Evaluation of Melanoma (SK-MEL-2) Cell Growth between Three-Dimensional (3D) and Two-Dimensional (2D) Cell Cultures with Fourier Transform Infrared (FTIR) Microspectroscopy



Abstract
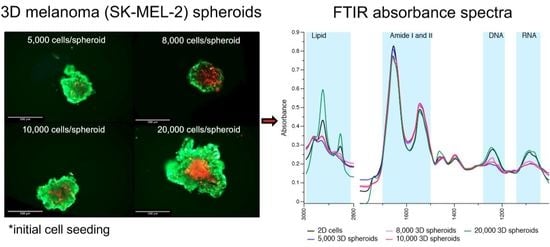
:
1. Introduction
2. Results
2.1. Discrimination of Morphology of 2D and 3D Cells’ Culture
2.2. 3D Cells’ Spheroid Variability
2.3. Principle Component Analyisis (PCA) between Melanoma 2D and All Shperoid Groups
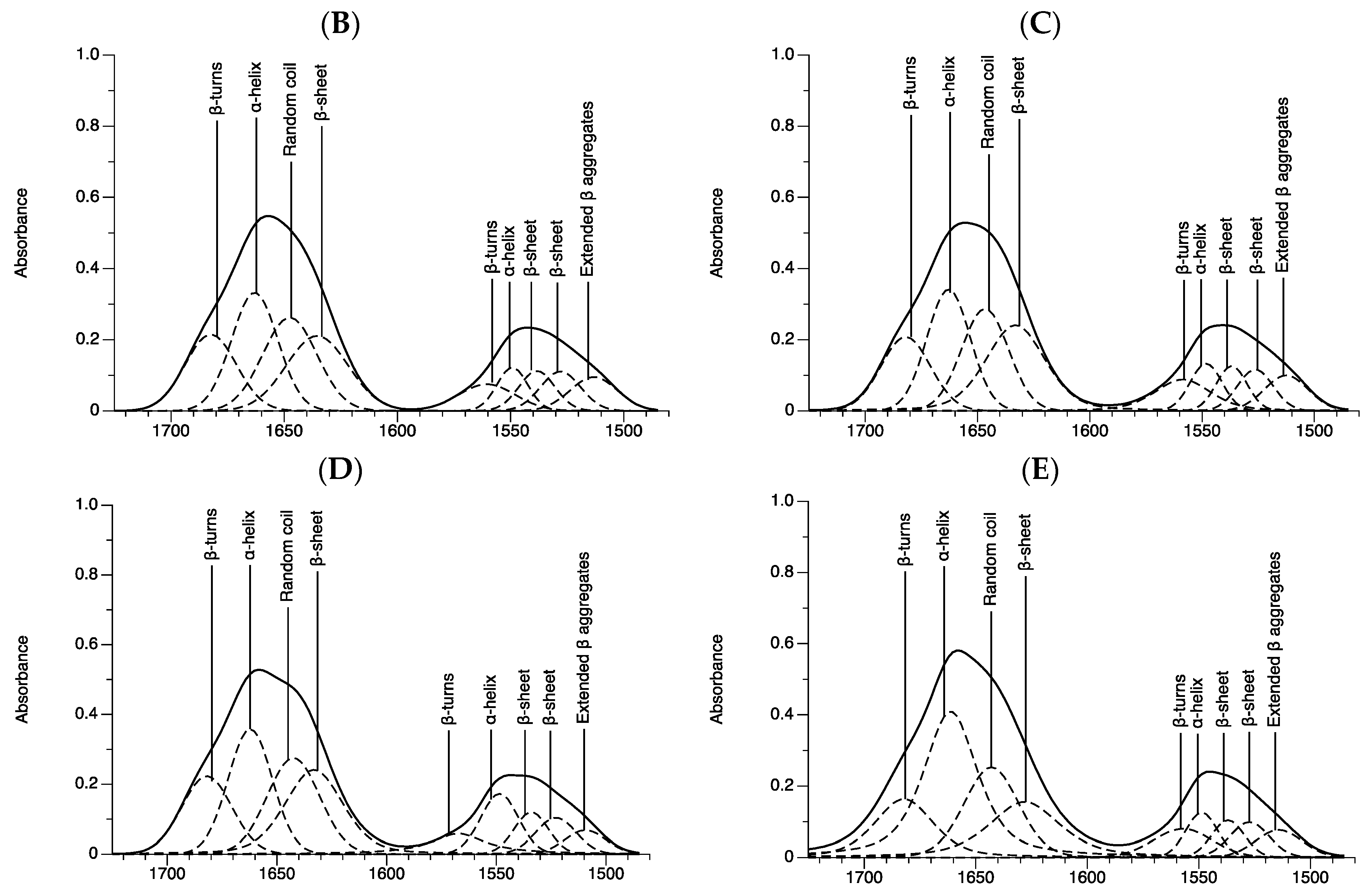
2.4. Protein Regions
3. Discussion
4. Materials and Methods
4.1. Cell Culture Reagents
4.2. Cell Line and Cell Culture
4.3. Spheroid Formation
4.4. Annexin V and Propidium Iodide Staining
- Dint = integrated density of the spheroid
- A = spheroid area
- F = mean fluorescence of background readings
4.5. FTIR Data Acquisition
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA. Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Sheikh, M.S. Melanoma: Molecular pathogenesis and therapeutic management. Mol. Cell. Pharmacol. 2014, 6, 228. [Google Scholar] [PubMed]
- Marconi, A.; Quadri, M.; Saltari, A.; Pincelli, C. Progress in melanoma modeling in vitro. Exp. Dermatol. 2018, 27, 578–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, Y.; Eglen, R.M. Three-dimensional cell cultures in drug discovery and development. Slas Discov. 2017, 22, 456–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edmondson, R.; Broglie, J.J.; Adcock, A.F.; Yang, L. Three-dimensional cell culture systems and their applications in drug discovery and cell-based biosensors. Assay Drug Dev. Technol. 2014, 12, 207–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sant, S.; Johnston, P.A. The production of 3D tumor spheroids for cancer drug discovery. Drug Discov. Today Technol. 2017, 23, 27–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanoni, M.; Piccinini, F.; Arienti, C.; Zamagni, A.; Santi, S.; Polico, R.; Bevilacqua, A.; Tesei, A. 3D tumor spheroid models for in vitro therapeutic screening: A systematic approach to enhance the biological relevance of data obtained. Sci. Rep. 2016, 6, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Smolina, M.; Goormaghtigh, E. Gene expression data and FTIR spectra provide a similar phenotypic description of breast cancer cell lines in 2D and 3D cultures. Analyst 2018, 143, 2520–2530. [Google Scholar] [CrossRef] [PubMed]
- Raza, A.; Colley, H.E.; Baggaley, E.; Sazanovich, I.V.; Green, N.H.; Weinstein, J.A.; Botchway, S.W.; MacNeil, S.; Haycock, J.W. Oxygen mapping of melanoma spheroids using small molecule platinum probe and phosphorescence lifetime imaging microscopy. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kessel, S.; Cribbes, S.; Bonasu, S.; Qiu, J.; Chan, L.L.-Y. Real-time apoptosis and viability high-throughput screening of 3D multicellular tumor spheroids using the Celigo image cytometer. SLAS Discov. Adv. Life Sci. RD 2018, 23, 202–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balan, V.; Mihai, C.-T.; Cojocaru, F.-D.; Uritu, C.-M.; Dodi, G.; Botezat, D.; Gardikiotis, I. Vibrational spectroscopy fingerprinting in medicine: From molecular to clinical practice. Materials 2019, 12, 2884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaccari, L.; Birarda, G.; Businaro, L.; Pacor, S.; Grenci, G. Infrared microspectroscopy of live cells in microfluidic devices (MD-IRMS): Toward a powerful label-free cell-based assay. Anal. Chem. 2012, 84, 4768–4775. [Google Scholar] [CrossRef] [PubMed]
- Ami, D.; Mereghetti, P.; Foli, A.; Tasaki, M.; Milani, P.; Nuvolone, M.; Palladini, G.; Merlini, G.; Lavatelli, F.; Natalello, A. ATR-FTIR spectroscopy supported by multivariate analysis for the characterization of adipose tissue aspirates from patients affected by systemic amyloidosis. Anal. Chem. 2019, 91, 2894–2900. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Beaumont, K.A.; Otte, N.J.; Font, J.; Bailey, C.G.; van Geldermalsen, M.; Sharp, D.M.; Tiffen, J.C.; Ryan, R.M.; Jormakka, M.; et al. Targeting glutamine transport to suppress melanoma cell growth. Int. J. Cancer 2014, 135, 1060–1071. [Google Scholar] [CrossRef] [PubMed]
- Shannan, B.; Chen, Q.; Watters, A.; Perego, M.; Krepler, C.; Thombre, R.; Li, L.; Rajan, G.; Peterson, S.; Gimotty, P.A.; et al. Enhancing the evaluation of PI3K inhibitors through 3D melanoma models. Pigment Cell Melanoma Res. 2016, 29, 317–328. [Google Scholar] [CrossRef] [PubMed]
- Smalley, K.S.M.; Haass, N.K.; Brafford, P.A.; Lioni, M.; Flaherty, K.T.; Herlyn, M. Multiple signaling pathways must be targeted to overcome drug resistance in cell lines derived from melanoma metastases. Mol. Cancer. Ther. 2006, 5, 1136–1144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Junhom, C.; Weerapreeyakul, N.; Tanthanuch, W.; Thumanu, K. FTIR microspectroscopy defines early drug resistant human hepatocellular carcinoma (HepG2) cells. Exp. Cell Res. 2016, 340, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Giorgini, E.; Sabbatini, S.; Rocchetti, R.; Notarstefano, V.; Rubini, C.; Conti, C.; Orilisi, G.; Mitri, E.; Bedolla, D.E.; Vaccari, L. In vitro FTIR microspectroscopy analysis of primary oral squamous carcinoma cells treated with cisplatin and 5-fluorouracil: A new spectroscopic approach for studying the drug–cell interaction. Analyst 2018, 143, 3317–3326. [Google Scholar] [CrossRef] [PubMed]
- Vidavsky, N.; Kunitake, J.A.M.R.; Diaz-Rubio, M.E.; Chiou, A.E.; Loh, H.-C.; Zhang, S.; Masic, A.; Fischbach, C.; Estroff, L.A. Mapping and profiling lipid distribution in a 3D model of breast cancer progression. ACS Cent. Sci. 2019, 5, 768–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruoß, M.; Kieber, V.; Rebholz, S.; Linnemann, C.; Rinderknecht, H.; Häussling, V.; Häcker, M.; Olde Damink, L.H.H.; Ehnert, S.; Nussler, A.K. Cell-type-specific quantification of a scaffold-based 3D liver co-culture. Methods Protoc. 2020, 3, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Notarstefano, V.; Sabbatini, S.; Conti, C.; Pisani, M.; Astolfi, P.; Pro, C.; Rubini, C.; Vaccari, L.; Giorgini, E. Investigation of human pancreatic cancer tissues by Fourier Transform infrared hyperspectral imaging. J. Biophotonics 2020, 13. [Google Scholar] [CrossRef] [PubMed]
- Goormaghtigh, E.; Raussens, V.; Ruysschaert, J.-M. Attenuated total reflection infrared spectroscopy of proteins and lipids in biological membranes. Biochim. Biophys. Acta 1999, 1422, 105–185. [Google Scholar] [CrossRef]
- Yang, W.; Xiao, X.; Tan, J.; Cai, Q. In situ evaluation of breast cancer cell growth with 3D ATR-FTIR spectroscopy. Vib. Spectrosc. 2009, 49, 64–67. [Google Scholar] [CrossRef]
- Machana, S.; Weerapreeyakul, N.; Barusrux, S.; Thumanu, K.; Tanthanuch, W. FTIR microspectroscopy discriminates anticancer action on human leukemic cells by extracts of Pinus kesiya; Cratoxylum formosum ssp. pruniflorum and melphalan. Talanta 2012, 93, 371–382. [Google Scholar] [CrossRef] [PubMed]
- Kong, J.; Yu, S. Fourier transform infrared spectroscopic analysis of protein secondary structures. Acta Biochim. Biophys. Sin. 2007, 39, 549–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siriwarin, B.; Weerapreeyakul, N.; Tanthanuch, W.; Thumanu, K. Biomolecular changes and DNA targeting effect of sesamol in human lung adenocarcinoma (SK-LU-1) cells by FTIR microscopy. Asian Pac. J. Trop. Biomed. 2018, 8, 377. [Google Scholar] [CrossRef]
- Han, Y.; Han, L.; Yao, Y.; Li, Y.; Liu, X. Key factors in FTIR spectroscopic analysis of DNA: The sampling technique, pretreatment temperature and sample concentration. Anal. Methods 2018, 10, 2436–2443. [Google Scholar] [CrossRef]
- Ribas, A.; Lawrence, D.; Atkinson, V.; Agarwal, S.; Miller, W.H.; Carlino, M.S.; Fisher, R.; Long, G.V.; Hodi, F.S.; Tsoi, J.; et al. Combined BRAF and MEK inhibition with PD-1 blockade immunotherapy in BRAF -mutant melanoma. Nat. Med. 2019, 25, 936. [Google Scholar] [CrossRef] [PubMed]
- Vörsmann, H.; Groeber, F.; Walles, H.; Busch, S.; Beissert, S.; Walczak, H.; Kulms, D. Development of a human three-dimensional organotypic skin-melanoma spheroid model for in vitro drug testing. Cell Death Dis. 2013, 4, e719. [Google Scholar] [CrossRef] [PubMed]
- Wufuer, M.; Lee, G.; Hur, W.; Jeon, B.; Kim, B.J.; Choi, T.H.; Lee, S. Skin-on-a-chip model simulating inflammation, edema and drug-based treatment. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, M.-Z.; Han, R.-R.; Qiu, G.-Z.; Ju, X.-C.; Lou, G.; Jin, W.-L. Organoids: An intermediate modeling platform in precision oncology. Cancer Lett. 2018, 414, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Spheroids Rapidly Becoming a Preferred 3D Cell Culture Format. Available online: https://www.ddw-online.com/therapeutics/p316729-spheroids-rapidly-becoming-a-preferred-3d-cell-culture-format.html (accessed on 22 February 2020).
- Langhans, S.A. Three-dimensional in vitro cell culture models in drug discovery and drug repositioning. Front. Pharmacol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Antoni, D.; Burckel, H.; Josset, E.; Noel, G. Three-dimensional cell culture: A breakthrough in vivo. Int. J. Mol. Sci. 2015, 16, 5517–5527. [Google Scholar] [CrossRef] [PubMed]
- Beaumont, K.A.; Anfosso, A.; Ahmed, F.; Weninger, W.; Haass, N.K. Imaging- and flow cytometry-based analysis of cell position and the cell cycle in 3D melanoma spheroids. J. Vis. Exp. JoVE 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Repetto, G.; del Peso, A.; Zurita, J.L. Neutral red uptake assay for the estimation of cell viability/cytotoxicity. Nat. Protoc. 2008, 3, 1125–1131. [Google Scholar] [CrossRef] [PubMed]
- Ho, W.Y.; Yeap, S.K.; Ho, C.L.; Rahim, R.A.; Alitheen, N.B. Development of multicellular tumor spheroid (MCTS) culture from breast cancer cell and a high throughput screening method using the MTT assay. PLoS ONE 2012, 7, e44640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riedl, A.; Schlederer, M.; Pudelko, K.; Stadler, M.; Walter, S.; Unterleuthner, D.; Unger, C.; Kramer, N.; Hengstschläger, M.; Kenner, L.; et al. Comparison of cancer cells in 2D vs 3D culture reveals differences in AKT–mTOR–S6K signaling and drug responses. J. Cell Sci. 2017, 130, 203–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Souza, A.G.; Silva, I.B.B.; Campos-Fernandez, E.; Barcelos, L.S.; Souza, J.B.; Marangoni, K.; Goulart, L.R.; Alonso-Goulart, V. Comparative assay of 2D and 3D cell culture models: Proliferation, gene expression and anticancer drug response. Curr. Pharm. Des. 2018, 24, 1689–1694. [Google Scholar] [CrossRef] [PubMed]
- Kapałczyńska, M.; Kolenda, T.; Przybyła, W.; Zajączkowska, M.; Teresiak, A.; Filas, V.; Ibbs, M.; Bliźniak, R.; Łuczewski, Ł.; Lamperska, K. 2D and 3D cell cultures – a comparison of different types of cancer cell cultures. Arch. Med. Sci. AMS 2018, 14, 910–919. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, B.D.; Grashoff, C.; Schwartz, M.A. Dynamic molecular processes mediate cellular mechanotransduction. Nature 2011, 475, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.L.A.; Altharawi, A.; Fale, P.; Song, C.L.; Kazarian, S.G.; Cinque, G.; Untereiner, V.; Sockalingum, G.D. Transmission Fourier transform infrared spectroscopic imaging, mapping, and synchrotron scanning microscopy with zinc sulfide hemispheres on living mammalian cells at sub-cellular resolution. Appl. Spectrosc. 2020, 74, 544–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lasch, P.; Naumann, D. Spatial resolution in infrared microspectroscopic imaging of tissues. BBA Biomembr. 2006, 1758, 814–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, K.L.A.; Kazarian, S.G.; Mavraki, A.; Williams, D.R. Fourier transform infrared imaging of human hair with a high spatial resolution without the use of a synchrotron. Appl. Spectrosc. 2005, 59, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Sandt, C.; Frederick, J.; Dumas, P. Profiling pluripotent stem cells and organelles using synchrotron radiation infrared microspectroscopy. J. Biophoton. 2013, 6, 60–72. [Google Scholar] [CrossRef] [PubMed]
- Diem, M.; Romeo, M.; Matthäus, C.; Miljkovic, M.; Miller, L.; Lasch, P. Comparison of Fourier transform infrared (FTIR) spectra of individual cells acquired using synchrotron and conventional sources. Infrared Phys. Technol. 2004, 45, 331–338. [Google Scholar] [CrossRef]
- Baker, M.J.; Trevisan, J.; Bassan, P.; Bhargava, R.; Butler, H.J.; Dorling, K.M.; Fielden, P.R.; Fogarty, S.W.; Fullwood, N.J.; Heys, K.A.; et al. Using Fourier transform IR spectroscopy to analyze biological materials. Nat. Protoc. 2014, 9, 1771–1791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smolina, M.; Goormaghtigh, E. Infrared imaging of MDA-MB-231 breast cancer cell line phenotypes in 2D and 3D cultures. Analyst 2015, 140, 2336–2343. [Google Scholar] [CrossRef] [PubMed]
- Gelfand, P.; Smith, R.J.; Stavitski, E.; Borchelt, D.R.; Miller, L.M. Characterization of protein structural changes in living cells using time-lapsed FTIR imaging. Anal. Chem. 2015, 87, 6025–6031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, K.L.A.; Kazarian, S.G. Correcting the effect of refraction and dispersion of light in FT-IR spectroscopic imaging in transmission through thick infrared windows. Anal. Chem. 2013, 85, 1029–1036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sroka-Bartnicka, A.; Kimber, J.A.; Borkowski, L.; Pawlowska, M.; Polkowska, I.; Kalisz, G.; Belcarz, A.; Jozwiak, K.; Ginalska, G.; Kazarian, S.G. The biocompatibility of carbon hydroxyapatite/β-glucan composite for bone tissue engineering studied with Raman and FTIR spectroscopic imaging. Anal. Bioanal. Chem. 2015, 407, 7775–7785. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Quaroni, L.; Casson, A.G. Fourier transform infrared (FTIR) spectromicroscopic characterization of stem-like cell populations in human esophageal normal and adenocarcinoma cell lines. Analyst 2010, 135, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Vazquez-Zapien, G.J.; Mata-Miranda, M.M.; Sanchez-Monroy, V.; Delgado-Macuil, R.J.; Perez-Ishiwara, D.G.; Rojas-Lopez, M. FTIR spectroscopic and molecular analysis during differentiation of pluripotent stem cells to pancreatic cells. Stem Cells Int. 2016, 2016, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Junhom, C.; Weerapreeyakul, N.; Tanthanuch, W.; Thumanu, K. Partial least squares regression and Fourier transform infrared (FTIR) microspectroscopy for prediction of resistance in hepatocellular carcinoma HepG2 cells. Exp. Cell Res. 2017, 351, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Ansari, N.; Müller, S.; Stelzer, E.H.K.; Pampaloni, F. Quantitative 3D cell-based assay performed with cellular spheroids and fluorescence microscopy. In Methods in Cell Biology; Elsevier: Amsterdam, The Netherlands, 2013; Volume 113, pp. 295–309. [Google Scholar]
- Pocasap, P.; Weerapreeyakul, N.; Thumanu, K. Structures of isothiocyanates attributed to reactive oxygen species generation and microtubule depolymerization in HepG2 cells. Biomed. Pharmacother. 2018, 101, 698–709. [Google Scholar] [CrossRef] [PubMed]
- Lorthongpanich, C.; Thumanu, K.; Tangkiettrakul, K.; Jiamvoraphong, N.; Laowtammathron, C.; Damkham, N.; U-pratya, Y.; Issaragrisil, S. YAP as a key regulator of adipo-osteogenic differentiation in human MSCs. Stem Cell Res. Ther. 2019, 10, 402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luanpitpong, S.; Janan, M.; Thumanu, K.; Poohadsuan, J.; Rodboon, N.; Klaihmon, P.; Issaragrisil, S. Deciphering the elevated lipid via CD36 in mantle cell lymphoma with bortezomib resistance using synchrotron-based Fourier transform infrared spectroscopy of single cells. Cancers 2019, 11, 576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goormaghtigh, E.; Ruysschaert, J.-M.; Raussens, V. Evaluation of the information content in infrared spectra for protein secondary structure determination. Biophys. J. 2006, 90, 2946–2957. [Google Scholar] [CrossRef] [PubMed] [Green Version]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Initial Cell Numbers | Volume (mm3) (n = 3) | Annexin V Intensity (× 107 CTCF 1) (n = 2) | Propidium Iodide Intensity (× 107 CTCF) (n = 2) |
|---|---|---|---|
| 5000 | 40.3 ± 5.4 a | 1.0 ± 0.2 a | 0.7 ± 0.0 a |
| 8000 | 68.8 ± 2.7 b | 0.8 ± 0.1 a | 1.5 ± 0.4 a |
| 10,000 | 86.3 ± 1.7 c | 2.5 ± 0.0 a | 1.4 ± 0.1 a |
| 20,000 | 97.7 ± 2.0 d | 2.3 ± 0.4 a | 4.0 ± 0.5 a |
| Group | Amide I | Amide II | |||||||
|---|---|---|---|---|---|---|---|---|---|
| β-Turns | α-Helix | Random Coil | β-Sheet | β-Turns | α-Helix | β-Sheet | β-Sheet | Extended β Aggregates | |
| Peak position (cm−1) | |||||||||
| 2D | 1681.5 ± 1.8 | 1662.0 ± 1.3 a,b | 1647.4 ± 1.1 b,c | 1631.8 ± 0.3 a,b | 1560.1 ± 2.6 a,b,c | 1549.7 ± 1.1 b | 1539.2 ± 1.2 b | 1528.6 ± 1.7 b | 1514.7 ± 1.2 b |
| 5000 | 1682.4 ± 0.2 | 1663.4 ± 0.6 b | 1648.4 ± 1.0 c | 1634.6 ± 0.8 b | 1560.1 ± 0.7 b,c | 1549.1 ± 0.3 a,b | 1538.0 ± 0.3 a,b | 1527.4 ± 0.5 a,b | 1513.1 ± 0.2 a,b |
| 8000 | 1681.9 ± 0.1 | 1662.7 ± 0.2 a,b | 1646.5 ± 0.1 b,c | 1633.1 ± 0.1 a,b | 1558.1 ± 0.5 a,b | 1548.1 ± 0.1 a,b | 1537.1 ± 0.3 a | 1526.7 ± 0.4 a,b | 1512.9 ± 0.3 a,b |
| 10,000 | 1682.6 ± 0.8 | 1662.2 ± 0.1 a | 1642.1 ± 0.4 a | 1631.6 ± 1.9 a,b | 1566.4 ± 0.8 c | 1549.4 ± 1.0 a,b | 1535.5 ± 1.8 a | 1524.0 ± 1.8 a | 1509.2 ± 0.9 a |
| 20,000 | 1681.8 ± 0.3 | 1660.9 ± 0.3 a | 1643.1 ± 0.6 b | 1627.6 ± 1.1 a | 1556.2 ± 0.9 a | 1548.3 ± 0.3 b | 1536.3 ± 0.6 a | 1526.3 ± 1.2 a,b | 1513.3 ± 1.1 a,b |
| Integral area (%) | |||||||||
| 2D | 13.2 ± 2.0 | 20.4 ± 0.4 a | 20.7 ± 0.3 b | 13.7 ± 1.8 a,b | 4.7 ± 0.6 a,b | 7.7 ± 0.1 a | 7.3 ± 0.2 | 6.8 ± 0.1 | 5.5 ± 0.3 c |
| 5000 | 13.3 ± 0.7 | 21.0 ± 0.8 a,b | 17.7 ± 0.5 a,b | 14.7 ± 1.0 b | 5.0 ± 0.1 a,b | 7.9 ± 0.2 a | 7.4 ± 0.1 | 7.1 ± 0.1 | 5.9 ± 0.3 c |
| 8000 | 13.0 ± 0.3 | 21.3 ± 0.4 a,b | 17.3 ± 0.2 a | 14.3 ± 0.3 b | 5.3 ± 0.1 b | 7.9 ± 0.2 a,b | 7.5 ± 0.1 | 7.1 ± 0.0 | 6.2 ± 0.1 c |
| 10,000 | 14.1 ± 0.7 | 25.1 ± 1.9 b,c | 18.4 ± 0.6 a,b | 9.7 ± 1.0 a | 4.1 ± 0.3 a | 9.9 ± 1.4 b | 7.5 ± 0.2 | 7.3 ± 0.5 | 3.9 ± 0.7 a |
| 20,000 | 11.6 ± 0.4 | 27.1 ± 0.9 c | 18.0 ± 0.8 a,b | 10.6 ± 1.3 a,b | 5.5 ± 0.1 c | 8.5 ± 0.2 a,b | 7.2 ± 0.2 | 6.6 ± 0.2 | 4.9 ± 0.4 b |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Srisongkram, T.; Weerapreeyakul, N.; Thumanu, K. Evaluation of Melanoma (SK-MEL-2) Cell Growth between Three-Dimensional (3D) and Two-Dimensional (2D) Cell Cultures with Fourier Transform Infrared (FTIR) Microspectroscopy. Int. J. Mol. Sci. 2020, 21, 4141. https://doi.org/10.3390/ijms21114141
Srisongkram T, Weerapreeyakul N, Thumanu K. Evaluation of Melanoma (SK-MEL-2) Cell Growth between Three-Dimensional (3D) and Two-Dimensional (2D) Cell Cultures with Fourier Transform Infrared (FTIR) Microspectroscopy. International Journal of Molecular Sciences. 2020; 21(11):4141. https://doi.org/10.3390/ijms21114141
Chicago/Turabian StyleSrisongkram, Tarapong, Natthida Weerapreeyakul, and Kanjana Thumanu. 2020. "Evaluation of Melanoma (SK-MEL-2) Cell Growth between Three-Dimensional (3D) and Two-Dimensional (2D) Cell Cultures with Fourier Transform Infrared (FTIR) Microspectroscopy" International Journal of Molecular Sciences 21, no. 11: 4141. https://doi.org/10.3390/ijms21114141