgen | Phase 1 | Solid Tumors | NCT01129349 | ||
| Deubiquitination (DUB) inhibitors | |||||
| VLX1570 | Vivolux, Mayo Clinic | Phase 1/2 | MM | NCT02372240 | Death of 2 patients receiving two doses at 1.2 mg/kg due to fatal pulmonary toxicity [152]. |
| NEDD8 activating enzyme (NAE) inhibitors | |||||
| Pevonedistat | Millennium Pharmaceuticals | Phase 3 | AML | NCT03268954 |
|
| NCI | Phase 2 | Metastatic Cholangiocarcinoma, HCC | NCT04175912 | ||
| University of Michigan Rogel Cancer Center | Phase 2 | NSCLC | NCT03228186 | ||
| NCI | Phase 2 | Myeloproliferative Neoplasm | NCT03238248 | ||
| Millennium Pharmaceuticals | Phase 1 | Advanced Solid Tumors and Neoplasms | NCT03057366 | ||
| TAS-4464 | Taiho Oncology | Phase 1/2; Terminated | MM, HNL | NCT02978235 | As of Dec 2019, discontinued for most indications. |
| TAK-243 | Takeda Pharmaceuticals | Phase 1 | Myelodysplastic Syndrome, AML, CMML | NCT03816319 | Phase 1 trial for AML, Myelodysplastic syndrome and CMML initiated in May 2019. |
| Millennium Pharmaceuticals | Phase 1; terminated | Advanced Malignant Solid Tumors | NCT02045095 | ||
4.2. Deubiquitination (DUB) Inhibitors
4.3. NAE (NEDD8 Activating Enzyme) Inhibitors
5. Molecules Inhibiting Nuclear Translocation, DNA Binding and Transcriptional Activation of NF-κB
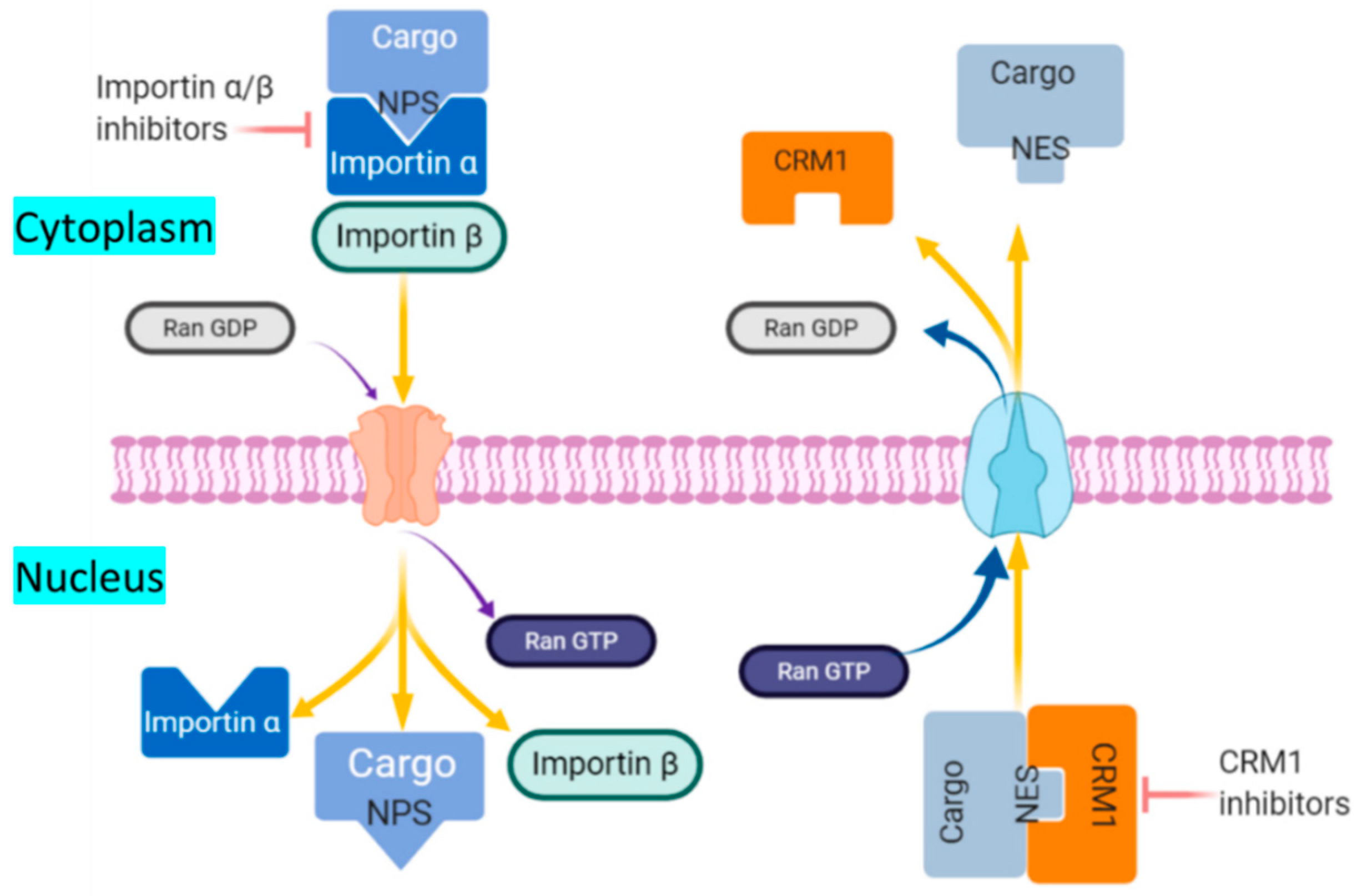
5.1. Nuclear Export Inhibitors
| Drug | Originator/Developer | Stage | Indication | Trail No | Purpose/Other Information |
|---|---|---|---|---|---|
| Selective inhibitors of nuclear export (SINE) | |||||
| Selinexor | Karyopharm Therapeutics | Launched at 2019 | RRMM | NCT03110562 |
|
| Phase 3 | Endometrial Cancer | NCT03555422 | |||
| Phase 2 | Thymoma, Advanced thymic epithelial tumor | NCT03193437 | |||
| Phase 2 | Coronavirus Infection | NCT04355676 | |||
| Phase 2 | Coronavirus Infection | NCT04349098 | |||
| Phase 2 | Myelofibrosis | NCT03627403 | |||
| Phase 2 | AML (Relapsed/Refractory) | NCT02249091 | |||
| Phase 2 | Ovarian, Endometrial and Cervical Carcinoma, Breast Cancer | NCT02025985 | |||
| Preregistered/Phase 2 | DLBCL | NCT02227251; NCT03992339 | |||
| Phase 1/2 | Diabetic Foot Ulcers | NCT02367690 | |||
| Phase 1/2 | NSCLC | NCT03095612 | |||
| Phase 1 | Colorectal Neoplasm | NCT02384850 | |||
| Phase 1 | BCL | NCT02741388 | |||
| Phase 1 | Solid Tumors | NCT02078349 | |||
| Phase 1 | Soft Tissue Sarcoma | NCT03042819 | |||
| NCI | Phase 1 | Gliosarcoma, Newly Diagnosed Glioblastoma | NCT04216329 | ||
| Phase 1 | Recurrent or Refractory Solid Tumors or High-Grade Gliomas | NCT02323880 | |||
| Eltanexor | Karyopharm Therapeutics | Phase 2 | RRMM, mCRC, mCRPC, HR-MDS | NCT02649790 | TEAE: Thrombocytopenia, neutropenia, anemia, leukopenia, and hyponatremia [172]. |
| Verdinexor | Karyopharm Therapeutics | Phase 1 | Healthy adults | NCT02431364 | Conditional approval by US-FDA to treat canine lymphoma [173]. |
| Felezonexor (SL-801) | Stemline Therapeutics | Phase 1 | Solid Tumors | NCT02667873 | Felezonexor showed partial response in the interim results for phase 1 study in microsatellite stable colorectal cancer. |
| Histone deacetylase inhibitors | |||||
| Vorinostat | Merck | Marketed | Cutaneous TCL | NCT00875056, NCT00091559 |
|
| Romidepsin | Celgene | Marketed | Cutaneous and Peripheral TCL | NCT00426764 | TEAE; nausea, fatigue, anemia, thrombocytopenia, ECG T-wave changes, neutropenia, and lymphopenia [175]. |
| Belinostat | Onxeo | Marketed | Peripheral TCL | NCT00274651 | TEAE: Nausea, fatigue, pyrexia, anemia, and vomiting [176]. |
| Panobinostat | Novartis/Secura Bio | Marketed | MM | NCT01023308 | TEAE: Thrombocytopenia, neutropenia, lymphopenia, anemia, diarrhea, fatigue, and nausea [177]. |
| Tucidinostat | HUYA Bioscience | Marketed | Peripheral TCL (different stages for other cancers) | NCT04040491 | TEAE: Thrombocytopenia, neutropenia, fatigue, leucopenia, vomiting, diarrhea, nausea, and anemia [178]. |
| DNA acetylation inhibitors | |||||
| Azacitidine | Pfizer/Celgene | Marketed | AML; CML; MDS | NCT03416179, NCT03416179, NCT01201811 |
|
| AbbVie/ | Marketed | MDS | NCT04401748 | ||
| Decitabine | Janssen-Cilag/Otsuka Pharmaceutical | Marketed | AML, MDS | NCT02472145, NCT01751867 | TEAE: Myelosuppression (neutropenia, thrombocytopenia, and anemia), Febrile neutropenia, pyrexia, fatigue, nausea, cough, petechiae, diarrhea, and constipation [180]. |
5.2. Nuclear Import Inhibitors
5.3. Inhibition of p65 Transactivation and DNA Binding
5.4. Inhibition of Post-Translational Modifications
6. Molecules that Can or May be Repurposed as NF-κB Pathway Inhibitors
7. Conclusions and Way Forward
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ADR | Adverse drug reaction |
| AIH | Autoimmune hepatitis |
| AKT | Protein Kinase B |
| ALT | Alanine aminotransferase |
| AML | Acute myeloid leukemia |
| ATM | Ataxia telangiectasia mutated |
| BAFF | B-cell activating factor |
| BAFFR | B-cell activating factor receptor |
| BCL | B-cell leukemia |
| BCR | B-cell receptors |
| BTK | Bruton’s tyrosine kinase |
| c-IAP | Cellular Inhibitor of Apoptosis Proteins |
| CLL | Chronic lymphocytic leukemia |
| CML | Chronic myeloid leukemia |
| CMML | Chronic myelomonocytic leukemia |
| COPD | Chronic obstructive pulmonary disease |
| DLBCL | Diffuse large B-cell lymphoma |
| DLT | Dose limiting toxicity |
| DMARD | Disease-modifying anti-rheumatic drug |
| DUB | Deubiquitination |
| FLT3 | FMS-like tyrosine kinase 3 |
| Fn14 | Fibroblast growth factor-inducible 14 |
| GVHD | Graft versus host disease |
| HCC | Hepatocellular carcinoma |
| IKK | IκB kinase |
| IκB | Inhibitor of κB |
| IKKγ | Inhibitor of nuclear factor kappa-B kinase subunit gamma |
| IL1R | Interleukin1 receptor |
| IRAK | Interleukin-1 Receptor-Associated Kinase |
| IRF | Interferon regulatory factor |
| JAK | Janus Kinase |
| JNK | c-jun N-terminal kinase |
| LT | Lymphotoxin |
| mAbs | Monoclonal antibodies |
| MAPK | Mitogen-activated protein kinase |
| MCL | Mantle cell lymphoma |
| MM | Multiple myeloma |
| MS | Multiple sclerosis |
| MTD | Maximum tolerated dose |
| MZL | Marginal zone lymphoma |
| NAE | NEDD8 activating enzyme |
| NASH | Non-alcoholic steatohepatitis |
| NBD | NEMO Binding Domain |
| NEMO | NF-κB essential modulator |
| NES | Nuclear export signal |
| NF-κB | Nuclear factor kappa B |
| NHL | Non-Hodgkin lymphoma |
| NIH | National Institutes of Health |
| NIK | NF-κB inducing kinase |
| NLS | Nuclear localization signals |
| NPCs | Nuclear pore complexes |
| NSCLC | Non-small cell lung cancer |
| OA | Osteoarthritis |
| ORR | Overall Response Rate |
| PI3K | Phosphatidyl Inositol-3 kinases |
| PK | Pharmacokinetic |
| PDn | Pharmacodynamic |
| RP2D | Recommended phase 2 dose |
| RA | Rheumatoid arthritis |
| RANK | Receptor activator of NF-κB |
| RANKL | Receptor activator of NF-κB ligand |
| RRCLL | Relapsed and refractory chronic lymphocytic leukemia |
| RRWM | Relapsed and refractory Multiple myeloma |
| RA | Rheumatoid arthritis |
| RANK | Receptor activator of nuclear factor κ B |
| SAR | Seasonal allergic rhinitis |
| siRNA | Small interfering RNA |
| SLL | Small lymphocytic lymphoma |
| SLE | Systemic lupus erythematosus |
| SMAC | Second mitochondria-derived activator of caspase |
| STING | Stimulator of interferon genes |
| TAK1 | Transforming growth factor beta-activated kinase 1 |
| TBK1 | IKKe and Tank binding kinase 1 |
| TCL | T-cell lymphoma |
| TCR | T-cell receptors |
| TEAE | Treatment-emergent adverse effects |
| TLR7 | Toll-like receptor 7 |
| TNF-α | Tumor necrosis factor alfa |
| TNFR | TNF-α receptor |
| TNFRSF | TNF receptor superfamily |
| TRAF | TNF receptor-associated factor |
| TWEAK | Tumor necrosis factor-like weak inducer of apoptosis |
| UPS | Ubiquitin-proteasome system |
| WM | Waldenstrom macroglobulinemia. |
| XIAP | X-linked inhibitor of apoptosis protein |
References
- Sen, R.; Baltimore, D. Multiple nuclear factors interact with the immunoglobulin enhancer sequences. Cell 1986, 46, 705–716. [Google Scholar] [CrossRef]
- Ang, H.L.; Tergaonkar, V. Notch and NFkappaB signaling pathways: Do they collaborate in normal vertebrate brain development and function? Bioessays 2007, 29, 1039–1047. [Google Scholar] [CrossRef]
- Correa, R.G.; Matsui, T.; Tergaonkar, V.; Rodriguez-Esteban, C.; Izpisua-Belmonte, J.C.; Verma, I.M. Zebrafish IkappaB kinase 1 negatively regulates NF-kappaB activity. Curr. Biol. 2005, 15, 1291–1295. [Google Scholar] [CrossRef] [Green Version]
- Garbati, M.R.; Gilmore, T.D. Inhibition of NF-kB signaling as a strategy in disease therapy. In NF-kB in Health and Disease. Current Topics in Microbiology and Immunology; Springer: Berlin, Heidelberg, 2011; Volume 349, pp. 245–263. [Google Scholar]
- Chew, C.L.; Conos, S.A.; Unal, B.; Tergaonkar, V. Noncoding RNAs: Master regulators of inflammatory signaling. Trends Mol. Med. 2018, 24, 66–84. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Cheng, H.S.; Chng, W.J.; Tergaonkar, V. Activation of mutant TERT promoter by RAS-ERK signaling is a key step in malignant progression of BRAF-mutant human melanomas. Proc. Natl. Acad. Sci. USA 2016, 113, 14402–14407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozturk, M.B.; Li, Y.; Tergaonkar, V. Current insights to regulation and role of telomerase in human diseases. Antioxidants 2017, 6, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puar, Y.R.; Shanmugam, M.K.; Fan, L.; Arfuso, F.; Sethi, G.; Tergaonkar, V. Evidence for the involvement of the master transcription factor NF-kappaB in cancer initiation and progression. Biomedicines 2018, 6, 82. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Li, Y.; Bharath, S.R.; Ozturk, M.B.; Bowler, M.W.; Loo, B.Z.L.; Tergaonkar, V.; Song, H. Structural basis for reactivating the mutant TERT promoter by cooperative binding of p52 and ETS1. Nat. Commun. 2018, 9, 3183. [Google Scholar] [CrossRef]
- Cildir, G.; Pant, H.; Lopez, A.F.; Tergaonkar, V. The transcriptional program, functional heterogeneity, and clinical targeting of mast cells. J. Exp. Med. 2017, 214, 2491–2506. [Google Scholar] [CrossRef]
- Khattar, E.; Maung, K.Z.Y.; Chew, C.L.; Ghosh, A.; Mok, M.M.H.; Lee, P.; Zhang, J.; Chor, W.H.J.; Cildir, G.; Wang, C.Q.; et al. Rap1 regulates hematopoietic stem cell survival and affects oncogenesis and response to chemotherapy. Nat. Commun. 2019, 10, 5349. [Google Scholar] [CrossRef] [Green Version]
- Herscovitch, M.; Gilmore, T.D. Inhibitors of NF-kB signaling: 785 and counting. Oncogene 2006, 25, 6887–6899. [Google Scholar]
- Zhang, Q.; Lenardo, M.J.; Baltimore, D. 30 Years of NF-kB: A blossoming of relevance to human pathobiology. Cell 2017, 168, 37–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayden, M.S.; Ghosh, S. NF-κB, the first quarter-century: Remarkable progress and outstanding questions. Genes Dev. 2012, 26, 203–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, S.C.; Ley, S.C. New insights into NF-κB regulation and function. Trends Immunol. 2008, 29, 469–478. [Google Scholar] [PubMed] [Green Version]
- Hayden, M.S.; Ghosh, S. Shared principles in NF-κB signaling. Cell 2008, 132, 344–362. [Google Scholar] [CrossRef] [Green Version]
- Vallabhapurapu, S.; Karin, M. Regulation and function of NF-κB transcription factors in the immune system. Annu. Rev. Immunol. 2009, 27, 693–733. [Google Scholar] [CrossRef]
- Cildir, G.; Low, K.C.; Tergaonkar, V. Noncanonical NF-kB Signaling in Health and Disease. Trends Mol. Med. 2016, 22, 414–429. [Google Scholar] [CrossRef]
- Sun, S.C. The noncanonical NF-κB pathway. Immunol. Rev. 2012, 246, 125–140. [Google Scholar] [CrossRef] [Green Version]
- Xiao, G.; Harhaj, E.W.; Sun, S.C. NF-κB-inducing kinase regulates the processing of NF-κB2 p100. Mol. Cell 2001, 7, 401–409. [Google Scholar] [CrossRef]
- Herrington, F.D.; Carmody, R.J.; Goodyear, C.S. Modulation of NF-κB signaling as a therapeutic target in autoimmunity. J. Biomol. Screen. 2016, 21, 223–242. [Google Scholar] [CrossRef] [Green Version]
- Fukuoka, M.; Yoshioka, K.; Hohjoh, H. NF-κB activation is an early event of changes in gene regulation for acquiring drug resistance in human adenocarcinoma PC-9 cells. PLoS ONE 2018, 13, e0201796. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Fang, Y.; Sun, W.; Xu, Z.; Zhang, Y.; Wei, X.; Ding, X.; Xu, Y. Endocrinotherapy resistance of prostate and breast cancer: Importance of the NF-κB pathway. Int. J. Oncol. 2020, 56, 1064–1074. [Google Scholar] [CrossRef]
- Nisr, R.B.; Shah, D.S.; Ganley, I.G.; Hundal, H.S. Proinflammatory NFkB signalling promotes mitochondrial dysfunction in skeletal muscle in response to cellular fuel overloading. Cell. Mol. Life Sci. 2019, 76, 4887–4904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennett, J.; Capece, D.; Begalli, F.; Verzella, D.; D’Andrea, D.; Tornatore, L.; Franzoso, G. NF-κB in the crosshairs: Rethinking an old riddle. Int. J. Biochem. Cell Biol. 2018, 95, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Kaltschmidt, C.; Banz-Jansen, C.; Benhidjeb, T.; Beshay, M.; Förster, C.; Greiner, J.; Hamelmann, E.; Jorch, N.; Mertzlufft, F.; Pfitzenmaier, F.; et al. A role for NF-κB in organ specific cancer and cancer stem cells. Cancers (Basel) 2019, 11, 655. [Google Scholar]
- Catrysse, L.; van Loo, G. Inflammation and the metabolic syndrome: The tissue-specific functions of NF-κB. Trends Cell Biol. 2017, 27, 417–429. [Google Scholar]
- Gupta, S.C.; Sundaram, C.; Reuter, S.; Aggarwal, B.B. Inhibiting NF-κB activation by small molecules as a therapeutic strategy. Biochim. Biophys. Acta 2010, 1799, 775–787. [Google Scholar] [CrossRef] [Green Version]
- Gurevich, E.V.; Gurevich, V.V. Therapeutic potential of small molecules and engineered proteins. Handb. Exp. Pharmacol. 2014, 219, 1–12. [Google Scholar]
- Nwibo, D.D.; Levi, C.A.; Nwibo, M.I. Small molecule drugs; down but not out: A future for medical research and therapeutics. IOSR JDMS 2015, 14, 70–77. [Google Scholar]
- Talevi, A. Multi-target pharmacology: Possibilities and limitations of the “skeleton key approach” from a medicinal chemist perspective. Front. Pharmacol. 2015, 6, 205. [Google Scholar] [CrossRef] [Green Version]
- Buvailo, A. Will Biologics Surpass Small Molecules in the Pharma Race? Available online: https://www.biopharmatrend.com/post/67-will-small-molecules-sustain-pharmaceutical-race-with-biologics/ (accessed on 13 May 2020).
- Begalli, F.; Bennett, J.; Capece, D.; Verzella, D.; D’Andrea, D.; Tornatore, L.; Franzoso, G. Unlocking the NF-κB Conundrum: Embracing Complexity to Achieve Specificity. Biomedicines 2017, 5, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tracey, D.; Klareskog, L.; Sasso, E.H.; Salfeld, J.G.; Tak, P.P. Tumor necrosis factor antagonist mechanisms of action: A comprehensive review. Pharmacol. Ther. 2008, 117, 244–279. [Google Scholar] [CrossRef] [PubMed]
- Ono, T.; Hayashi, M.; Sasaki, F.; Nakashima, T. RANKL biology: Bone metabolism, the immune system, and beyond. Inflamm. Regen. 2020, 40, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Weyden, C.A.; Pileri, S.A.; Feldman, A.L.; Whisstock, J.; Prince, H.M. Understanding CD30 biology and therapeutic targeting: A historical perspective providing insight into future directions. Blood Cancer J. 2017, 7, e603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Connell, J.; Porter, J.; Kroeplien, B.; Norman, T.; Rapecki, S.; Davis, R.; McMillan, D.; Arakaki, T.; Burgin, A.; III, D.F.; et al. Small molecules that inhibit TNF signalling by stabilising an asymmetric form of the trimer. Nat. Commun. 2019, 10, 1–12. [Google Scholar]
- Anwar, M.A.; Shah, M.; Kim, J.; Choi, S. Recent clinical trends in Toll-like receptor. Med. Res. Rev. 2019, 39, 1053–1090. [Google Scholar] [CrossRef] [Green Version]
- Idera Announces FDA Prphan Dryg Designation for IMO-8400 for the Treatment of Diffuse Large B-Cell Lymphoma. Available online: http://ir.iderapharma.com/news-releases/news-release-details/idera-announces-fda-orphan-drug-designation-imo-8400-treatment (accessed on 5 May 2020).
- FORM 10-K, Annual Report: Fiscal Year Ended. 31 December 2016. Available online: https://www.sec.gov/Archives/edgar/data/861838/000155837017001845/idra-20161231x10k.htm#Item16Form10KSummary_498298 (accessed on 5 May 2020).
- Pharmaceuticals, T. Clinical Trial Arena. Available online: https://www.clinicaltrialsarena.com/comment/taiwanjs-jkb-121-falls-victim-unexpected-placebo-response-nash-patients/ (accessed on 12 May 2020).
- JKB-122. Clinical Trial Arena. Available online: https://www.clinicaltrialsarena.com/news/taiwanj-reports-positive-data/ (accessed on 22 May 2020).
- Novack, D.V.; Yin, L.; Hagen-Stapleton, A.; Schreiber, R.D.; Goeddel, D.V.; Ross, F.P.; Teitelbaum, S.L. The IkappaB function of NF-kappaB2 p100 controls stimulated osteoclastogenesis. J. Exp. Med. 2003, 198, 771–781. [Google Scholar] [CrossRef]
- FDA Approves Amgens XGEVA Denosumab for the Treatment of Hypercalcemia of Malignancy Refractory to Bisposphonate Therapy. Available online: https://www.amgen.com/media/news-releases/2014/12/fda-approves-amgens-xgeva-denosumab-for-the-treatment-of-hypercalcemia-of-malignancy-refractory-to-bisphosphonate-therapy/ (accessed on 13 May 2020).
- FDA Approves XGEVA Denosumab for the Prevention of Skeletal Relevated Events in Patients with Multiple Myeloma. Available online: https://www.amgen.com/media/news-releases/2018/01/fda-approves-xgeva-denosumab-for-the-prevention-of-skeletalrelated-events-in-patients-with-multiple-myeloma/ (accessed on 13 May 2020).
- FDA Approves PROLIA Denosumab for Glucocorticoid Induced Osteoporosis. Available online: https://www.amgen.com/media/news-releases/2018/05/fda-approves-prolia-denosumab-for-glucocorticoidinduced-osteoporosis/ (accessed on 13 May 2020).
- Tyan, A.; Patel, S.P.; Block, S.; Hughes, T.; McCowen, K.C. Rebound vertebral fractures in a patient with lung cancer after oncology-dose Denosumab discontinuation: A cautionary tale. MCP IQ O 2019, 3, 235–237. [Google Scholar] [CrossRef] [Green Version]
- Jiang, M.; Peng, L.; Yang, K.; Wang, T.; Yan, X.; Jiang, T.; Xu, J.; Qi, J.; Zhou, H.; Qian, N.; et al. Development of small-molecules targeting Receptor Activator of Nuclear Factor-κB Ligand (RANKL)—Receptor Activator of Nuclear Factor-κB (RANK) protein–protein interaction by structure-based virtual screening and hit optimization. J. Med. Chem. 2019, 62, 5370–5381. [Google Scholar] [CrossRef]
- VBL Therapeutics Reports Topline Results From Phase 2 Studies of VB-201 in Psoriasis and Ulcerative Colitis. Available online: https://www.vblrx.com/vbl-therapeutics-reports-topline-results-from-phase-2-studies-of-vb-201-in-psoriasis-and-ulcerative-colitis/ (accessed on 1 July 2020).
- Kaptein, A.; Bruin, G.D.; Hoek, M.E.-V.; Kar, B.V.D.; Jong, A.D.; Gulrajani, M.; Demont, D.; Covey, T.; Mittag, D.; Barf, T. Potency and selectivity of BTK inhibitors in clinical development for B-cell malignancies. Blood 2018, 132, 1871. [Google Scholar]
- Owen, C.; Berinstein, N.L.; Christofides, A.; Sehn, L.H. Review of Bruton tyrosine kinase inhibitors for the treatment of relapsed or refractory mantle cell lymphoma. Curr. Oncol. 2019, 26, e233–e240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Astor, L. FDA Approves Acalabrutinib for CLL/SLL. Available online: https://www.targetedonc.com/view/fda-approves-acalabrutinib-for-cllsll (accessed on 5 May 2020).
- FDA Grants Accelerated Approval to Zanubrutinib for Mantle Cell Lymphoma. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-zanubrutinib-mantle-cell-lymphoma (accessed on 5 May 2020).
- Smith, J. Orelabrutinib could be ‘Preferred’ BTK Inhibitor for MCL. Available online: https://www.mdedge.com/hematology-oncology/article/214174/mantle-cell-lymphoma/orelabrutinib-could-be-preferred-btk (accessed on 5 May 2020).
- Wankhade, S. Ono Pharma Seeks Japanese Approval for Tirabrutinib. Available online: https://www.biospectrumasia.com/news/50/14985/ono-pharma-seeks-japanese-approval-for-tirabrutinib-.html (accessed on 5 May 2020).
- Kutsch, N.; Pallasch, C.; Decker, T.; Hebart, H.; Chow, K.U.; Graeven, U.; Kisro, J.; Kroeber, A.; Tausch, E.; Wendtner, C.M.; et al. A prospective, open-label, multicenter, phase 2 trial to evaluate the safety and efficacy of the combination of Tirabrutinib (ONO/GS-4059) and Idelalisib with and without Obinutuzumab in patients with relapsed/refractory chronic lymphocytic leukemia (CLL). Blood 2019, 134, 3047. [Google Scholar]
- Rule, S.A.; Cartron, G.; Fegan, C.; Morschhauser, F.; Han, L.; Mitra, S.; Salles, G.; Dyer, M.J.S. Long-term follow-up of patients with mantle cell lymphoma (MCL) treated with the selective Bruton’s tyrosine kinase inhibitor tirabrutinib (GS/ONO-4059). Leukemia Vol. 2020, 34, 1458–1461. [Google Scholar] [CrossRef] [Green Version]
- Sanofi Brain-Penetrant BTK Inhibitor Meets Primary Endpoint of Phase 2 Trial in Relapsing Multiple Sclerosis. Available online: https://www.sanofi.com/en/media-room/press-releases/2020/2020-02-06-07-00-00 (accessed on 6 May 2020).
- Chee, C.E.; Krishnamurthi, S.; Nock, C.J.; Meropol, N.J.; Gibbons, J.; Fu, P.; Bokar, J.; Teston, L.; O’Brien, T.; Gudena, V.; et al. Phase II study of Dasatinib (BMS-354825) in patients with metastatic adenocarcinoma of the pancreas. Oncologist 2013, 18, 1091–1092. [Google Scholar] [CrossRef] [Green Version]
- Tsou, P.-S.; Haak, A.J.; Khanna, D.; Neubig, R.R. Cellular Mechanisms of Tissue Fibrosis. 8. Current and future drug targets in fibrosis: Focus on Rho GTPase-regulated gene transcription. Am. J. Physiol. Cell Physiol. 2014, 307, C2–C13. [Google Scholar]
- Acalabrutinib. In LiverTox: Clinical and Research Information on Drug-Induced Liver Injury; Bethesda (MD): Rockville, MD, USA, 2012.
- Levy, I.; Polliack, A.; Tadmor, T. Five Ibrutinib-associated side effects that all clinicians should be aware of. Acta Haematol. 2019, 141, 254–255. [Google Scholar] [CrossRef]
- Conchon, M.; Freitas, C.M.; Rego, M.A.; Braga Junior, J.W. Dasatinib - clinical trials and management of adverse events in imatinib resistant/intolerant chronic myeloid leukemia. Rev. Bras. Hematol. Hemoter. 2011, 33, 131–139. [Google Scholar] [CrossRef] [Green Version]
- Nagane, M.; Narita, Y.; Mishima, K.; Terui, Y.; Arakawa, Y.; Yonezawa, H.; Asai, K.; Fukuhara, N.; Sugiyama, K.; Shinojima, N.; et al. Phase 1/2 study of Tirabrutinib (ONO/GS-4059), a next-generation Bruton’s Tyrosine Kinase (BTK) inhibitor, monotherapy in patients with relapsed/refractory Primary Central Nervous System Lymphoma (PCNSL). Blood 2019, 134, 1586. [Google Scholar] [CrossRef]
- New late-breaking data at EAN indicate Evobrutinib is the first BTK inhibitor to report efficacy and safety in MS over 108 Weeks. Available online: https://www.prnewswire.com/news-releases/new-late-breaking-data-at-ean-indicate-evobrutinib-is-the-first-btk-inhibitor-to-report-efficacy-and-safety-in-ms-over-108-weeks-301064619.html (accessed on 2 July 2020).
- Song, Y.; Song, Y.; Liu, L.; Zhang, M.; Li, Z.; Ji, C.; Xu, W.; Liu, T.; Xu, B.; Wang, X.; et al. Safety and efficacy of Orelabrutinib monotherapy in Chinese patients with relapsed or refractory mantle celllLymphoma: A multicenter, open-label, phase II study. Blood 2019, 134, 755. [Google Scholar] [CrossRef]
- Sanofi’s BTK inhibitor SAR442168 significantly reduced disease activity in Phase 2b trial in relapsing multiple sclerosis. Available online: https://www.trialsitenews.com/sanofis-btk-inhibitor-sar442168-significantly-reduced-disease-activity-in-phase-2b-trial-in-relapsing-multiple-sclerosis/ (accessed on 2 July 2020).
- Catlett, I.M.; Nowak, M.; Kundu, S.; Zheng, N.; Liu, A.; He, B.; Girgis, I.G.; Grasela, D.M. Safety, pharmacokinetics and pharmacodynamics of branebrutinib (BMS-986195), a covalent, irreversible inhibitor of Bruton’s tyrosine kinase: Randomised phase I, placebo-controlled trial in healthy participants. Br. J. Clin. Pharmacol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Kumagai, Y.; Tanaka, Y.; Murata, M.; Takeuchi, T. FRI0156 a phase 1, single and multiple ascending dose study of TAS5315—A novel highly selective inhibitor of Bruton’s tyrosine kinase—in healthy male volunteers. Ann. Rheum. Dis. 2019, 78, 750. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.K.; Xing, J.; Catlett, I.M.; Adamczyk, R.; Griffies, A.; Liu, A.; Murthy, B.; Nowak, M. Safety, pharmacokinetics, and pharmacodynamics of BMS-986142, a novel reversible BTK inhibitor, in healthy participants. Eur. J. Clin. Pharmacol. 2017, 73, 689–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, S.B. Efficacy and safety of Fenebrutinib, a BTK inhibitor, compared to placebo in rheumatoid arthritis patients with active disease despite TNF inhibitor treatment: Randomized, double blind, phase 2 study. In Proceedings of the American College of Rheumatology, Atlanta, GA, USA, 10 November 2019. [Google Scholar]
- Elvidge, S. $690M deal in peril after Lilly ends mid-stage studies. Available online: https://www.biopharmadive.com/news/690m-deal-in-peril-after-lilly-ends-mid-stage-studies/517372/ (accessed on 2 July 2020).
- Jurczak, W.R.; Townsend, W.; Tucker, D.; Sarholz, B.; Scheele, J.; Gribben, J.; Zinzani, P.L. PDPhase I/II, first in human trial with M7583, a Bruton’s tyrosine kinase inhibitor (BTKi), in patients with B cell malignancies. In Proceedings of the European Society for Medical Oncology (ESMO) congress, Munich, Germany, 19–23 October 2018; pp. viii359–viii371. [Google Scholar]
- ArQule announces final phase 1 clinical data for its reversible BTK inhibitor, ARQ 531, at the American Society of Hematology. Available online: https://www.bloomberg.com/press-releases/2019-12-09/arqule-announces-final-phase-1-clinical-data-for-its-reversible-btk-inhibitor-arq-531-at-the-american-society-of-hematology (accessed on 2 July 2020).
- Danto, S.I.; Shojaee, N.; Singh, R.S.P.; Li, C.; Gilbert, S.A.; Manukyan, Z.; Kilty, I. Safety, tolerability, pharmacokinetics, and pharmacodynamics of PF-06650833, a selective interleukin-1 receptor-associated kinase 4 (IRAK4) inhibitor, in single and multiple ascending dose randomized phase 1 studies in healthy subjects. Arthritis Res. Ther. 2019, 21, 269–285. [Google Scholar] [PubMed] [Green Version]
- Younes, A.; Nowakowski, G.; Rosenthal, A.C.; Leslie, L.A.; Tun, H.W.; Lunning, M.A.; Isufi, I.; Martell, R.; Patel, K. Phase 1 dose-finding study investigating CA-4948, an IRAK4 kinase inhibitor, in patients with R/R NHL: Report of initial efficacy and updated safety information. Blood 2019, 134, 5327. [Google Scholar] [CrossRef]
- Rigel Initiates Phase 1 Clinical Trial of R835, an IRAK1/4 Inhibitor for Autoimmune and Inflammatory Diseases. Available online: https://www.prnewswire.com/news-releases/rigel-initiates-phase-1-clinical-trial-of-r835-an-irak14-inhibitor-for-autoimmune-and-inflammatory-diseases-300672042.html (accessed on 1 July 2020).
- Medivir, A.B. Futility analysis performed of the phase II combination study with Birinapant and Keytruda® in colorectal cancer patients. Available online: https://www.prnewswire.com/news-releases/futility-analysis-performed-of-the-phase-ii-combination-study-with-birinapant-and-keytruda-in-colorectal-cancer-patients-300975351.html (accessed on 6 May 2020).
- Rasco, D.W.; Li, Y.; Tang, Y.; Men, L.; Wang, H.; Ji, J.; Liang, Z.; Sun, J.; Amaya, A.; Huang, Y.; et al. A phase I study of a novel IAP inhibitor APG-1387 as a monotherapy or in combination with pembrolizumab in treatments of patients with advanced solid tumors. J. Clin. Oncol. 2019, 37, 3125. [Google Scholar] [CrossRef]
- Pemmaraju, N.; Carter, B.Z.; Kantarjian, H.M.; Cortes, J.E.; Bose, P.; Kadia, T.M.; Garcia-Manero, G.; Bueso-Ramos, C.E.; DiNardo, C.D.; Bledsoe, S.; et al. Final results of phase 2 clinical trial of LCL161, a novel oral SMAC mimetic/IAP antagonist, for patients with intermediate to high risk myelofibrosis. Blood 2019, 134, 555. [Google Scholar] [CrossRef]
- Mita, M.M.; LoRusso, P.M.; Papadopoulos, K.P.; Gordon, M.S.; Mita, A.C.; Ferraldeschi, R.; Keer, H.; Oganesian, A.; Su, X.Y.; Jueliger, S.; et al. A phase I study of ASTX660, an antagonist of inhibitors of apoptosis proteins, in adults with advanced cancers or lymphoma. Clin. Cancer Res. 2020, 26, 2819–2826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tucker, N. FDA grants Breakthrough Therapy Designation to Debio 1143 in front-line head and neck cancer. Available online: https://www.targetedonc.com/view/fda-grants-breakthrough-therapy-designation-to-debio-1143-in-frontline-head-and-neck-cancer (accessed on 2 July 2020).
- Tolcher, A.W.; Bendell, J.C.; Papadopoulos, K.P.; Burris, H.A.; Patnaik, A.; Fairbrother, W.J.; Wong, H.; Budha, N.; Darbonne, W.C.; Peale, F.; et al. A Phase I Dose-Escalation Study Evaluating the Safety Tolerability and Pharmacokinetics of CUDC-427, a Potent, Oral, Monovalent IAP Antagonist, in Patients with Refractory Solid Tumors. Clin. Cancer Res. 2016, 22, 4567–4573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhyasen, G.W.; Starczynowski, D.T. IRAK signalling in cancer. Br. J. Cancer 2015, 112, 232–237. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Wesche, H.; Stevens, T.; Walker, N.; Yeh, W.C. IRAK-4 inhibitors for inflammation. Curr. Top. Med. Chem. 2009, 9, 724–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singer, J.W.; Fleischman, A.; Al-Fayoumi, S.; Mascarenhas, O.J.; Yu, Q.; Agarwal, A. Inhibition of interleukin-1 receptor-associated kinase 1 (IRAK1) as a therapeutic strategy. Oncotarget 2018, 9, 33416–33439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Press Release on CU-4948 Phase 1 Study. Available online: http://investors.curis.com/2019-12-06-Curis-Provides-First-Ever-Demonstration-that-Targeting-IRAK4-in-Patients-with-Relapsed-Refractory-Non-Hodgkins-Lymphoma-Results-in-Anti-Cancer-Activity-in-Ongoing-Phase-1-Study (accessed on 5 May 2020).
- Manning, B.D.; Tok, A. AKT/PKB signaling: Navigating the network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozes, O.N.; Mayo, L.D.; Gustin, J.A.; Pfeffer, S.R.; Pfeffer, L.M.; Donner, D.B. NF-kappaB activation by tumour necrosis factor requires the Akt serine-threonine kinase. Nature 1999, 401, 82–85. [Google Scholar] [CrossRef] [PubMed]
- Reddy, S.A.; Huang, J.H.; Liao, W.S. Phosphatidylinositol 3-kinase in interleukin 1 signaling. Physical interaction with the interleukin 1 receptor and requirement in NFkappaB and AP-1 activation. J. Biol. Chem. 1997, 272, 29167–29173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fruman, D.A.; Rommel, C. PI3K and cancer: Lessons, challenges and opportunities. Nat. Rev. Drug Discov. 2014, 13, 140–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghoneum, A.; Said, N. PI3K-AKT-mTOR and NFkappaB pathways in ovarian cancer: Implications for targeted therapeutics. Cancers 2019, 11, 949. [Google Scholar] [CrossRef] [Green Version]
- Pacold, M.E.; Suire, S.; Perisic, O.; Lara-Gonzalez, S.; Davis, C.T.; Walker, E.H.; Hawkins, P.T.; Stephens, L.; Eccleston, J.F.; Williams, R.L. Crystal structure and functional analysis of Ras binding to its effector phosphoinositide 3-kinase gamma. Cell 2000, 103, 931–943. [Google Scholar] [CrossRef] [Green Version]
- Guo, H.; German, P.; Bai, S.; Barnes, S.; Guo, W.; Qi, X.; Lou, H.; Liang, J.; Jonasch, E.; Mills, G.B.; et al. The PI3K/AKT pathway and renal cell carcinoma. J. Genet. Genomics 2015, 42, 343–353. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Nie, J.; Ma, X.; Wei, Y.; Peng, Y.; Wei, X. Targeting PI3K in cancer: Mechanisms and advances in clinical trials. Mol. Cancer 2019, 18, 26. [Google Scholar] [CrossRef] [Green Version]
- Manning, B.D.; Cantley, L.C. AKT/PKB signaling: Navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, K.; Tang, W.; Zhuo, H.; Zhao, P.G. Recent Advance of Akt Inhibitors in Clinical Trials. Chemistry Select 2019, 4, 9040–9044. [Google Scholar] [CrossRef]
- Mayer, I.A.; Arteaga, C.L. The PI3K/AKT pathway as a target for cancer treatment. Annu. Rev. Med. 2016, 67, 11–28. [Google Scholar] [CrossRef] [PubMed]
- Michie, J.; Kearney, C.J.; Hawkins, E.D.; Silke, J.; Oliaro, J. The immuno-modulatory effects of inhibitor of apoptosis protein antagonists in cancer immunotherapy. Cells 2020, 9, 207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Webster, J.D.; Dugger, D.L.; Goncharov, T.; Roose-Girma, M.; Hung, J.; Kwon, Y.C.; Vucic, D.; Newton, K.; Dixit, V.M. Ubiquitin ligases cIAP1 and cIAP2 limit cell death to prevent inflammation. Cell Rep. 2019, 27, 2679.e2673–2689.e2673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahoney, D.J.; Cheung, H.H.; Mrad, R.L.; Plenchette, S.; Simard, C.; Enwere, E.; Arora, V.; Mak, T.W.; Lacasse, E.C.; Waring, J.; et al. Both cIAP1 and cIAP2 regulate TNFalpha-mediated NF-kappaB activation. Proc. Natl. Acad. Sci. USA 2008, 105, 11778–11783. [Google Scholar] [CrossRef] [Green Version]
- Ea, C.K.; Deng, L.; Xia, Z.P.; Pineda, G.; Chen, Z.J. Activation of IKK by TNFalpha requires site-specific ubiquitination of RIP1 and polyubiquitin binding by NEMO. Mol. Cell 2006, 22, 245–257. [Google Scholar] [CrossRef]
- Guicciardi, M.E.; Werneburg, N.W.; Bronk, S.F.; Franke, A.; Yagita, H.; Thomas, G.; Gores, G.J. Cellular inhibitor of apoptosis (cIAP)-mediated ubiquitination of phosphofurin acidic cluster sorting protein 2 (PACS-2) negatively regulates tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) cytotoxicity. PLoS ONE 2014, 9, e92124. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; Lu, J.; Liu, L.; Yang, C.Y.; Wang, S. Potent and selective small-molecule inhibitors of cIAP1/2 proteins reveal that the binding of Smac mimetics to XIAP BIR3 is not required for their effective induction of cell death in tumor cells. ACS Chem. Biol. 2014, 9, 994–1002. [Google Scholar] [CrossRef]
- Clinical Pipeline: ASTX660. Available online: https://astx.com/research-development/clinical-pipeline/astx660-dual-iap-antagonist-solid-tumors-lymphomas/ (accessed on 6 May 2020).
- EMA grants Orphan Drug Designation to Debiopharm International SA’sIAP inhibitor Debio 1143 in the Treatment of Ovarian Cancer. Available online: https://www.debiopharm.com/drug-development/press-releases/ema-grants-orphan-drug-designation-to-debiopharm-international-sas-iap-inhibitor-debio-1143-in-the-treatment-of-ovarian-cancer/ (accessed on 6 May 2020).
- Ascentage Pharma Announces Approval for the Phase Ib/II Clinical Trial of APG-1387 in Combination with Chemotherapy for the Treatment of Advanced Pancreatic Cancer in China. Available online: https://en.prnasia.com/releases/global/ascentage-pharma-announces-approval-for-the-phase-ib-ii-clinical-trial-of-apg-1387-in-combination-with-chemotherapy-for-the-treatment-of-advanced-pancreatic-cancer-in-china-273315.shtml (accessed on 6 May 2020).
- Terry, M. TetraLogic Pharmaceuticals’s New IPO Takes A Hit As Company Halts Hepatitis B Trial. Available online: https://www.biospace.com/article/tetralogic-pharmaceuticals-s-new-ipo-takes-a-hit-as-company-halts-hepatitis-b-trial-/ (accessed on 6 May 2020).
- Prescott, J.A.; Cook, S.J. Targeting IKKβ in cancer: Challenges and opportunities for the therapeutic utilisation of IKKβ inhibitors. Cell 2018, 7, 115. [Google Scholar] [CrossRef] [Green Version]
- Xu, M.; Skaug, B.; Zeng, W.; Chen, Z.J. A ubiquitin replacement strategy in human cells reveals distinct mechanisms of IKK activation by TNFalpha and IL-1beta. Mol. Cell 2009, 36, 302–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Deng, L.; Hong, M.; Akkaraju, G.R.; Inoue, J.; Chen, Z.J. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature 2001, 412, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Perkins, N.D.; Gilmore, T.D. Good cop, bad cop: The different faces of NF-kappaB. Cell Death Diff. 2006, 13, 759–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, C.K.; Liptay, S.; Wirth, T.; Adler, G.; Schmid, R.M. Suppression of NF-κB activity by sulfasalazine is mediated by direct inhibition of IκB kinases α and β. Gastroenterology 2000, 119, 1209–1218. [Google Scholar] [CrossRef] [PubMed]
- Yin, M.J.; Yamamoto, Y.; Gaynor, R.B. The anti-inflammatory agents aspirin and salicylate inhibit the activity of I(kappa)B kinase-beta. Nature 1998, 396, 77–80. [Google Scholar]
- Goldfine, A.B.; Silver, R.; Aldhahi, W.; Cai, D.; Tatro, E.; Lee, J.; Shoelson, S.E. Use of Salsalate to Target Inflammation in the Treatment of Insulin Resistance and Type 2 Diabetes. Clin. Transl. Sci. 2008, 1, 36–43. [Google Scholar] [CrossRef]
- Sepulveda, P.; Jobin, C.; Baldwin, A.S.; Li, P.D.F.; Karrasch, T.; Uno, J.K.; Sepulveda, A.R.; Jobin, C.; Baldwin, A.S.; Robbins, P.D.; et al. Amelioration of chronic murine colitis by peptide-mediated transduction of the IkB kinase inhibitor NEMO binding domain peptide. J. Immunol. 2007, 179, 7852–7859. [Google Scholar]
- Rhodes, C.A.; Dougherty, P.G.; Cooper, J.K.; Qian, Z.; Lindert, S.; Wang, Q.-E.; Pei, D. Cell-permeable bicyclic peptidyl inhibitors against NEMO-IκB kinase interaction directly from a combinatorial library. J. Am. Chem. Soc. 2018, 140, 12102–12110. [Google Scholar] [CrossRef]
- Verhelst, K.; Verstrepen, L.; Carpentier, I.; Beyaert, R. IκB kinase ε (IKKε): A therapeutic target in inflammation and cancer. Biochem. Pharmacol. 2013, 85, 873–880. [Google Scholar] [CrossRef]
- Hasana, M.; Yan, N. Therapeutic potential of targeting TBK1 in autoimmune diseases and interferonopathies. Pharmacol. Res. 2016, 111, 336–342. [Google Scholar] [CrossRef] [Green Version]
- Hovstadius, P.; Larsson, R.; Jonsson, E.; Skov, T.; Kissmeyer, A.M.; Krasilnikoff, K.; Bergh, J.; Karlsson, M.O.; Lonnebo, A.; Ahlgren, J. A Phase I study of CHS 828 in patients with solid tumor malignancy. Clin. Cancer Res. 2002, 8, 2843–2850. [Google Scholar] [PubMed]
- Grothe, K.; Flechsenhar, K.; Paehler, T.; Ritzeler, O.; Beninga, J.; Saas, J.; Herrmann, M.; Rudolphi, K. IkappaB kinase inhibition as a potential treatment of osteoarthritis—Results of a clinical proof-of-concept study. Osteoarthr. Cartilage 2017, 25, 46–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- TRACON Pharmaceuticals reports fourth quarter and year-end 2018 financial results and provides corporate update. Available online: https://traconpharma.gcs-web.com/news-releases/news-release-details/tracon-pharmaceuticals-reports-fourth-quarter-and-year-end-2018 (accessed on 6 May 2020).
- Stansfield, I.; Querolle, O.A.G.; Poncelet, V.S.; Gross, G.M.; Jacoby, E.; Meerpoel, L.; Kulagowski, J.J.; Macleod, C.; Mann, S.E.; Green, S.R.; et al. New Substituted Cyanoindoline Derivatives as MAP3K14 Kinase Inhibitors for the Treatment of Cancer and Autoimmune Disorders. ACS Med. Chem. Lett. 2017, 8, 908–910. [Google Scholar]
- Pohl, C.; Dikic, I. Cellular quality control by the ubiquitin-proteasome system and autophagy. Science 2019, 366, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Thibaudeau, T.A.; Smith, D.M. A Practical Review of Proteasome Pharmacology. Pharmacol. Rev. 2019, 71, 170–197. [Google Scholar] [CrossRef] [Green Version]
- Clague, M.J.; Heride, C.; Urbe, S. The demographics of the ubiquitin system. Trends Cell Biol. 2015, 25, 417–426. [Google Scholar] [CrossRef]
- Myung, J.; Kim, K.B.; Crews, C.M. The ubiquitin-proteasome pathway and proteasome inhibitors. Med. Res. Rev. 2001, 21, 245–273. [Google Scholar] [CrossRef]
- Ohtake, F.; Tsuchiya, H. The emerging complexity of ubiquitin architecture. J. Biochem. 2017, 161, 125–133. [Google Scholar] [CrossRef] [Green Version]
- Meyer, H.J.; Rape, M. Enhanced protein degradation by branched ubiquitin chains. Cell 2014, 157, 910–921. [Google Scholar] [CrossRef] [Green Version]
- Ohtake, F.; Saeki, Y.; Ishido, S.; Kanno, J.; Tanaka, K. The K48-K63 branched ubiquitin chain regulates NF-kappaB signaling. Mol. Cell 2016, 64, 251–266. [Google Scholar] [CrossRef] [Green Version]
- Sherman, D.J.; Li, J. Proteasome inhibitors: Harnessing proteostasis to combat disease. Molecules 2020, 25, 671. [Google Scholar] [CrossRef] [Green Version]
- Velcade (Bortezomib). Available online: https://myelomaresearchnews.com/velcade-bortezomib/ (accessed on 8 May 2020).
- Huang, Z.; Wu, Y.; Zhou, X.; Xu, J.; Zhu, W.; Shu, Y.; Liu, P. Efficacy of therapy with bortezomib in solid tumors: A review based on 32 clinical trials. Future Oncol. Lond. Engl. 2014, 10, 1795–1807. [Google Scholar] [CrossRef]
- Gupta, N.; Hanley, M.J.; Xia, C.; Labotka, R.; Harvey, R.D.; Venkatakrishnan, K. Clinical Pharmacology of Ixazomib: The First Oral Proteasome Inhibitor. Clin. Pharmacokinet. 2019, 58, 431–449. [Google Scholar] [CrossRef] [PubMed]
- Ninlaro (ixazomib). Available online: https://www.myeloma.org/treatment/current-fda-approved-medications/ninlaro-ixazomib (accessed on 8 May 2020).
- Takeda Provides Update on TOURMALINE-AL1 Phase 3 Trial in AL Amyloidosis. Available online: https://www.takeda.com/newsroom/newsreleases/2019/takeda-provides-update-on-tourmaline-al1-phase-3-trial/ (accessed on 8 May 2020).
- Perel, G.; Bliss, J.; Thomas, C.M. Carfilzomib (Kyprolis): A novel proteasome inhibitor for relapsed and/or refractory multiple myeloma. P T 2016, 41, 303–307. [Google Scholar] [PubMed]
- Waxman, A.J.; Clasen, S.; Hwang, W.T.; Garfall, A.; Vogl, D.T.; Carver, J.; O’Quinn, R.; Cohen, A.D.; Stadtmauer, E.A.; Ky, B.; et al. Carfilzomib-Associated Cardiovascular Adverse Events: A Systematic Review and Meta-analysis. JAMA Oncol. 2018, 4, e174519. [Google Scholar] [CrossRef] [PubMed]
- Camargo, M.; Farouk, S.; Campbell, K. Help or hindrance? An atypical presentation of Carfilzomib-induced nephrotoxicity. In Proceedings of the 19th International Conference on Dialysis, Advances in CKD, Las Vegas, NV, USA, 1–3 February 2017. [Google Scholar]
- Oprozomib. Available online: https://myelomaresearchnews.com/oprozomib/ (accessed on 8 May 2020).
- Marizomib. Available online: https://myelomaresearchnews.com/marizomib/ (accessed on 8 May 2020).
- Roth, P.; Reijneveld, J.C.; Gorlia, T.; Dhermain, F.; Vos, F.Y.F.L.D.; Vanlancker, M.; O’Callaghan, C.J.; Rhun, E.L.; Bent, M.J.V.D.; Mason, W.P.; et al. EORTC 1709/CCTG CE.8: A phase III trial of marizomib in combination with standard temozolomide-based radiochemotherapy versus standard temozolomide-based radiochemotherapy alone in patients with newly diagnosed glioblastoma. J. Clin. Oncol. 2019, 37, TPS2072. [Google Scholar] [CrossRef]
- Cvek, B.; Dvorak, Z. The value of proteasome inhibition in cancer: Can the old drug, disulfiram, have a bright new future as a novel proteasome inhibitor? Drug Discov. Today 2008, 13, 716–722. [Google Scholar]
- Ekinci, E.; Rohondia, S.; Khan, R.; Dou, Q.P. Repurposing Disulfiram as An Anti-Cancer Agent: Updated Review on Literature and Patents. Recent Pat. Anti-Canc. 2019, 14, 113–132. [Google Scholar]
- Cunha, J.P. Antabuse side effects center. Available online: https://www.rxlist.com/antabuse-side-effects-drug-center.htm#overview (accessed on 2 July 2020).
- Field-Smith, A.; Morgan, G.J.; Davies, F.E. Bortezomib (Velcadetrade mark) in the treatment of multiple myeloma. Ther. Clin. Risk Manag. 2006, 2, 271–279. [Google Scholar] [CrossRef] [Green Version]
- Cunha, J.P. Ninlaro side effects center. Available online: https://www.rxlist.com/ninlaro-side-effects-drug-center.htm (accessed on 2 July 2020).
- Siegel, D.S. From clinical trials to clinical practice: Single-agent carfilzomib adverse events and their management in patients with relapsed and/or refractory multiple myeloma. Ther. Adv. Hematol. 2013, 4, 354–365. [Google Scholar] [CrossRef] [Green Version]
- McBride, L.; Samuel, C.O. The side effect profile of Carfilzomib: From clinical trials to clinical practice. J. Adv. Pract. Oncol. 2013, 4, 22–30. [Google Scholar]
- Potts, B.C.; Albitar, M.X.; Anderson, K.C.; Baritaki, S.; Berkers, C.; Bonavida, B.; Chandra, J.; Chauhan, D.; Cusack, J.C., Jr.; Fenical, W.; et al. Marizomib, a proteasome inhibitor for all seasons: Preclinical profile and a framework for clinical trials. Curr. Cancer Drug Targets 2011, 11, 254–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajan, A.M.; Kumar, S. New investigational drugs with single-agent activity in multiple myeloma. Blood Cancer J. 2016, 6, e451. [Google Scholar] [CrossRef] [Green Version]
- Baker, C. Results from a phase I study of VLX1570 for patients with relapsed/refractory multiple myeloma. Available online: https://multiplemyelomahub.com/medical-information/results-from-a-phase-i-study-of-vlx1570-for-patients-with-relapsedrefractory-multiple-myeloma (accessed on 2 July 2020).
- Swords, R.T.; Watts, J.; Erba, H.P.; Altman, J.K.; Maris, M.; Anwer, F.; Hua, Z.; Stein, H.; Faessel, H.; Sedarati, F.; et al. Expanded safety analysis of pevonedistat, a first-in-class NEDD8-activating enzyme inhibitor, in patients with acute myeloid leukemia and myelodysplastic syndromes. Blood Cancer J. 2017, 7, e520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soucy, T.A.; Dick, L.R.; Smith, P.G.; Milhollen, M.A.; Brownell, J.E. The NEDD8 Conjugation Pathway and Its Relevance in Cancer Biology and Therapy. Genes Cancer 2010, 1, 708–716. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Jiang, Y.; Luo, Q.; Li, L.; Jia, L. Neddylation: A novel modulator of the tumor microenvironment. Mol. Cancer 2019, 18, 77–87. [Google Scholar] [CrossRef] [Green Version]
- Pevonedistat (TAK-924): A Potential New Treatment for HR-MDS and AML. Available online: https://www.takeda.com/siteassets/system/investors/report/quarterlyannouncements/fy2019/4_tak924_20191114.pdf (accessed on 9 May 2020).
- LianLiang, P.; Zhang, H.; Wang, G.; Li, S.; Cong, S.; Luo, Y.; Zhang., B. KPNB1, XPO7 and IPO8 mediate the translocation of NF-κB/p65 into the nucleus. Traffic 2013, 14, 1132–1143. [Google Scholar]
- Fagerlund, R.; Kinnunen, L.; Kohler, M.; Julkunen, I.; Melen, K. NF-{kappa} B is transported into the nucleus by importin {alpha}3 and importin {alpha}4. J. Biol. Chem. 2005, 280, 15942–15951. [Google Scholar] [CrossRef] [Green Version]
- Giridharan, S.; Srinivasan, M. Mechanisms of NF-κB p65 and strategies for therapeutic manipulation. J. Inflamm. Res. 2018, 11, 407–419. [Google Scholar] [CrossRef] [Green Version]
- Viatour, P.; Merville, M.P.; Bours, V.; Chariot, A. Phosphorylation of NF-kappaB and IkappaB proteins: Implications in cancer and inflammation. Trends Biochem. Sci. 2005, 30, 43–52. [Google Scholar] [CrossRef]
- Huang, B.; Yang, X.D.; Lamb, A.; Chen, L.F. Posttranslational modifications of NF-kappaB: Another layer of regulation for NF-kappaB signaling pathway. Cell Signal 2010, 22, 1282–1290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.W.; Jang, S.M.; Kim, C.H.; An, J.H.; Kang, E.J.; Choi, K.H. New molecular bridge between RelA/p65 and NF-κB target genes via histone acetyltransferase TIP60 cofactor. J. Biol. Chem. 2012, 7780-91, 287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiernan, R.; Bres, V.; Ng, R.W.; Coudart, M.P.; El Messaoudi, S.; Sardet, C.; Jin, D.Y.; Emiliani, S.; Benkirane, M. Post-activation turn-off of NF-kappa B-dependent transcription is regulated by acetylation of p65. J. Biol. Chem. 2003, 278, 2758–2766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hajmirza, A.; Emadali, A.; Gauthier, A.; Casasnovas, O.; Gressin, R.; Callanan, M.B. BET family protein BRD4: An emerging actor in NFκB signaling in inflammation and cancer. Biomedicines 2018, 6, 16. [Google Scholar] [CrossRef] [Green Version]
- Mathew, C.; Ghildyal, R. CRM1 inhibitors for antiviral therapy. Front. Microbiol. 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Ossareh-Nazari, B.; Bachelerie, F.; Dargemont, C. Evidence for a role of CRM1 in signal-mediated nuclear protein export. Science 1997, 278, 141–144. [Google Scholar] [CrossRef]
- Wolff, B.; Sangher, J.-J.; Wang, A.Y. Leptomycin B is an inhibitor of nuclear export: Inhibition of nucleo-cytoplasmic translocation of the human immunodeficiency virus type 1 (HIV-l) Rev protein and Rev-dependent mRNA. Chem. Biol. 1997, 4, 139–147. [Google Scholar] [CrossRef] [Green Version]
- Kosyna, F.K.; Depping, R. Controlling the gatekeeper: Therapeutic targeting of nuclear transport. Cells 2018, 7, 221. [Google Scholar] [CrossRef] [Green Version]
- Sadowski, A.R.; Gardner, H.L.; Borgatti, A.; Wilson, H.; Vail, D.M.; Lachowicz, J.; Manley, C.; Turner, A.; Klein, M.K.; Waite, A.; et al. Phase II study of the oral selective inhibitor of nuclear export (SINE) KPT-335 (verdinexor) in dogs with lymphoma. BMC Vet. Res. 2018, 14, 1–7. [Google Scholar] [CrossRef]
- Wang, A.Y.; Liu, H. The past, present, and future of CRM1/XPO1 inhibitors. Stem Cell Investig. 2019, 6, 6. [Google Scholar] [CrossRef]
- Gavriatopoulou, M.; Chari, A.; Chen, C.; Bahlis, N.; Vogl, D.T.; Jakubowiak, A.; Dingli, D.; Cornell, R.F.; Hofmeister, C.C.; Siegel, D.; et al. Integrated safety profile of selinexor in multiple myeloma: Experience from 437 patients enrolled in clinical trials. Leukemia 2020. [Google Scholar] [CrossRef] [PubMed]
- Karyopharm presents positive phase 1/2 Eltanexor data at the American Society of Hematology 2017 annual meeting. Available online: https://investors.karyopharm.com/news-releases/news-release-details/karyopharm-presents-positive-phase-12-eltanexor-data-american (accessed on 2 July 2020).
- Karyopharm Announces: FDA Considers the Effectiveness and Safety Technical Sections Complete to Support Conditional Approval for the New Animal Drug Application for Verdinexor (KPT-335) to Treat Lymphoma in Client Owned Dogs. Available online: https://investors.karyopharm.com/news-releases/news-release-details/karyopharm-announces-fda-considers-effectiveness-and-safety (accessed on 2 July 2020).
- Bubna, A.K. Vorinostat-An Overview. Indian J. Dermatol. 2015, 60, 419. [Google Scholar] [CrossRef] [PubMed]
- ISTODAX® (romidepsin) for Injection: Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2009/022393lbl.pdf (accessed on 2 July 2020).
- BELEODAQ® (Belinostat) for Injection, for Intravenous Administration: Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/206256lbl.pdf (accessed on 2 July 2020).
- Liu, J.D.; Sun, C.Y.; Tang, L.; Wu, Y.Y.; Wang, Q.Y.; Hu, B.; Hu, Y. Efficacy and safety of Panobinostat in relapsed or/and refractory multiple myeloma: Meta analyses of clinical trials and systematic review. Sci. Rep. 2016, 6, 27361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Wang, T.; Geng, C.; Zhang, Y.; Zhang, J.; Ning, Z.; Jiang, Z. Exploratory clinical study of chidamide, an oral subtype-selective histone deacetylase inhibitor, in combination with exemestane in hormone receptor-positive advanced breast cancer. Chin. J. Cancer Res. 2018, 30, 605–612. [Google Scholar] [CrossRef]
- Khan, C.; Pathe, N.; Fazal, S.; Lister, J.; Rossetti, J.M. Azacitidine in the management of patients with myelodysplastic syndromes. Ther. Adv. Hematol. 2012, 3, 355–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saba, H.I. Decitabine in the treatment of myelodysplastic syndromes. Ther. Clin. Risk Manag. 2007, 3, 807–817. [Google Scholar] [PubMed]
- Wagstaff, K.M.; Sivakumaran, H.; Heaton, S.M.; Harrich, D.; Jans, D.A. Ivermectin is a specific inhibitor of importin α/β-mediated nuclear import able to inhibit replication of HIV-1 and dengue virus. Biochem. J. 2012, 443, 851–856. [Google Scholar] [CrossRef] [Green Version]
- Soderholm, J.F.; Bird, S.L.; Kalab, P.; Sampathkumar, Y.; Hasegawa, K.; Uehara-Bingen, M.; Weis, K.; Heald, R. Importazole, a small molecule inhibitor of the Transport Receptor Importin-β. ACS Chem. Biol. 2011, 6, 700–708. [Google Scholar]
- Lin, Y.-Z.; Yao, S.; Veach, R.A.; Torgerson, T.R.; Hawiger, J. Inhibition of nuclear translocation of transcription factor NF-κB by a synthetic peptide containing a cell membrane-permeable motif and nuclear localization sequence. J. Biol. Chem. 1995, 270, 14255–14258. [Google Scholar]
- Wang, Y.F.; Xu, X.; Fan, X.; Zhang, C.; Wei, Q.; Wang, X.; Guo, W.; Xing, W.; Yu, J.; Yan, J.-L.; et al. A Cell-penetrating peptide suppresses inflammation by inhibiting NF-κB signaling. Mol. Ther. 2011, 19, 1849–1857. [Google Scholar]
- O’Shea, J.M.; Perkins, N.D. Regulation of the RelA (p65) transactivation domain. Biochem. Soc. Trans. 2008, 36, 603–608. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.F.; Mu, Y.; Greene, W.C. Acetylation of RelA at discrete sites regulates distinct nuclear functions of NF-kappaB. EMBO J. 2002, 21, 6539–6548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lecoq, L.; Raiola, L.; Chabot, P.R.; Cyr, N.; Arseneault, G.; Legault, P.; Omichinski, J.G. Structural characterization of interactions between transactivation domain 1 of the p65 subunit of NF-kappaB and transcription regulatory factors. Nucleic Acids Res. 2017, 45, 5564–5576. [Google Scholar] [CrossRef] [PubMed]
- Rothgiesser, K.M.; Fey, M.; Hottiger, M.O. Acetylation of p65 at lysine 314 is important for late NF-kappaB-dependent gene expression. BMC Genom. 2010, 11, 22. [Google Scholar] [CrossRef] [Green Version]
- Virág, D.; Dalmadi-Kiss, B.; Vékey, K.; Drahos, L.; Klebovich, I.; Antal, I.; Ludányi, K. Current trends in the analysis of post-translational modifications. Chromatographia 2020, 83, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Yuksel, M.; Okajima, K.; Uchiba, M.; Okabe, H. Gabexate mesilate, a synthetic protease inhibitor, inhibits lipopolysaccharide-induced tumor necrosis factor-alpha production by inhibiting activation of both nuclear factor-kappaB and activator protein-1 in human monocytes. J. Pharmacol. Exp. Ther. 2003, 305, 298–305. [Google Scholar] [CrossRef] [Green Version]
- Hoffman, E.P.; Schwartz, B.D.; Mengle-Gaw, L.J.; Smith, E.C.; Castro, D.; Mah, J.K.; McDonald, C.M.; Kuntz, N.L.; Finkel, R.S.; Guglieri, M.; et al. Vamorolone trial in Duchenne muscular dystrophy shows dose-related improvement of muscle function. Neurology 2019, 93, e1312–e1323. [Google Scholar] [CrossRef] [Green Version]
- Zimmermann, P.; Ziesenitz, V.C.; Curtis, N.; Ritz, N. The immunomodulatory effects of macrolides—A systematic review of the underlying mechanisms. Front. Immunol. 2018, 9, 302–315. [Google Scholar] [CrossRef] [Green Version]
- Sugawara, A.; Shima, H.; Sueki, A.; Hirose, T.; Matsui, H.; Nakano, H.; Hanaki, H.; Akagawa, K.S.; Ōmura, S.; Sunazuka, T. Non-antibiotic 12-membered macrolides: Design, synthesis and biological evaluation in a cigarette-smoking model. J. Antibiot. (Tokyo) 2016, 69, 319–326. [Google Scholar] [CrossRef]
- Niloo, K.; Ritesh, R.; Erika, G.; Gerald, M. Plant polyphenols as inhibitors of NF-κB induced cytokine production—a potential anti-inflammatory treatment for Alzheimer’s disease? Front. Mol. Neurosci. 2015, 8, 24–28. [Google Scholar]
- Shimizu, K.; Funamoto, M.; Sunagawa, Y.; Shimizu, S.; Katanasaka, Y.; Miyazaki, Y.; Wada, H.; Hasegawa, K.; Morimoto, T. Anti-inflammatory action of Curcumin and its use in the treatment of lifestyle-related diseases. Eur. Cardiol. 2019, 14, 117–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharifi-Rad, M.; Pezzani, R.; Redaelli, M.; Zorzan, M.; Imran, M.; Ahmed Khalil, A.; Salehi, B.; Sharopov, F.; Cho, W.C.; Sharifi-Rad, J. Preclinical activities of Epigallocatechin gallate in signaling pathways in cancer. Molecules 2020, 25, 467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, S.; Li, S.; Tian, J.; Li, F. Iguratimod as a new drug for rheumatoid arthritis: Current landscape. Front. Pharmacol. 2020, 11, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Yamasaki, R.; Fang, M.; Masaki, K.; Ochi, H.; Matsushita, T.; Kira, J.I. Novel disease-modifying anti-rheumatic drug iguratimod suppresses chronic experimental autoimmune encephalomyelitis by down-regulating activation of macrophages/microglia through an NF-κB pathway. Sci. Rep. 2018, 8, 1933–1948. [Google Scholar] [CrossRef] [PubMed]
- Ehsanian, R.; Van Waes, C.; Feller, S.M. Beyond DNA binding—A review of the potential mechanisms mediating quinacrine’s therapeutic activities in parasitic infections, inflammation, and cancers. Cell Commun. Signal 2011, 9, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, R.; Mrowietz, U.; Wiegrebe, W.; Jirovsky, D.; Loewe, R.; Pillinger, M.; Gröger, M.; Holnthoner, W.; Wolff, K.; Petzelbauer, P. Dimethylfumarate inhibits Tumor-Necrosis-Factor-induced CD62E expression in an NF-κB-dependent manner. J. Invest. Dermatol. 2001, 117, 1363–1368. [Google Scholar] [CrossRef] [Green Version]
- Yan, B.; Kuick, C.H.; Lim, M.; Venkataraman, K.; Tennakoon, C.; Loh, E.; Lian, D.; Leong, M.Y.; Lakshmanan, M.; Tergaonkar, V.; et al. Platform comparison for evaluation of ALK protein immunohistochemical expression, genomic copy number and hotspot mutation status in neuroblastomas. PLoS ONE 2014, 9, e106575. [Google Scholar] [CrossRef] [Green Version]
- Phase I/IIa Study of DTP3 in Patients with Advanced Multiple Myeloma. Available online: https://www.hra.nhs.uk/planning-and-improving-research/application-summaries/research-summaries/phase-iiia-study-of-dtp3-in-patients-with-advanced-multiple-myeloma/ (accessed on 10 May 2020).




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramadass, V.; Vaiyapuri, T.; Tergaonkar, V. Small Molecule NF-κB Pathway Inhibitors in Clinic. Int. J. Mol. Sci. 2020, 21, 5164. https://doi.org/10.3390/ijms21145164
Ramadass V, Vaiyapuri T, Tergaonkar V. Small Molecule NF-κB Pathway Inhibitors in Clinic. International Journal of Molecular Sciences. 2020; 21(14):5164. https://doi.org/10.3390/ijms21145164
Chicago/Turabian StyleRamadass, Venkataramanan, Thamilselvan Vaiyapuri, and Vinay Tergaonkar. 2020. "Small Molecule NF-κB Pathway Inhibitors in Clinic" International Journal of Molecular Sciences 21, no. 14: 5164. https://doi.org/10.3390/ijms21145164