Influence of the First Chromophore-Forming Residue on Photobleaching and Oxidative Photoconversion of EGFP and EYFP

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Results
2.1. Mutants General Description (Spectral Characteristics)
2.2. Photostability
2.3. Redding
2.4. Lifetimes
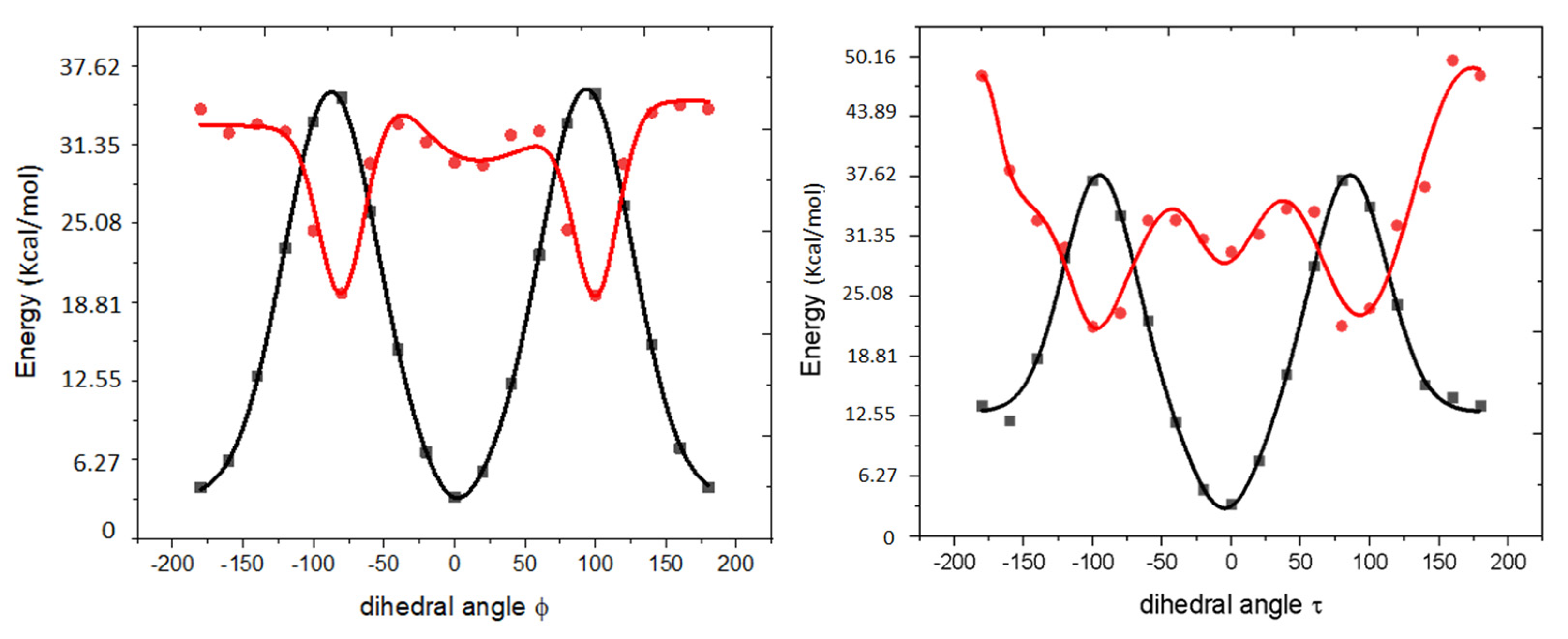
2.5. Computational Results
- Quantum-chemical calculations of the isolated model chromophores (structures, excitation energies, and oscillator strengths);
- Molecular dynamics simulations of the model proteins in the ground and electronically excited states;
- Hybrid QM/MM (quantum mechanics/molecular mechanics) calculations of the spectral properties of the model proteins (excitation energies and oscillator strengths for the structures taken from the ground-state molecular dynamics simulations).
3. Discussion
- -
- EGFP-T65G has a larger extinction coefficient than EGFP because of its larger oscillator strength;
- -
- Because of the faster radiationless relaxation, EGFP-T65G has lower FQY than EGFP, as per Equation (5); likewise, EYFP has lower FQY than EYFP-G65T;
- -
- Having glycine in position 65 leads to faster radiationless relaxation (shorter half-life of A), thus suppressing the bleaching and leading to an increased photostability;
- -
- Larger FQY in EYFP relative to EGFP-T65G arises due to the suppression of torsional motions by the π-stacking interactions, which is reflected in longer radiationless half-life.
4. Materials and Methods
4.1. Spectroscopy and Fluorescence Brightness Evaluation
4.2. Microscopy
4.3. Protein Expression and Purification
4.4. Site-Directed Mutagenesis
4.5. Fluorescence Lifetime Imaging Microscopy of the Purified Proteins upon Single-Photon Excitation
4.6. Computational Details
4.6.1. Protonation State and Crystal Structures
4.6.2. Molecular Dynamics Setup
- Minimization for 2000 steps with 2 fs time step.
- Equilibration of the solvent (keeping the protein frozen) for 500 ps with 1 fs time step.
- Equilibration of the system for 2 ns (with 1 fs time step) with periodic boundary condition (PBC) under the isobaric–isothermal NPT ensemble.
- Production run for 2 ns with 1 fs time step in an NPT ensemble.
4.6.3. QM/MM Setup
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| avGFP | Aequorea victoria green fluorescent protein |
| EC | Extinction coefficient |
| EGFP | Enhanced green fluorescent protein |
| ET | Electron transfer |
| EYFP | Enhanced yellow fluorescent protein |
| FL | Fluorescence lifetime |
| FP | Fluorescent protein |
| FQY | Fluorescence quantum yield |
| GFP | Green fluorescent protein |
| GYG | Glycine-tyrosine-glycine |
| PES | Potential energy surface |
| PB | Phosphate buffer |
| PBS | Phosphate buffered saline |
| QY | Quantum yield |
| SYG | Serine-tyrosine-glycine |
| TYG | Threonine-tyrosine-glycine |
| QM/MM | Quantum mechanics/molecular mechanics |
References
- Cormack, B.P.; Valdivia, R.H.; Falkow, S. FACS-optimized mutants of the green fluorescent protein (GFP). Gene 1996, 173, 33–38. [Google Scholar] [CrossRef]
- Ormo, M.; Cubitt, A.B.; Kallio, K.; Gross, L.A.; Tsien, R.Y.; Remington, S.J. Crystal Structure of the Aequorea victoria Green Fluorescent Protein. Science 1996, 273, 1392–1395. [Google Scholar] [CrossRef] [PubMed]
- Barondeau, D.P.; Kassmann, C.J.; Tainer, J.A.; Getzoff, E.D. Understanding GFP posttranslational chemistry: Structures of designed variants that achieve backbone fragmentation, hydrolysis, and decarboxylation. J. Am. Chem. Soc. 2006, 128, 4685–4693. [Google Scholar] [CrossRef] [PubMed]
- Pletneva, N.V.; Pletnev, V.Z.; Lukyanov, K.A.; Gurskaya, N.G.; Goryacheva, E.A.; Martynov, V.I.; Wlodawer, A.; Dauter, Z.; Pletnev, S. Structural evidence for a dehydrated intermediate in green fluorescent protein chromophore biosynthesis. J. Biol. Chem. 2010, 285, 15978–15984. [Google Scholar] [CrossRef] [PubMed]
- Grigorenko, B.L.; Kryloy, A.I.; Nemukhin, A.V. Molecular modeling clarifies the mechanism of chromophore maturation in the green fluorescent protein. J. Am. Chem. Soc. 2017, 139, 10239–10249. [Google Scholar] [CrossRef] [PubMed]
- Chudakov, D.M.; Matz, M.V.; Lukyanov, S.; Lukyanov, K.A. Fluorescent proteins and their applications in imaging living cells and tissues. Physiol. Rev. 2010, 90, 1103–1163. [Google Scholar] [CrossRef] [PubMed]
- Acharya, A.; Bogdanov, A.M.; Grigorenko, B.L.; Bravaya, K.B.; Nemukhin, A.V.; Lukyanov, K.A.; Krylov, A.I. Photoinduced Chemistry in Fluorescent Proteins: Curse or Blessing? Chem. Rev. 2017, 25, 758–795. [Google Scholar] [CrossRef]
- Tsien, R.Y. The green fluorescent protein. Annu. Rev. Biochem. 1998, 67, 509–544. [Google Scholar] [CrossRef] [PubMed]
- Heim, R.; Cubitt, A.B.; Tsien, R.Y. Improved green fluorescence. Nature 1995, 373, 663–664. [Google Scholar] [CrossRef] [PubMed]
- Delagrave, S.; Hawtin, R.E.; Silva, C.M.; Yang, M.M.; Youvan, D.C. Red-shifted excitation mutants of the green fluorescent protein. Biotechnology 1995, 13, 151–154. [Google Scholar] [CrossRef]
- Royant, A.; Noirclerc-Savoye, M. Stabilizing role of glutamic acid 222 in the structure of Enhanced Green Fluorescent Protein. J. Struct. Biol. 2011, 174, 385–390. [Google Scholar] [CrossRef] [PubMed]
- Arpino, J.A.J.; Rizkallah, P.J.; Jones, D.D. Crystal Structure of Enhanced Green Fluorescent Protein to 1.35 Å Resolution Reveals Alternative Conformations for Glu222. PLoS ONE 2012, 7, e47032. [Google Scholar] [CrossRef] [PubMed]
- Barondeau, D.P.; Kassmann, C.J.; Tainer, J.A.; Getzoff, E.D. The Case of the Missing Ring Radical Cleavage of a Carbon−Carbon Bond and Implications for GFP Chromophore Biosynthesis. J. Am. Chem. Soc. 2007, 129, 3118–3126. [Google Scholar] [CrossRef] [PubMed]
- Wachter, R.M.; Elsliger, M.A.; Kallio, K.; Hanson, G.T.; Remington, S.J. Structural basis of spectral shifts in the yellow-emission variants of green fluorescent protein. Structure 1998, 6, 1267–1277. [Google Scholar] [CrossRef] [Green Version]
- Wachter, R.M.; Remington, S.J. Sensitivity of the yellow variant of green fluorescent protein to halides and nitrate. Curr. Biol. 1999, 9, 628–629. [Google Scholar] [CrossRef]
- Kremers, G.J.; Goedhart, J.; Van Den Heuvel, D.J.; Gerritsen, H.C.; Gadella, T.W.J. Improved green and blue fluorescent proteins for expression in bacteria and mammalian cells. Biochemistry 2007, 46, 3775–3783. [Google Scholar] [CrossRef]
- Mamontova, A.V.; Solovyev, I.D.; Savitsky, A.P.; Shakhov, A.; Lukyanov, K.A.; Bogdanov, A.M. Bright GFP with subnanosecond fluorescence lifetime. Sci. Rep. 2018, 8, 1–5. [Google Scholar] [CrossRef]
- Bogdanov, A.M.; Mishin, A.S.; Yampolsky, I.V.; Belousov, V.V.; Chudakov, D.M.; Subach, F.V.; Verkhusha, V.V.; Lukyanov, S.; Lukyanov, K.A. Green fluorescent proteins are light-induced electron donors. Nat. Chem. Biol. 2009, 5, 459–461. [Google Scholar] [CrossRef]
- Bogdanov, A.M.; Acharya, A.; Titelmayer, A.V.; Mamontova, A.V.; Bravaya, K.B.; Kolomeisky, A.B.; Lukyanov, K.A.; Krylov, A.I. Turning on and off Photoinduced Electron Transfer in Fluorescent Proteins by π-Stacking, Halide Binding, and Tyr145 Mutations. J. Am. Chem. Soc. 2016, 138, 4807–4817. [Google Scholar] [CrossRef]
- Sarkisyan, K.S.; Goryashchenko, A.S.; Lidsky, P.V.; Gorbachev, D.A.; Bozhanova, N.G.; Gorokhovatsky, A.Y.; Pereverzeva, A.R.; Ryumina, A.P.; Zherdeva, V.V.; Savitsky, A.P.; et al. Green Fluorescent Protein with Anionic Tryptophan-Based Chromophore and Long Fluorescence Lifetime. Biophys. J. 2015, 109, 380–389. [Google Scholar] [CrossRef] [Green Version]
- Herman, P.; Holoubek, A.; Brodska, B. Lifetime-based photoconversion of EGFP as a tool for FLIM. Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 266–277. [Google Scholar] [CrossRef] [PubMed]
- Suhling, K.; Siegel, J.; Phillips, D.; French, P.M.W.; Le, S.; Webb, S.E.D.; Davis, D.M. Imaging the Environment of Green Fluorescent Protein. Biophys. J. 2002, 83, 3589–3595. [Google Scholar] [CrossRef]
- Nakabayashi, T.; Ohta, N. Sensing of Intracellular Environments by Fluorescence Lifetime Imaging of Exogenous Fluorophores. Anal. Sci. 2015, 31, 275–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arosio, D.; Garau, G.; Ricci, F.; Marchetti, L.; Bizzarri, R.; Nifosì, R.; Beltram, F. Spectroscopic and structural study of proton and halide ion cooperative binding to GFP. Biophys. J. 2007, 93, 232–244. [Google Scholar] [CrossRef]
- Jayaraman, S.; Haggie, P.; Wachter, R.M.; Remington, S.J.; Verkman, A.S. Mechanism and cellular applications of a green fluorescent protein-based halide sensor. J. Biol. Chem. 2000, 275, 6047–6050. [Google Scholar] [CrossRef]
- Cheng, C.W.; Huang, G.; Hsu, H.Y.; Prabhakar, C.; Lee, Y.P.; Diau, E.W.; Yang, J.S. Effects of hydrogen bonding on internal conversion of GFP-like chromophores. II. The meta-amino systems. J. Phys. Chem. B 2013, 117, 2705–2716. [Google Scholar] [CrossRef]
- Bas, D.C.; Rogers, D.M.; Jensen, J.H. Very fast prediction and rationalization of pKa values for protein–ligand complexes. Proteins Struct. Funct. Bioinform. 2008, 73, 765–783. [Google Scholar] [CrossRef]
- Olsson, M.H.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent treatment of internal and surface residues in empirical pKa predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef]
- Dolinsky, T.J.; Nielsen, J.E.; McCammon, J.A.; Baker, N.A. PDB2PQR: An automated pipeline for the setup of Poisson–Boltzmann electrostatics calculations. Nucleic Acids Res. 2004, 32, W665–W667. [Google Scholar] [CrossRef]
- Gosnell, T.R. Fundamentals of Spectroscopy and Laser Physics. Camb. Univ. Press 2002, 3–37. [Google Scholar]
- Hand, D.B. The refractivity of protein solutions. J. Biol. Chem. 1935, 108, 703–707. [Google Scholar]
- Hipp, N.J. Refractive Indices of Amino Acids, Proteins, and Related Substances. Adv. Chem. 1964, 44, 54–66. [Google Scholar]
- Polyakov, I.V.; Grigorenko, B.L.; Epifanovsky, E.M.; Krylov, A.I.; Nemukhin, A.V. Potential energy landscape of the electronic states of the GFP chromophore in different protonation forms: Electronic transition energies and conical intersections. J. Chem. Theory Comput. 2010, 6, 2377–2387. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.E.; Negri, F.; Olivucci, M. Origin, nature, and fate of the fluorescent state of the green fluorescent protein chromophore at the CASPT2//CASSCF resolution. J. Am. Chem. Soc. 2004, 126, 5452–5464. [Google Scholar] [CrossRef] [PubMed]
- Altoe’, P.; Bernardi, F.; Garavelli, M.; Orlandi, G.; Negri, F. Solvent effects on the vibrational activity and photodynamics of the green fluorescent protein chromophore: A quantum-chemical study. J. Am. Chem. Soc. 2005, 127, 3952–3963. [Google Scholar] [CrossRef] [PubMed]
- Ko, C.; Levine, B.; Toniolo, A.; Manohar, L.; Olsen, S.; Werner, H.J.; Martínez, T.J. Ab initio excited-state dynamics of the photoactive yellow protein chromophore. J. Am. Chem. Soc. 2003, 125, 12710–12711. [Google Scholar] [CrossRef] [PubMed]
- Belousov, V.V.; Fradkov, A.F.; Lukyanov, K.A.; Staroverov, D.B.; Shakhbazov, K.S.; Terskikh, A.V.; Lukyanov, S. Genetically encoded fluorescent indicator for intracellular hydrogen peroxide. Nat. Methods 2006, 4, 281–286. [Google Scholar] [CrossRef]
- Bilan, D.S.; Matlashov, M.E.; Gorokhovatsky, A.Y.; Schultz, C.; Enikolopov, G.; Belousov, V.V. Genetically encoded fluorescent indicator for imaging NAD(+)/NADH ratio changes in different cellular compartments. Biochim. Biophys. Acta 2014, 1840, 951–957. [Google Scholar] [CrossRef]
- Matlashov, M.E.; Bogdanova, Y.A.; Ermakova, G.V.; Mishina, N.M.; Ermakova, Y.G.; Nikitin, E.S.; Balaban, P.M.; Okabe, S.; Lukyanov, S.; Enikolopov, G.; et al. Fluorescent ratiometric pH indicator SypHer2: Applications in neuroscience and regenerative biology. Biochim. Biophys. Acta 2015, 1850, 2318–2328. [Google Scholar] [CrossRef] [Green Version]
- Shaner, N.C.; Steinbach, P.A.; Tsien, R.Y. A guide to choosing fluorescent proteins. Nat. Methods 2005, 2, 905. [Google Scholar] [CrossRef]
- MacKerell, A.D., Jr.; Banavali, N.; Foloppe, N. Development and current status of the CHARMM force field for nucleic acids. Biopolym. Orig. Res. Biomol. 2000, 56, 257–265. [Google Scholar] [CrossRef]
- Reuter, N.; Lin, H.; Thiel, W. Green fluorescent proteins: Empirical force field for the neutral and deprotonated forms of the chromophore. Molecular dynamics simulations of the wild type and S65T mutant. J. Phys. Chem. B 2002, 106, 6310–6321. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kale, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chai, J.D.; Head-Gordon, M. Systematic optimization of long-range corrected hybrid density functionals. J. Chem. Phys. 2008, 128, 084106. [Google Scholar] [CrossRef] [PubMed]
- Chai, J.D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.; Gan, Z.; Epifanovsky, E.; Gilbert, A.T.; Wormit, M.; Kussmann, J.; Lange, A.W.; Behn, A.; Deng, J.; Feng, X.; et al. Advances in molecular quantum chemistry contained in the Q-Chem 4 program package. Mol. Phys. 2015, 113, 184–215. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fluorescent Protein | λex/λem, nm | EC, M−1cm−1 | FQY | Relative Brightness, % * | Fluorescence Lifetime, ns ** | Photobleaching, s # | Redding Rate | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PB | PBS | PB | PBS | PB + Ox | PBS + Ox | PB | PBS | |||||
| EGFP | 489/509 | 55000 | 0.60 | 100 | n/d | 2.8 | n/d | 80 ± 10 | n/d | 5 ± 2.5 | n/d | strong |
| EGFP-T65G | 488/508 | 70000 | 0.06 | 13 | n/d | 1.3 | n/d | 170 ± 25 | n/d | 85 ± 15 | n/d | weak |
| EYFP | 514/526 | 83400 | 0.61 | 154 | 3.18 ± 0.07 | 3.0 ± 0.08 | 21 ± 2 | 35 ± 2 | 3 ± 2 | 2 ± 1 | no | moderate |
| EYFP-G65T | 510/525 | n/d | 0.78 | n/d | 3.7 ± 0.06/4.0 ± 0.09 | 3.5 ± 0.07 and 0.5 ± 0.07/ 3.9 ± 0.08 | 25 ± 8 | 180 ± 8 | 10 ± 2 | 32 ± 7 | moderate | moderate |
| Protein/Chro | EGFP/ TYG | EGFP-T65G/ GYG | EYFP/ GYG | EYFP-G65T/ TYG | EYFP+Cl–/ GYG |
|---|---|---|---|---|---|
| Average No. of hbond | 2.81 | 2.31 | 1.34 | 1.93 | 1.45 |
| STD (hbond) | 1.12 | 1.03 | 0.83 | 1.06 | 0.87 |
| Δ | 7.40 | 6.44 | 4.41 | 5.50 | 7.02 |
| STD (Δ) | 16.7 | 8.2 | 7.8 | 8.0 | 7.29 |
| Protein | Eex, eV (fl) | τfl, ns | τfl,rel | ||||
|---|---|---|---|---|---|---|---|
| Gas phase | QM/MM | Gas phase (n = 1) | QM/MM (n = 1) | QM/MM (n = 1.6) | Gas phase | QM/MM | |
| EGFP | 3.101 (1.02) | 3.081 (0.97) | 29.50 | 31.24 | 7.63 | 1.00 | 1.00 |
| EGFP-T65G | 3.123 (1.05) | 3.142 (1.04) | 28.25 | 28.18 | 6.88 | 0.95 | 0.90 |
| EYFP | 3.123 (1.05) | 3.097 (1.05) | 28.25 | 28.71 | 7.02 | 0.95 | 0.92 |
| EYFP-G65T | 3.101 (1.02) | 3.015 (0.98) | 29.50 | 32.49 | 7.94 | 1.00 | 1.04 |
| EYFP+Cl– | 3.123 (1.05) | 3.077 (1.07) | 28.25 | 28.57 | 6.97 | 0.95 | 0.91 |
| Protein | τfl, ns (τfl,rel) a | τnr, ns (τnr,rel) | τ, ns (τrel) b | FQY | Ybl, rel c |
|---|---|---|---|---|---|
| EGFP | 7.63 (1.00) | 5.92 (1.00) | 3.33 (1.0) | 0.44 | 1.0 |
| EGFP-T65G | 6.88 (0.90) | 0.25 (0.04) | 0.24 (0.1) | 0.04 | 0.1 |
| EYFP | 7.02 (0.92) | 1.73 (0.29) | 1.39 (0.4) | 0.20 | 0.4 |
| EYFP-G65T | 7.94 (1.04) | 10.8 (1.82) | 4.58 (1.4) | 0.58 | 1.4 |
| EYFP+Cl– | 6.97 (0.91) | 0.57 (0.10) | 0.53 (0.2) | 0.07 | 0.2 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sen, T.; Mamontova, A.V.; Titelmayer, A.V.; Shakhov, A.M.; Astafiev, A.A.; Acharya, A.; Lukyanov, K.A.; Krylov, A.I.; Bogdanov, A.M. Influence of the First Chromophore-Forming Residue on Photobleaching and Oxidative Photoconversion of EGFP and EYFP. Int. J. Mol. Sci. 2019, 20, 5229. https://doi.org/10.3390/ijms20205229
Sen T, Mamontova AV, Titelmayer AV, Shakhov AM, Astafiev AA, Acharya A, Lukyanov KA, Krylov AI, Bogdanov AM. Influence of the First Chromophore-Forming Residue on Photobleaching and Oxidative Photoconversion of EGFP and EYFP. International Journal of Molecular Sciences. 2019; 20(20):5229. https://doi.org/10.3390/ijms20205229
Chicago/Turabian StyleSen, Tirthendu, Anastasia V. Mamontova, Anastasia V. Titelmayer, Aleksander M. Shakhov, Artyom A. Astafiev, Atanu Acharya, Konstantin A. Lukyanov, Anna I. Krylov, and Alexey M. Bogdanov. 2019. "Influence of the First Chromophore-Forming Residue on Photobleaching and Oxidative Photoconversion of EGFP and EYFP" International Journal of Molecular Sciences 20, no. 20: 5229. https://doi.org/10.3390/ijms20205229