NF-κB and Its Role in Checkpoint Control

 , ,
, , {kind=link}
{kind=link}
Abstract
:1. Introduction
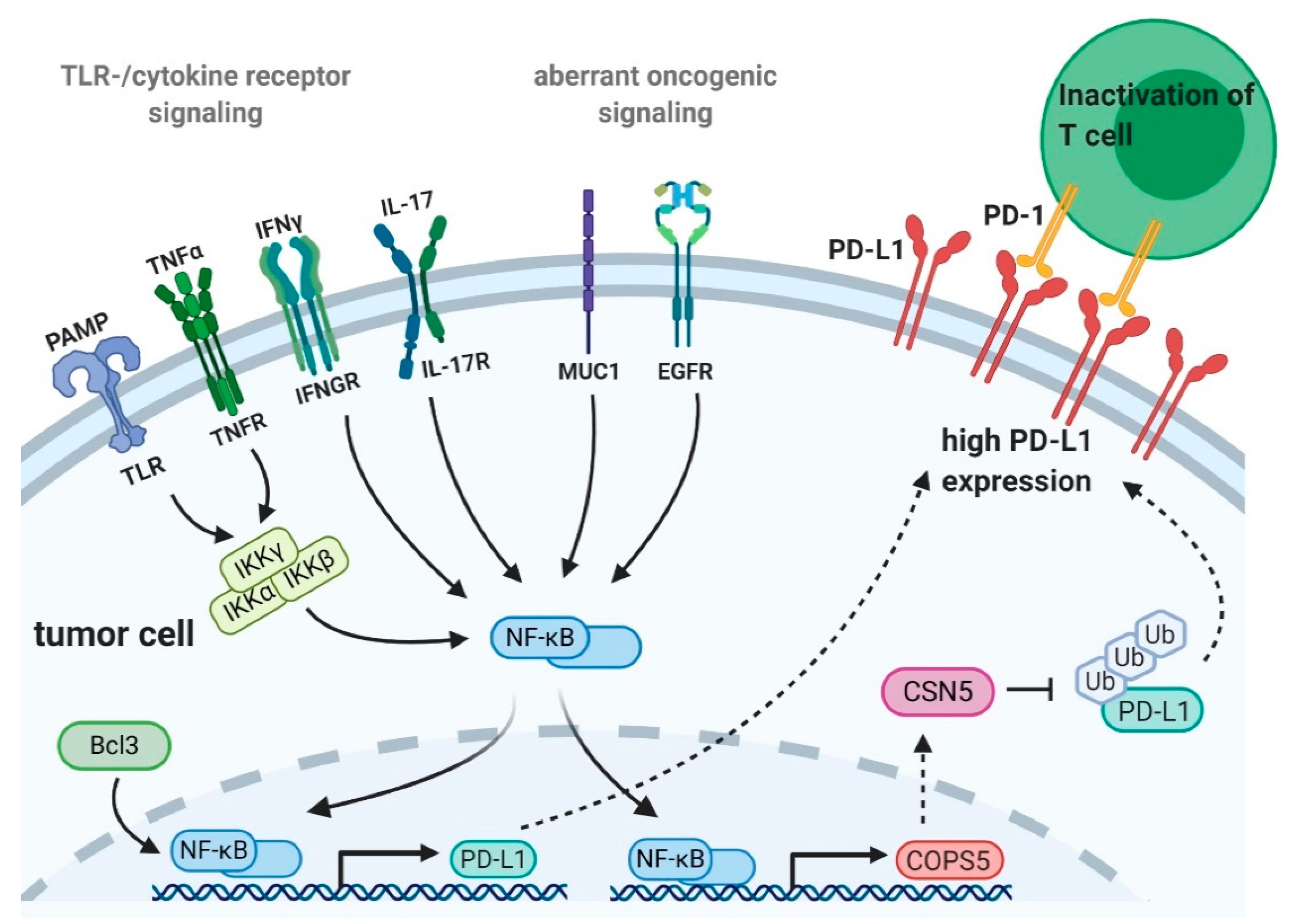
2. NF-κB and Tumor Immune Checkpoint Expression
2.1. Transcriptional Regulation of PD-L1 Expression by NF-κB
2.1.1. Regulation of PD-L1 Expression by Activation of NF-κB upon Toll-Like Receptor- or Cytokine Receptor-Mediated Signaling
2.1.2. Control of PD-L1 Expression by NF-κB and Oncogene- or Tumor Suppressor Mediated Transcriptional Regulation
2.1.3. Regulation of PD-L1 Expression by NF-κB and Epidermal Growth Factor Receptor Signaling
2.2. Posttranslational Regulation of PD-L1 Expression by NF-κB
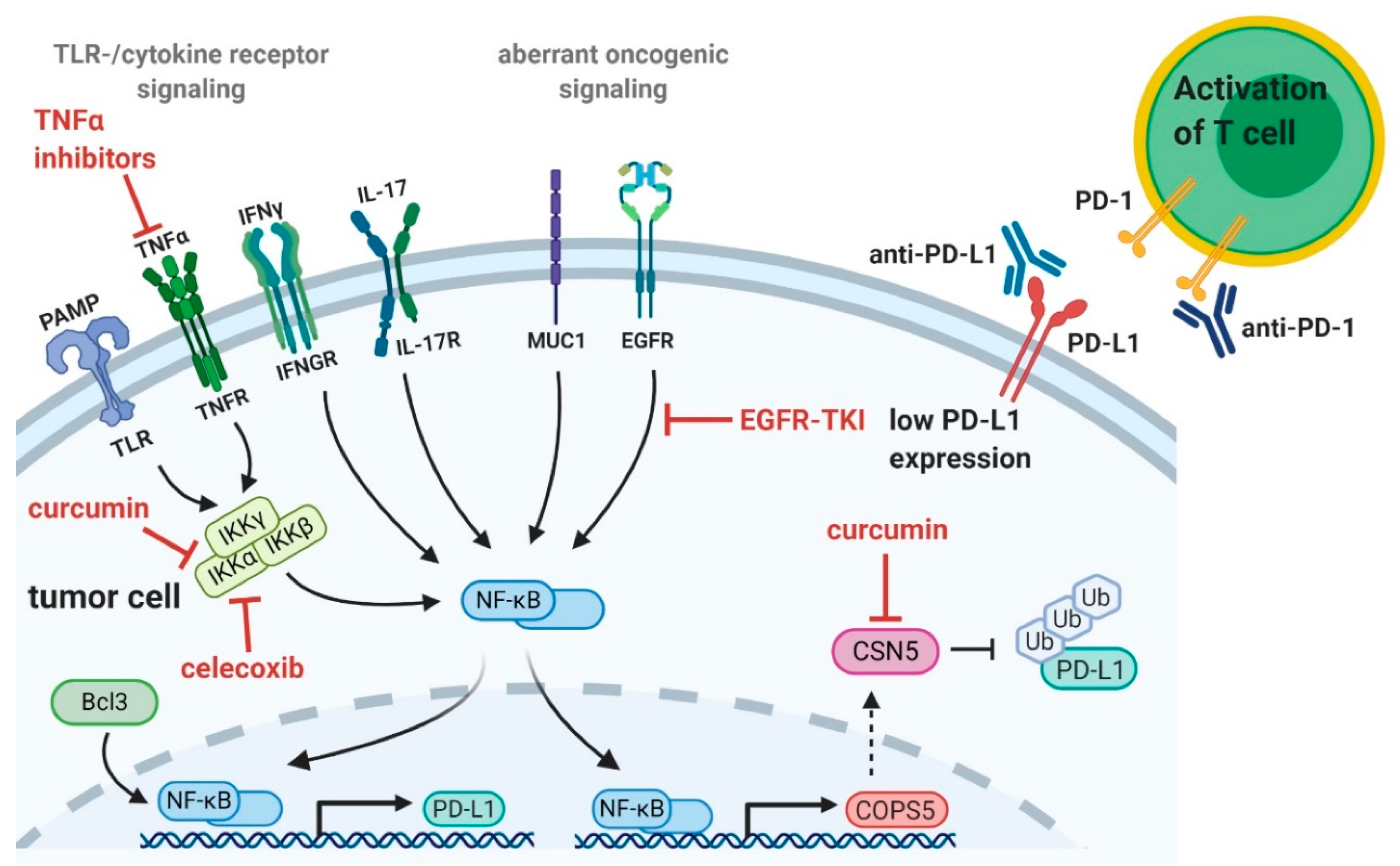
3. NF-κB as Therapeutic Target in Cancer
3.1. Natural Compounds
3.2. Pentoxifylline
3.3. TNFα Inhibitors
3.4. Cyclooxygenase 2 Inhibitors
3.5. EGFR-Tyrosine Kinase Inhibitors
3.6. CDK4/6 Inhibitors
4. Conclusions and Outlook
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| NF-κB | Nuclear factor κB |
| IKK | IκB kinase |
| Perez-Ruiz | T-lymphocyte associated protein |
| B7 | Ligand for CTLA-4 |
| Treg | Regulatory T cell |
| PD-1 | Programmed cell death protein 1 |
| PD-L1 | programmed-death ligand 1 |
| TME | Tumor microenvironment |
| IFN | Interferon |
| IL-17 | Interleukin-17 |
| TNFα | Tumor necrosis factor α |
| TLR | Toll-like receptor |
| PAMPs | Pathogen-associated molecular patterns |
| LPS | Lipopolysaccharide |
| DC | Dendritic cell |
| JAK | Janus kinase |
| STAT | Signal transducer and activation of transcription |
| EBV | Epstein–Barr virus |
| LMP1 | Latent membrane protein 1 |
| HCC | Hepatocellular carcinoma cells |
| NSCLC | Non-small cell lung carcinoma |
| Bcl3 | B cell lymphoma 3 |
| Muc1 | Mucin1 |
| RB | Retinoblastoma |
| CDK | Cyclin-dependent kinase |
| EGFR | Epidermal growth factor receptor |
| HIF-1α | Hypoxia-induced factor 1α |
| EGFR-TKI | EGFR-tyrosine kinase inhibitors |
| CSN5 | Fifth element of the COP9 signalosome |
| COP9 | Constitutive photomorphogenesis 9 |
| COPS5 | COP9 signalosome complex subunit 5 |
| mAb | Monoclonal antibody |
| PTXF | Pentoxifylline |
| COX-2 | Cyclooxygenase-2 |
| Andro | Andrographolide |
References
- Taniguchi, K.; Karin, M. NF-κB, inflammation, immunity and cancer: Coming of age. Nat. Rev. Immunol. 2018, 18, 309–324. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, Inflammation, and Cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karin, M.; Greten, F.R. NF-κB: Linking inflammation and immunity to cancer development and progression. Nat. Rev. Immunol. 2005, 5, 749–759. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; Ghosh, S. Shared principles in NF-kappaB signaling. Cell 2008, 132, 344–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, S.-C. The non-canonical NF-κB pathway in immunity and inflammation. Nat. Rev. Immunol. 2017, 17, 545–558. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [Green Version]
- Bonizzi, G.; Karin, M. The two NF-κB activation pathways and their role in innate and adaptive immunity. Trends Immunol. 2004, 25, 280–288. [Google Scholar] [CrossRef]
- Pires, B.R.B.; Silva, R.C.M.C.; Ferreira, G.M.; Abdelhay, E. NF-kappaB: Two Sides of the Same Coin. Genes 2018, 9, 24. [Google Scholar] [CrossRef] [Green Version]
- Xia, Y.; Shen, S.; Verma, I.M. NF-κB, an active player in human cancers. Cancer Immunol. Res. 2014, 2, 823–830. [Google Scholar] [CrossRef] [Green Version]
- Karin, M. Nuclear factor-κB in cancer development and progression. Nature 2006, 441, 431–436. [Google Scholar] [CrossRef]
- Park, M.H.; Hong, J.T. Roles of NF-κB in Cancer and Inflammatory Diseases and Their Therapeutic Approaches. Cells 2016, 5, 15. [Google Scholar] [CrossRef] [PubMed]
- Baud, V.; Karin, M. Is NF-κB a good target for cancer therapy? Hopes and pitfalls. Nat. Rev. Drug Discov. 2009, 8, 33–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.; Pardoll, D.; Jove, R. STATs in cancer inflammation and immunity: A leading role for STAT3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Karin, M. Dangerous liaisons: STAT3 and NF-κB collaboration and crosstalk in cancer. Cytokine Growth Factor Rev. 2010, 21, 11–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerondakis, S.; Siebenlist, U. Roles of the NF-kappaB pathway in lymphocyte development and function. Cold Spring Harb. Perspect. Biol. 2010, 2, a000182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, H.; Ghosh, S. NF-κB: Roles and regulation in different CD4+ T-cell subsets. Immunol. Rev. 2013, 252, 41–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishikawa, H.; Sakaguchi, S. Regulatory T cells in tumor immunity. Int. J. Cancer 2010, 127, 759–767. [Google Scholar] [CrossRef]
- Sakaguchi, S. Naturally Arising CD4+ Regulatory T Cells for Immunologic Self-Tolerance and Negative Control of Immune Responses. Annu. Rev. Immunol. 2004, 22, 531–562. [Google Scholar] [CrossRef]
- Grinberg-Bleyer, Y.; Oh, H.; Desrichard, A.; Bhatt, D.M.; Caron, R.; Chan, T.A.; Schmid, R.M.; Klein, U.; Hayden, M.S.; Ghosh, S. NF-κB c-Rel Is Crucial for the Regulatory T Cell Immune Checkpoint in Cancer. Cell 2017, 170, 1096–1108.e1013. [Google Scholar] [CrossRef]
- Topalian, S.L.; Weiner, G.J.; Pardoll, D.M. Cancer Immunotherapy Comes of Age. J. Clin. Oncol. 2011, 29, 4828–4836. [Google Scholar] [CrossRef] [Green Version]
- Dong, H.; Strome, S.E.; Salomao, D.R.; Tamura, H.; Hirano, F.; Flies, D.B.; Roche, P.C.; Lu, J.; Zhu, G.; Tamada, K.; et al. Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat. Med. 2002, 8, 793–800. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Crabill, G.A.; Pritchard, T.S.; McMiller, T.L.; Wei, P.; Pardoll, D.M.; Pan, F.; Topalian, S.L. Mechanisms regulating PD-L1 expression on tumor and immune cells. J. Immunother. Cancer 2019, 7, 305. [Google Scholar] [CrossRef] [PubMed]
- Taube, J.M.; Young, G.D.; McMiller, T.L.; Chen, S.; Salas, J.T.; Pritchard, T.S.; Xu, H.; Meeker, A.K.; Fan, J.; Cheadle, C.; et al. Differential Expression of Immune-Regulatory Genes Associated with PD-L1 Display in Melanoma: Implications for PD-1 Pathway Blockade. Clin. Cancer Res. 2015, 21, 3969–3976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seidel, J.A.; Otsuka, A.; Kabashima, K. Anti-PD-1 and Anti-CTLA-4 Therapies in Cancer: Mechanisms of Action, Efficacy, and Limitations. Front. Oncol. 2018, 8, 86. [Google Scholar] [CrossRef]
- Kythreotou, A.; Siddique, A.; Mauri, F.A.; Bower, M.; Pinato, D.J. PD-L1. J. Clin. Pathol. 2018, 71, 189–194. [Google Scholar] [CrossRef]
- Rowshanravan, B.; Halliday, N.; Sansom, D.M. CTLA-4: A moving target in immunotherapy. Blood 2018, 131, 58–67. [Google Scholar] [CrossRef]
- Harlin, H.; Hwang, K.W.; Palucki, D.A.; Kim, O.; Thompson, C.B.; Boothby, M.; Alegre, M.L. CTLA-4 engagement regulates NF-kappaB activation in vivo. Eur. J. Immunol. 2002, 32, 2095–2104. [Google Scholar] [CrossRef]
- Qureshi, O.S.; Zheng, Y.; Nakamura, K.; Attridge, K.; Manzotti, C.; Schmidt, E.M.; Baker, J.; Jeffery, L.E.; Kaur, S.; Briggs, Z.; et al. Trans-endocytosis of CD80 and CD86: A molecular basis for the cell-extrinsic function of CTLA-4. Science 2011, 332, 600–603. [Google Scholar] [CrossRef] [Green Version]
- Soskic, B.; Qureshi, O.S.; Hou, T.; Sansom, D.M. A transendocytosis perspective on the CD28/CTLA-4 pathway. Adv. Immunol. 2014, 124, 95–136. [Google Scholar]
- Darvin, P.; Toor, S.M.; Sasidharan Nair, V.; Elkord, E. Immune checkpoint inhibitors: Recent progress and potential biomarkers. Exp. Mol. Med. 2018, 50, 165. [Google Scholar] [CrossRef] [Green Version]
- Zou, W.; Wolchok, J.D.; Chen, L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci. Transl. Med. 2016, 8, 328rv324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalbasi, A.; Ribas, A. Tumour-intrinsic resistance to immune checkpoint blockade. Nat. Rev. Immunol. 2020, 20, 25–39. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.C.; Duffy, C.R.; Allison, J.P. Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov. 2018, 8, 1069–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritprajak, P.; Azuma, M. Intrinsic and extrinsic control of expression of the immunoregulatory molecule PD-L1 in epithelial cells and squamous cell carcinoma. Oral Oncol. 2015, 51, 221–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Wang, K.; Xu, J.; Huang, J.; Zhang, T. Prognostic Significance of Circulating Tumor Cells in Non-Small-Cell Lung Cancer Patients: A Meta-Analysis. PLoS ONE 2013, 8, e78070. [Google Scholar] [CrossRef] [Green Version]
- Peng, J.; Hamanishi, J.; Matsumura, N.; Abiko, K.; Murat, K.; Baba, T.; Yamaguchi, K.; Horikawa, N.; Hosoe, Y.; Murphy, S.K.; et al. Chemotherapy Induces Programmed Cell Death-Ligand 1 Overexpression via the Nuclear Factor-κB to Foster an Immunosuppressive Tumor Microenvironment in Ovarian Cancer. Cancer Res. 2015, 75, 5034–5045. [Google Scholar] [CrossRef] [Green Version]
- Gowrishankar, K.; Gunatilake, D.; Gallagher, S.J.; Tiffen, J.; Rizos, H.; Hersey, P. Inducible but Not Constitutive Expression of PD-L1 in Human Melanoma Cells Is Dependent on Activation of NF-κB. PLoS ONE 2015, 10, e0123410. [Google Scholar] [CrossRef] [Green Version]
- Lim, S.-O.; Li, C.-W.; Xia, W.; Cha, J.-H.; Chan, L.-C.; Wu, Y.; Chang, S.-S.; Lin, W.-C.; Hsu, J.-M.; Hsu, Y.-H.; et al. Deubiquitination and Stabilization of PD-L1 by CSN5. Cancer Cell 2016, 30, 925–939. [Google Scholar] [CrossRef] [Green Version]
- Bouillez, A.; Rajabi, H.; Jin, C.; Samur, M.; Tagde, A.; Alam, M.; Hiraki, M.; Maeda, T.; Hu, X.; Adeegbe, D.; et al. MUC1-C integrates PD-L1 induction with repression of immune effectors in non-small-cell lung cancer. Oncogene 2017, 36, 4037–4046. [Google Scholar] [CrossRef] [Green Version]
- Maeda, T.; Hiraki, M.; Jin, C.; Rajabi, H.; Tagde, A.; Alam, M.; Bouillez, A.; Hu, X.; Suzuki, Y.; Miyo, M.; et al. MUC1-C Induces PD-L1 and Immune Evasion in Triple-Negative Breast Cancer. Cancer Res. 2018, 78, 205–215. [Google Scholar] [CrossRef] [Green Version]
- Lucas, J.; Hsieh, T.C.; Halicka, H.D.; Darzynkiewicz, Z.; Wu, J.M. Upregulation of PDL1 expression by resveratrol and piceatannol in breast and colorectal cancer cells occurs via HDAC3/p300mediated NFkappaB signaling. Int. J. Oncol. 2018, 53, 1469–1480. [Google Scholar] [PubMed] [Green Version]
- Jin, X.; Ding, D.; Yan, Y.; Li, H.; Wang, B.; Ma, L.; Ye, Z.; Ma, T.; Wu, Q.; Rodrigues, D.N.; et al. Phosphorylated RB Promotes Cancer Immunity by Inhibiting NF-κB Activation and PD-L1 Expression. Mol. Cell 2019, 73, 22–35.e26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, R.; Li, Y.; Wang, Z.; Bai, H.; Duan, J.; Wang, S.; Wang, L.; Wang, J. Hypoxia-inducible factor-1alpha and nuclear factor-kappaB play important roles in regulating programmed cell death ligand 1 expression by epidermal growth factor receptor mutants in non-small-cell lung cancer cells. Cancer Sci. 2019, 110, 1665–1675. [Google Scholar] [CrossRef] [PubMed]
- Azuma, T.; Yao, S.; Zhu, G.; Flies, A.S.; Flies, S.J.; Chen, L. B7-H1 is a ubiquitous antiapoptotic receptor on cancer cells. Blood 2008, 111, 3635–3643. [Google Scholar] [CrossRef] [PubMed]
- Clark, C.A.; Gupta, H.B.; Sareddy, G.; Pandeswara, S.; Lao, S.; Yuan, B.; Drerup, J.M.; Padron, A.; Conejo-Garcia, J.; Murthy, K.; et al. Tumor-Intrinsic PD-L1 Signals Regulate Cell Growth, Pathogenesis, and Autophagy in Ovarian Cancer and Melanoma. Cancer Res. 2016, 76, 6964–6974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Li, Y.; Liu, X.; Chen, C.; Harrington, S.M.; Cao, S.; Xie, T.; Pham, T.; Mansfield, A.S.; Yan, Y.; et al. Targeting B7-H1 (PD-L1) sensitizes cancer cells to chemotherapy. Heliyon 2018, 4, e01039. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.-H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; van der Windt, G.J.W.; et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef] [Green Version]
- Manguso, R.T.; Pope, H.W.; Zimmer, M.D.; Brown, F.D.; Yates, K.B.; Miller, B.C.; Collins, N.B.; Bi, K.; LaFleur, M.W.; Juneja, V.R.; et al. In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature 2017, 547, 413–418. [Google Scholar] [CrossRef] [Green Version]
- Pan, D.; Kobayashi, A.; Jiang, P.; Ferrari de Andrade, L.; Tay, R.E.; Luoma, A.M.; Tsoucas, D.; Qiu, X.; Lim, K.; Rao, P.; et al. A major chromatin regulator determines resistance of tumor cells to T cell–mediated killing. Science 2018, 359, 770–775. [Google Scholar] [CrossRef] [Green Version]
- Huang, G.; Wen, Q.; Zhao, Y.; Gao, Q.; Bai, Y. NF-κB Plays a Key Role in Inducing CD274 Expression in Human Monocytes after Lipopolysaccharide Treatment. PLoS ONE 2013, 8, e61602. [Google Scholar] [CrossRef] [Green Version]
- Zou, Y.; Uddin, M.M.; Padmanabhan, S.; Zhu, Y.; Bu, P.; Vancura, A.; Vancurova, I. The proto-oncogene Bcl3 induces immune checkpoint PD-L1 expression, mediating proliferation of ovarian cancer cells. J. Biol. Chem. 2018, 293, 15483–15496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imai, D.; Yoshizumi, T.; Okano, S.; Itoh, S.; Ikegami, T.; Harada, N.; Aishima, S.; Oda, Y.; Maehara, Y. IFN-γ Promotes Epithelial-Mesenchymal Transition and the Expression of PD-L1 in Pancreatic Cancer. J. Surg. Res. 2019, 240, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Wang, J.; Zhang, N.; Zhuang, M.; Zong, Z.; Zou, J.; Li, G.; Wang, X.; Zhou, H.; Zhang, L.; et al. Cross-talk between TNF-α and IFN-γ signaling in induction of B7-H1 expression in hepatocellular carcinoma cells. Cancer Immunol. Immunother. 2018, 67, 271–283. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Diaz, A.; Shin, D.S.; Moreno, B.H.; Saco, J.; Escuin-Ordinas, H.; Rodriguez, G.A.; Zaretsky, J.M.; Sun, L.; Hugo, W.; Wang, X.; et al. Interferon Receptor Signaling Pathways Regulating PD-L1 and PD-L2 Expression. Cell Rep. 2017, 19, 1189–1201. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Yang, L.; Huang, F.; Zhang, Q.; Liu, S.; Ma, L.; You, Z. Inflammatory cytokines IL-17 and TNF-α up-regulate PD-L1 expression in human prostate and colon cancer cells. Immunol. Lett. 2017, 184, 7–14. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Hamrouni, A.; Wolowiec, D.; Coiteux, V.; Kuliczkowski, K.; Hetuin, D.; Saudemont, A.; Quesnel, B. Plasma cells from multiple myeloma patients express B7-H1 (PD-L1) and increase expression after stimulation with IFN-γ and TLR ligands via a MyD88-, TRAF6-, and MEK-dependent pathway. Blood 2007, 110, 296–304. [Google Scholar] [CrossRef] [Green Version]
- Qian, Y.; Deng, J.; Geng, L.; Xie, H.; Jiang, G.; Zhou, L.; Wang, Y.; Yin, S.; Feng, X.; Liu, J.; et al. TLR4 Signaling Induces B7-H1 Expression Through MAPK Pathways in Bladder Cancer Cells. Cancer Investig. 2008, 26, 816–821. [Google Scholar] [CrossRef]
- Li, H.; Xia, J.-Q.; Zhu, F.-S.; Xi, Z.-H.; Pan, C.-Y.; Gu, L.-M.; Tian, Y.-Z. LPS promotes the expression of PD-L1 in gastric cancer cells through NF-κB activation. J. Cell. Biochem. 2018, 119, 9997–10004. [Google Scholar] [CrossRef]
- Mühlbauer, M.; Fleck, M.; Schütz, C.; Weiss, T.; Froh, M.; Blank, C.; Schölmerich, J.; Hellerbrand, C. PD-L1 is induced in hepatocytes by viral infection and by interferon-α and -γ and mediates T cell apoptosis. J. Hepatol. 2006, 45, 520–528. [Google Scholar] [CrossRef]
- Chen, L.; Zhang, Z.; Chen, W.; Zhang, Z.; Li, Y.; Shi, M.; Zhang, J.; Chen, L.; Wang, S.; Wang, F.-S. B7-H1 Up-Regulation on Myeloid Dendritic Cells Significantly Suppresses T Cell Immune Function in Patients with Chronic Hepatitis B. J. Immunol. 2007, 178, 6634–6641. [Google Scholar] [CrossRef] [Green Version]
- Bazhin, A.V.; von Ahn, K.; Fritz, J.; Werner, J.; Karakhanova, S. Interferon-α Up-Regulates the Expression of PD-L1 Molecules on Immune Cells Through STAT3 and p38 Signaling. Front. Immunol 2018, 9, 2129. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, Y.; Kishida, T.; Kotani, S.I.; Takayama, K.; Mazda, O. Interferon-β signal may up-regulate PD-L1 expression through IRF9-dependent and independent pathways in lung cancer cells. Biochem. Biophys. Res. Commun. 2018, 507, 330–336. [Google Scholar] [CrossRef] [PubMed]
- Jacquelot, N.; Yamazaki, T.; Roberti, M.P.; Duong, C.P.M.; Andrews, M.C.; Verlingue, L.; Ferrere, G.; Becharef, S.; Vétizou, M.; Daillère, R.; et al. Sustained Type I interferon signaling as a mechanism of resistance to PD-1 blockade. Cell Res. 2019, 29, 846–861. [Google Scholar] [CrossRef] [PubMed]
- Schreiner, B.; Mitsdoerffer, M.; Kieseier, B.C.; Chen, L.; Hartung, H.-P.; Weller, M.; Wiendl, H. Interferon-β enhances monocyte and dendritic cell expression of B7-H1 (PD-L1), a strong inhibitor of autologous T-cell activation: Relevance for the immune modulatory effect in multiple sclerosis. J. Neuroimmunol. 2004, 155, 172–182. [Google Scholar] [CrossRef]
- Du, Z.; Wei, L.; Murti, A.; Pfeffer, S.R.; Fan, M.; Yang, C.H.; Pfeffer, L.M. Non-conventional signal transduction by type 1 interferons: The NF-kappaB pathway. J Cell Biochem 2007, 102, 1087–1094. [Google Scholar] [CrossRef]
- de Weerd, N.A.; Nguyen, T. The interferons and their receptors—Distribution and regulation. Immunol. Cell Biol. 2012, 90, 483–491. [Google Scholar] [CrossRef]
- Lee, S.-K.; Seo, S.-H.; Kim, B.-S.; Kim, C.-D.; Lee, J.-H.; Kang, J.-S.; Maeng, P.J.; Lim, J.-S. IFN-gamma regulates the expression of B7-H1 in dermal fibroblast cells. J. Dermatol. Sci. 2005, 40, 95–103. [Google Scholar] [CrossRef]
- Kowanetz, M.; Zou, W.; Gettinger, S.N.; Koeppen, H.; Kockx, M.; Schmid, P.; Kadel, E.E.; Wistuba, I.; Chaft, J.; Rizvi, N.A.; et al. Differential regulation of PD-L1 expression by immune and tumor cells in NSCLC and the response to treatment with atezolizumab (anti–PD-L1). Proc. Natl. Acad. Sci. USA 2018, 115, E10119–E10126. [Google Scholar] [CrossRef] [Green Version]
- Thiem, A.; Hesbacher, S.; Kneitz, H.; di Primio, T.; Heppt, M.V.; Hermanns, H.M.; Goebeler, M.; Meierjohann, S.; Houben, R.; Schrama, D. IFN-gamma-induced PD-L1 expression in melanoma depends on p53 expression. J. Exp. Clin. Cancer Res. 2019, 38, 397. [Google Scholar] [CrossRef] [Green Version]
- Fang, W.; Zhang, J.; Hong, S.; Zhan, J.; Chen, N.; Qin, T.; Tang, Y.; Zhang, Y.; Kang, S.; Zhou, T.; et al. EBV-driven LMP1 and IFN-γ up-regulate PD-L1 in nasopharyngeal carcinoma: Implications for oncotargeted therapy. Oncotarget 2014, 5, 12189–12202. [Google Scholar] [CrossRef]
- Wang, L.W.; Jiang, S.; Gewurz, B.E. Epstein-Barr Virus LMP1-Mediated Oncogenicity. J. Virol. 2017, 91, e01718-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruddy, M.J.; Wong, G.C.; Liu, X.K.; Yamamoto, H.; Kasayama, S.; Kirkwood, K.L.; Gaffen, S.L. Functional Cooperation between Interleukin-17 and Tumor Necrosis Factor-α Is Mediated by CCAAT/Enhancer-binding Protein Family Members. J. Biol. Chem. 2004, 279, 2559–2567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asgarova, A.; Asgarov, K.; Godet, Y.; Peixoto, P.; Nadaradjane, A.; Boyer-Guittaut, M.; Galaine, J.; Guenat, D.; Mougey, V.; Perrard, J.; et al. PD-L1 expression is regulated by both DNA methylation and NF-kB during EMT signaling in non-small cell lung carcinoma. OncoImmunology 2018, 7, e1423170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MaruYama, T. The nuclear IκB family of proteins controls gene regulation and immune homeostasis. Int. Immunopharmacol. 2015, 28, 836–840. [Google Scholar] [CrossRef]
- Thornburg, N.J.; Pathmanathan, R.; Raab-Traub, N. Activation of Nuclear Factor-κB p50 Homodimer/Bcl-3 Complexes in Nasopharyngeal Carcinoma. Cancer Res. 2003, 63, 8293–8301. [Google Scholar]
- Puvvada, S.D.; Funkhouser, W.K.; Greene, K.; Deal, A.; Chu, H.; Baldwin, A.S.; Tepper, J.E.; O’Neil, B.H. NF-ĸB and Bcl-3 Activation Are Prognostic in Metastatic Colorectal Cancer. Oncology 2010, 78, 181–188. [Google Scholar] [CrossRef] [Green Version]
- Maldonado, V.; Melendez-Zajgla, J. Role of Bcl-3 in solid tumors. Mol. Cancer 2011, 10, 152. [Google Scholar] [CrossRef] [Green Version]
- Wakefield, A.; Soukupova, J.; Montagne, A.; Ranger, J.; French, R.; Muller, W.J.; Clarkson, R.W.E. Bcl3 Selectively Promotes Metastasis of ERBB2-Driven Mammary Tumors. Cancer Res. 2013, 73, 745–755. [Google Scholar] [CrossRef] [Green Version]
- Dimitrakopoulos, F.-I.D.; Antonacopoulou, A.G.; Kottorou, A.; Marousi, S.; Koukourikou, I.; Kalofonou, M.; Panagopoulos, N.; Scopa, C.; Dougenis, D.; Papadaki, H.; et al. Variant of BCL3 gene is strongly associated with five-year survival of non-small-cell lung cancer patients. Lung Cancer 2015, 89, 311–319. [Google Scholar] [CrossRef]
- Urban, B.C.; Collard, T.J.; Eagle, C.J.; Southern, S.L.; Greenhough, A.; Hamdollah-Zadeh, M.; Ghosh, A.; Poulsom, R.; Paraskeva, C.; Silver, A.; et al. BCL-3 expression promotes colorectal tumorigenesis through activation of AKT signalling. Gut 2016, 65, 1151–1164. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Wang, W.; Zhao, Q.; Hu, G.; Deng, K.; Liu, Y. BCL3 exerts an oncogenic function by regulating STAT3 in human cervical cancer. OncoTargets 2016, 9, 6619–6629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buerki, C.; Rothgiesser, K.M.; Valovka, T.; Owen, H.R.; Rehrauer, H.; Fey, M.; Lane, W.S.; Hottiger, M.O. Functional relevance of novel p300-mediated lysine 314 and 315 acetylation of RelA/p65. Nucleic Acids Res. 2008, 36, 1665–1680. [Google Scholar] [CrossRef] [PubMed]
- Rothgiesser, K.M.; Fey, M.; Hottiger, M.O. Acetylation of p65 at lysine 314 is important for late NF-κB-dependent gene expression. BMC Genom. 2010, 11, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farahmand, L.; Merikhian, P.; Jalili, N.; Darvishi, B.; Majidzadeh, A.K. Significant Role of MUC1 in Development of Resistance to Currently Existing Anti-cancer Therapeutic Agents. Curr. Cancer Drug Targets 2018, 18, 737–748. [Google Scholar] [CrossRef]
- Ahmad, R.; Raina, D.; Joshi, M.D.; Kawano, T.; Ren, J.; Kharbanda, S.; Kufe, D. MUC1-C Oncoprotein Functions as a Direct Activator of the Nuclear Factor-κB p65 Transcription Factor. Cancer Res. 2009, 69, 7013–7021. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, R.; Raina, D.; Trivedi, V.; Ren, J.; Rajabi, H.; Kharbanda, S.; Kufe, D. MUC1 oncoprotein activates the IkappaB kinase beta complex and constitutive NF-kappaB signalling. Nat. Cell Biol. 2007, 9, 1419–1427. [Google Scholar] [CrossRef]
- Takahashi, H.; Jin, C.; Rajabi, H.; Pitroda, S.; Alam, M.; Ahmad, R.; Raina, D.; Hasegawa, M.; Suzuki, Y.; Tagde, A.; et al. MUC1-C activates the TAK1 inflammatory pathway in colon cancer. Oncogene 2015, 34, 5187–5197. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.-T.; Chen, W.-C.; Chang, Y.-H.; Lin, W.-Y.; Chen, M.-F. The role of PD-L1 in the radiation response and clinical outcome for bladder cancer. Sci. Rep. 2016, 6, 19740. [Google Scholar] [CrossRef] [Green Version]
- Sato, H.; Niimi, A.; Yasuhara, T.; Permata, T.B.M.; Hagiwara, Y.; Isono, M.; Nuryadi, E.; Sekine, R.; Oike, T.; Kakoti, S.; et al. DNA double-strand break repair pathway regulates PD-L1 expression in cancer cells. Nat. Commun. 2017, 8, 1751. [Google Scholar] [CrossRef]
- Permata, T.B.M.; Hagiwara, Y.; Sato, H.; Yasuhara, T.; Oike, T.; Gondhowiardjo, S.; Held, K.D.; Nakano, T.; Shibata, A. Base excision repair regulates PD-L1 expression in cancer cells. Oncogene 2019, 38, 4452–4466. [Google Scholar] [CrossRef]
- Sun, L.L.; Yang, R.Y.; Li, C.W.; Chen, M.K.; Shao, B.; Hsu, J.M.; Chan, L.C.; Yang, Y.; Hsu, J.L.; Lai, Y.J.; et al. Inhibition of ATR downregulates PD-L1 and sensitizes tumor cells to T cell-mediated killing. Am. J. Cancer Res. 2018, 8, 1307–1316. [Google Scholar] [PubMed]
- Vendetti, F.P.; Karukonda, P.; Clump, D.A.; Teo, T.; Lalonde, R.; Nugent, K.; Ballew, M.; Kiesel, B.F.; Beumer, J.H.; Sarkar, S.N.; et al. ATR kinase inhibitor AZD6738 potentiates CD8+ T cell-dependent antitumor activity following radiation. J. Clin. Investig. 2018, 128, 3926–3940. [Google Scholar] [CrossRef] [PubMed]
- Azuma, K.; Ota, K.; Kawahara, A.; Hattori, S.; Iwama, E.; Harada, T.; Matsumoto, K.; Takayama, K.; Takamori, S.; Kage, M.; et al. Association of PD-L1 overexpression with activating EGFR mutations in surgically resected nonsmall-cell lung cancer. Ann. Oncol. 2014, 25, 1935–1940. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Fang, W.; Zhang, Y.; Hong, S.; Kang, S.; Yan, Y.; Chen, N.; Zhan, J.; He, X.; Qin, T.; et al. The association between PD-L1 and EGFR status and the prognostic value of PD-L1 in advanced non-small cell lung cancer patients treated with EGFR-TKIs. Oncotarget 2015, 6. [Google Scholar] [CrossRef] [Green Version]
- D’Incecco, A.; Andreozzi, M.; Ludovini, V.; Rossi, E.; Capodanno, A.; Landi, L.; Tibaldi, C.; Minuti, G.; Salvini, J.; Coppi, E.; et al. PD-1 and PD-L1 expression in molecularly selected non-small-cell lung cancer patients. Br. J. Cancer 2015, 112, 95–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, Z.-Y.; Zhang, J.-T.; Liu, S.-Y.; Su, J.; Zhang, C.; Xie, Z.; Zhou, Q.; Tu, H.-Y.; Xu, C.-R.; Yan, L.-X.; et al. EGFR mutation correlates with uninflamed phenotype and weak immunogenicity, causing impaired response to PD-1 blockade in non-small cell lung cancer. OncoImmunology 2017, 6, e1356145. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Li, G.; Wang, Y.; Wang, Y.; Zhao, S.; Haihong, P.; Zhao, H.; Wang, Y. PD-L1 expression in lung cancer and its correlation with driver mutations: A meta-analysis. Sci. Rep. 2017, 7, 10255. [Google Scholar] [CrossRef] [Green Version]
- Takada, K.; Okamoto, T.; Shoji, F.; Shimokawa, M.; Akamine, T.; Takamori, S.; Katsura, M.; Suzuki, Y.; Fujishita, T.; Toyokawa, G.; et al. Clinical Significance of PD-L1 Protein Expression in Surgically Resected Primary Lung Adenocarcinoma. J. Thorac. Oncol. 2016, 11, 1879–1890. [Google Scholar] [CrossRef] [Green Version]
- Jiang, K.; Sun, Y.; Wang, C.; Ji, J.; Li, Y.; Ye, Y.; Lv, L.; Guo, Y.; Guo, S.; Li, H.; et al. Genome-wide association study identifies two new susceptibility loci for colorectal cancer at 5q23.3 and 17q12 in Han Chinese. Oncotarget 2015, 6, 40327–40336. [Google Scholar] [CrossRef] [Green Version]
- Yao, G.; Zhang, Q.; Doeppner, T.R.; Niu, F.; Li, Q.; Yang, Y.; Kuckelkorn, U.; Hagemann, N.; Li, W.; Hermann, D.M.; et al. LDL suppresses angiogenesis through disruption of the HIF pathway via NF-κB inhibition which is reversed by the proteasome inhibitor BSc2118. Oncotarget 2015, 6, 30251–30262. [Google Scholar] [CrossRef]
- Mak, P.; Li, J.; Samanta, S.; Mercurio, A.M. ERβ regulation of NF-kB activation in prostate cancer is mediated by HIF-1. Oncotarget 2015, 6, 40247–40254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walmsley, S.R.; Print, C.; Farahi, N.; Peyssonnaux, C.; Johnson, R.S.; Cramer, T.; Sobolewski, A.; Condliffe, A.M.; Cowburn, A.S.; Johnson, N.; et al. Hypoxia-induced neutrophil survival is mediated by HIF-1alpha-dependent NF-kappaB activity. J. Exp. Med. 2005, 201, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.; Cheng, J.; Yang, T.; Li, Y.; Zhu, B. EGFR-TKI down-regulates PD-L1 in EGFR mutant NSCLC through inhibiting NF-κB. Biochem. Biophys. Res. Commun. 2015, 463, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Li, C.W.; Lim, S.O.; Xia, W.; Lee, H.H.; Chan, L.C.; Kuo, C.W.; Khoo, K.H.; Chang, S.S.; Cha, J.H.; Kim, T.; et al. Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nat. Commun. 2016, 7, 12632. [Google Scholar] [CrossRef] [Green Version]
- Pan, Y.; Wang, S.; Su, B.; Zhou, F.; Zhang, R.; Xu, T.; Zhang, R.; Leventaki, V.; Drakos, E.; Liu, W.; et al. Stat3 contributes to cancer progression by regulating Jab1/Csn5 expression. Oncogene 2017, 36, 1069–1079. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Yao, Z.; Wang, J.; Zhang, W.; Yang, Y.; Zhang, Y.; Qu, X.; Zhu, Y.; Zou, J.; Peng, S.; et al. Macrophage-derived CCL5 facilitates immune escape of colorectal cancer cells via the p65/STAT3-CSN5-PD-L1 pathway. Cell Death Differ. 2019. [Google Scholar] [CrossRef]
- Lin, Y.; Bai, L.; Chen, W.; Xu, S. The NF-kappaB activation pathways, emerging molecular targets for cancer prevention and therapy. Expert. Opin. Targets 2010, 14, 45–55. [Google Scholar] [CrossRef] [Green Version]
- Shen, H.-M.; Tergaonkar, V. NFκB signaling in carcinogenesis and as a potential molecular target for cancer therapy. Apoptosis 2009, 14, 348–363. [Google Scholar] [CrossRef]
- Greten, F.R.; Arkan, M.C.; Bollrath, J.; Hsu, L.-C.; Goode, J.; Miething, C.; Göktuna, S.I.; Neuenhahn, M.; Fierer, J.; Paxian, S.; et al. NF-κB Is a Negative Regulator of IL-1β Secretion as Revealed by Genetic and Pharmacological Inhibition of IKKβ. Cell 2007, 130, 918–931. [Google Scholar] [CrossRef] [Green Version]
- Gilmore, T.D.; Herscovitch, M. Inhibitors of NF-κB signaling: 785 and counting. Oncogene 2006, 25, 6887–6899. [Google Scholar] [CrossRef] [Green Version]
- Calzado, M.A.; Bacher, S.; Schmitz, M.L. NF-kappaB inhibitors for the treatment of inflammatory diseases and cancer. Curr. Med. Chem. 2007, 14, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Akinleye, A.; Rasool, Z. Immune checkpoint inhibitors of PD-L1 as cancer therapeutics. J. Hematol. Oncol. 2019, 12, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, X.; Wang, J.; Deng, X.; Xiong, F.; Ge, J.; Xiang, B.; Wu, X.; Ma, J.; Zhou, M.; Li, X.; et al. Role of the tumor microenvironment in PD-L1/PD-1-mediated tumor immune escape. Mol. Cancer 2019, 18, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.M.; Chen, D.S. Immune escape to PD-L1/PD-1 blockade: Seven steps to success (or failure). Ann. Oncol. 2016, 27, 1492–1504. [Google Scholar] [CrossRef]
- Zou, W.; Chen, L. Inhibitory B7-family molecules in the tumour microenvironment. Nat. Rev. Immunol. 2008, 8, 467–477. [Google Scholar] [CrossRef]
- Nowicki, T.S.; Hu-Lieskovan, S.; Ribas, A. Mechanisms of Resistance to PD-1 and PD-L1 Blockade. Cancer J. 2018, 24, 47–53. [Google Scholar] [CrossRef]
- Carlsson, J.; Sundqvist, P.; Kosuta, V.; Fält, A.; Giunchi, F.; Fiorentino, M.; Davidsson, S. PD-L1 Expression is Associated With Poor Prognosis in Renal Cell Carcinoma. Appl. Immunohistochem. Mol. Morphol. 2020, 28, 213–220. [Google Scholar] [CrossRef]
- Chang, Y.-L.; Yang, C.-Y.; Huang, Y.-L.; Wu, C.-T.; Yang, P.-C. High PD-L1 expression is associated with stage IV disease and poorer overall survival in 186 cases of small cell lung cancers. Oncotarget 2017, 8, 18021–18030. [Google Scholar] [CrossRef] [Green Version]
- Jobin, C.; Bradham, C.A.; Russo, M.P.; Juma, B.; Narula, A.S.; Brenner, D.A.; Sartor, R.B. Curcumin Blocks Cytokine-Mediated NF-κB Activation and Proinflammatory Gene Expression by Inhibiting Inhibitory Factor I-κB Kinase Activity. J. Immunol. 1999, 163, 3474–3483. [Google Scholar]
- Aggarwal, B.B.; Kumar, A.; Bharti, A.C. Anticancer potential of curcumin: Preclinical and clinical studies. Anticancer Res. 2003, 23, 363–398. [Google Scholar]
- Uhle, S.; Medalia, O.; Waldron, R.; Dumdey, R.; Henklein, P.; Bech-Otschir, D.; Huang, X.; Berse, M.; Sperling, J.; Schade, R.; et al. Protein kinase CK2 and protein kinase D are associated with the COP9 signalosome. EMBO J. 2003, 22, 1302–1312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, Z.; Su, Z.; Han, S.; Huang, J.; Lin, L.; Shuai, X. Dual pH-sensitive nanodrug blocks PD-1 immune checkpoint and uses T cells to deliver NF-kappaB inhibitor for antitumor immunotherapy. Sci. Adv. 2020, 6, eaay7785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shishodia, S.; Koul, D.; Aggarwal, B.B. Cyclooxygenase (COX)-2 inhibitor celecoxib abrogates TNF-induced NF-kappa B activation through inhibition of activation of I kappa B alpha kinase and Akt in human non-small cell lung carcinoma: Correlation with suppression of COX-2 synthesis. J. Immun. 2004, 173, 2011–2022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef] [PubMed]
- Ahn, M.-J.; Sun, J.-M.; Lee, S.-H.; Ahn, J.S.; Park, K. EGFR TKI combination with immunotherapy in non-small cell lung cancer. Expert Opin. Drug Saf. 2017, 16, 465–469. [Google Scholar] [CrossRef] [PubMed]
- Perez-Ruiz, E.; Minute, L.; Otano, I.; Alvarez, M.; Ochoa, M.C.; Belsue, V.; de Andrea, C.; Rodriguez-Ruiz, M.E.; Perez-Gracia, J.L.; Marquez-Rodas, I.; et al. Prophylactic TNF blockade uncouples efficacy and toxicity in dual CTLA-4 and PD-1 immunotherapy. Nature 2019, 569, 428–432. [Google Scholar] [CrossRef]
- Berman, A.Y.; Motechin, R.A.; Wiesenfeld, M.Y.; Holz, M.K. The therapeutic potential of resveratrol: A review of clinical trials. NPJ Precis. Oncol. 2017, 1, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Trung, L.Q.; An, D.T.T. Is Resveratrol a Cancer Immunomodulatory Molecule? Front. Pharmacol. 2018, 9. [Google Scholar] [CrossRef]
- Quoc Trung, L.; Espinoza, J.L.; Takami, A.; Nakao, S. Resveratrol induces cell cycle arrest and apoptosis in malignant NK cells via JAK2/STAT3 pathway inhibition. PLoS ONE 2013, 8, e55183. [Google Scholar] [CrossRef] [Green Version]
- Trung, L.Q.; Espinoza, J.L.; An, D.T.; Viet, N.H.; Shimoda, K.; Nakao, S. Resveratrol selectively induces apoptosis in malignant cells with the JAK2V617F mutation by inhibiting the JAK2 pathway. Mol. Nutr. Food Res. 2015, 59, 2143–2154. [Google Scholar] [CrossRef]
- Wang, W.; Tam, W.F.; Hughes, C.C.W.; Rath, S.; Sen, R. c-Rel Is a Target of Pentoxifylline-Mediated Inhibition of T Lymphocyte Activation. Immunity 1997, 6, 165–174. [Google Scholar] [CrossRef] [Green Version]
- Rotte, A. Combination of CTLA-4 and PD-1 blockers for treatment of cancer. J. Exp. Clin. Cancer Res. 2019, 38, 255. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.C.; Anang, N.-A.A.S.; Sharma, R.; Andrews, M.C.; Reuben, A.; Levine, J.H.; Cogdill, A.P.; Mancuso, J.J.; Wargo, J.A.; Pe’er, D.; et al. Combination anti–CTLA-4 plus anti–PD-1 checkpoint blockade utilizes cellular mechanisms partially distinct from monotherapies. Proc. Natl. Acad. Sci. USA 2019, 116, 22699–22709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2019, 381, 1535–1546. [Google Scholar] [CrossRef] [Green Version]
- Spain, L.; Diem, S.; Larkin, J. Management of toxicities of immune checkpoint inhibitors. Cancer Treat. Rev. 2016, 44, 51–60. [Google Scholar] [CrossRef]
- Bertrand, F.; Montfort, A.; Marcheteau, E.; Imbert, C.; Gilhodes, J.; Filleron, T.; Rochaix, P.; Andrieu-Abadie, N.; Levade, T.; Meyer, N.; et al. TNFα blockade overcomes resistance to anti-PD-1 in experimental melanoma. Nat. Commun. 2017, 8, 2256. [Google Scholar] [CrossRef] [Green Version]
- Postow, M.A.; Hellmann, M.D. Adverse Events Associated with Immune Checkpoint Blockade. N. Engl. J. Med. 2018, 378, 1165. [Google Scholar] [CrossRef]
- Wei, Y.; Zhao, Q.; Gao, Z.; Lao, X.-M.; Lin, W.-M.; Chen, D.-P.; Mu, M.; Huang, C.-X.; Liu, Z.-Y.; Li, B.; et al. The local immune landscape determines tumor PD-L1 heterogeneity and sensitivity to therapy. J. Clin. Investig. 2019, 129, 3347–3360. [Google Scholar] [CrossRef] [Green Version]
- Mir, H.; Kapur, N.; Singh, R.; Sonpavde, G.; Lillard, J.W.; Singh, S. Andrographolide inhibits prostate cancer by targeting cell cycle regulators, CXCR3 and CXCR7 chemokine receptors. Cell Cycle 2016, 15, 819–826. [Google Scholar] [CrossRef]
- Peng, Y.; Wang, Y.; Tang, N.; Sun, D.; Lan, Y.; Yu, Z.; Zhao, X.; Feng, L.; Zhang, B.; Jin, L.; et al. Andrographolide inhibits breast cancer through suppressing COX-2 expression and angiogenesis via inactivation of p300 signaling and VEGF pathway. J. Exp. Clin. Cancer Res. 2018, 37, 248. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Hao, Q.; Cao, W.; Vadgama, J.V.; Wu, Y. Celecoxib in breast cancer prevention and therapy. Cancer Manag. Res. 2018, 10, 4653–4667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuo, C.; Hong, Y.; Qiu, X.; Yang, D.; Liu, N.; Sheng, X.; Zhou, K.; Tang, B.; Xiong, S.; Ma, M.; et al. Celecoxib suppresses proliferation and metastasis of pancreatic cancer cells by down-regulating STAT3 / NF-kB and L1CAM activities. Pancreatology 2018, 18, 328–333. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Fang, M.; Zhang, J.; Wang, J.; Song, Y.; Shi, J.; Li, W.; Wu, G.; Ren, J.; Wang, Z.; et al. Hydrogel dual delivered celecoxib and anti-PD-1 synergistically improve antitumor immunity. Oncoimmunology 2015, 5, e1074374. [Google Scholar] [CrossRef] [PubMed]
- Zelenay, S.; van der Veen, A.G.; Böttcher, J.P.; Snelgrove, K.J.; Rogers, N.; Acton, S.E.; Chakravarty, P.; Girotti, M.R.; Marais, R.; Quezada, S.A.; et al. Cyclooxygenase-Dependent Tumor Growth through Evasion of Immunity. Cell 2015, 162, 1257–1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.A.; Arcila, M.E.; Rekhtman, N.; Sima, C.S.; Zakowski, M.F.; Pao, W.; Kris, M.G.; Miller, V.A.; Ladanyi, M.; Riely, G.J. Analysis of Tumor Specimens at the Time of Acquired Resistance to EGFR-TKI Therapy in 155 Patients with EGFR-Mutant Lung Cancers. Clin. Cancer Res. 2013, 19, 2240–2247. [Google Scholar] [CrossRef] [Green Version]
- Jänne, P.A.; Yang, J.C.; Kim, D.W.; Planchard, D.; Ohe, Y.; Ramalingam, S.S.; Ahn, M.J.; Kim, S.W.; Su, W.C.; Horn, L.; et al. AZD9291 in EGFR inhibitor-resistant non-small-cell lung cancer. N. Engl. J. Med. 2015, 372, 1689–1699. [Google Scholar] [CrossRef] [PubMed]
- Thress, K.S.; Paweletz, C.P.; Felip, E.; Cho, B.C.; Stetson, D.; Dougherty, B.; Lai, Z.; Markovets, A.; Vivancos, A.; Kuang, Y.; et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nat. Med. 2015, 21, 560–562. [Google Scholar] [CrossRef] [Green Version]
- Akbay, E.A.; Koyama, S.; Carretero, J.; Altabef, A.; Tchaicha, J.H.; Christensen, C.L.; Mikse, O.R.; Cherniack, A.D.; Beauchamp, E.M.; Pugh, T.J.; et al. Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer Discov. 2013, 3, 1355–1363. [Google Scholar] [CrossRef] [Green Version]
- Chen, N.; Fang, W.; Zhan, J.; Hong, S.; Tang, Y.; Kang, S.; Zhang, Y.; He, X.; Zhou, T.; Qin, T.; et al. Upregulation of PD-L1 by EGFR Activation Mediates the Immune Escape in EGFR-Driven NSCLC: Implication for Optional Immune Targeted Therapy for NSCLC Patients with EGFR Mutation. J. Thorac. Oncol. 2015, 10, 910–923. [Google Scholar] [CrossRef] [Green Version]
- Gettinger, S.; Rizvi, N.; Chow, L.Q.; Borghaei, H.; Brahmer, J.R.; Juergens, R.; Shepherd, F.A.; Laurie, S.A.; Gerber, D.E.; Goldman, J.; et al. Nivolumab (Anti-Pd-1; Bms-936558, Ono-4538) in Combination with Platinum-Based Doublet Chemotherapy (Pt-Dc) or Erlotinib (Erl) in Advanced Non-Small Cell Lung Cancer (Nsclc). Ann. Oncol. 2014, 25, iv363. [Google Scholar] [CrossRef]
- Ahn, M.J.; Yang, J.; Yu, H.; Saka, H.; Ramalingam, S.; Goto, K.; Kim, S.W.; Yang, L.; Walding, A.; Oxnard, G.R. 136O: Osimertinib combined with durvalumab in EGFR-mutant non-small cell lung cancer: Results from the TATTON phase Ib trial. J. Thorac. Oncol. 2016, 11, S115. [Google Scholar] [CrossRef]
- Kim, S.J.; Asfaha, S.; Dick, F.A. CDK4 Inhibitors Thwart Immunity by Inhibiting Phospho-RB-NF-κB Complexes. Mol. Cell 2019, 73, 1–2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goel, S.; DeCristo, M.J.; Watt, A.C.; BrinJones, H.; Sceneay, J.; Li, B.B.; Khan, N.; Ubellacker, J.M.; Xie, S.; Metzger-Filho, O.; et al. CDK4/6 inhibition triggers anti-tumour immunity. Nature 2017, 548, 471–475. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Bu, X.; Wang, H.; Zhu, Y.; Geng, Y.; Nihira, N.T.; Tan, Y.; Ci, Y.; Wu, F.; Dai, X.; et al. Cyclin D–CDK4 kinase destabilizes PD-L1 via cullin 3–SPOP to control cancer immune surveillance. Nature 2018, 553, 91–95. [Google Scholar] [CrossRef] [Green Version]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Betzler, A.C.; Theodoraki, M.-N.; Schuler, P.J.; Döscher, J.; Laban, S.; Hoffmann, T.K.; Brunner, C. NF-κB and Its Role in Checkpoint Control. Int. J. Mol. Sci. 2020, 21, 3949. https://doi.org/10.3390/ijms21113949
Betzler AC, Theodoraki M-N, Schuler PJ, Döscher J, Laban S, Hoffmann TK, Brunner C. NF-κB and Its Role in Checkpoint Control. International Journal of Molecular Sciences. 2020; 21(11):3949. https://doi.org/10.3390/ijms21113949
Chicago/Turabian StyleBetzler, Annika C., Marie-Nicole Theodoraki, Patrick J. Schuler, Johannes Döscher, Simon Laban, Thomas K. Hoffmann, and Cornelia Brunner. 2020. "NF-κB and Its Role in Checkpoint Control" International Journal of Molecular Sciences 21, no. 11: 3949. https://doi.org/10.3390/ijms21113949