Insights into Effects/Risks of Chronic Hypergastrinemia and Lifelong PPI Treatment in Man Based on Studies of Patients with Zollinger–Ellison Syndrome


Abstract
:1. Introduction
2. Chronic Hypergastrinemia: General
3. Why is Chronic Hypergastrinemia from PPI Use Receiving Increased Attention and Generating Continued Debate?
4. Why is Prolonged PPI Use Receiving Increased Attention and Generating Continued Debate?
5. Why Would Results from the Long-Term Study of Zollinger–Ellison Syndrome (ZES) Provide Useful Insights into the Issue of Safety of Chronic Hypergastrinemia from Lifelong Use of PPIs in nonZES Patients?
6. Insights of Effects of Chronic Hypergastrinemia in Patients with ZES
6.1. Gastric Mucosal Effects in ZES Patients
6.1.1. Gastric Mucosal Effects in ZES Patients; Non-Endocrine Cells
6.1.2. Gastric Mucosal Effects in ZES Patients: ECL Cells and Gastric Carcinoids
Gastric Mucosal Effects in Sporadic ZES Patients: ECL Cells and Gastric Carcinoids (Table 1)
Gastric Mucosal Effects in MEN1/ZES Patients: ECL Cells and Gastric Carcinoids (Table 1)
Gastric Mucosal Effects in ZES Patients: ECL Cells and Gastric Carcinoids-Sporadic ZES versus MEN1/ZES
Gastric Mucosal Effects in ZES Patients: Comparison with Results from Studies of Chronic Hypergastrinemia in Animals, ECL Cell Changes Reported with Chronic PPI/Potent Anti-Secretory Drug Use, and ECL Cell Changes in Other Common Chronic Hypergastrinemic States (CAG/PA) in Man
6.2. Other Effects of Chronic Hypergastrinemia in ZES
6.2.1. Other Effects of Chronic Hypergastrinemia in ZES: Gastrin and Esophageal adenocarcinoma (Table 1)
6.2.2. Other Effects of Chronic Hypergastrinemia in ZES: Gastrin and Gastric Adenocarcinoma (Table 1)
6.2.3. Other Effects of Chronic Hypergastrinemia in ZES: Gastrin and Colorectal Cancer (CRC) (Table 1)
6.2.4. Other Effects of Chronic Hypergastrinemia in ZES: Gastrin and Pancreatic Cancer (Table 1)
6.3. ZES as Model for Long-Term PPI Effects (Safety, Side-Effects, Effectiveness)
6.3.1. Why is There a Need for Assessment of Long PPI Effects Particularly Related to Safety and Side-Effects?
6.3.2. Why Could Results from the Long-Term Study of Zollinger–Ellison Syndrome (ZES) Provide Useful Insights into the Issues of Long-Term PPI Effects (Safety, Side-Effects, Effectiveness) in nonZES Patients?
6.3.3. Chronic PPI Treatment and Nutrient Malabsorption (vitamin B12 (VB12), Iron, Calcium, Magnesium [Mg]) (Table 1)
Chronic PPI Treatment and Nutrient Malabsorption (VB12, Iron, Calcium, Mg): General
Chronic PPI Treatment and Nutrient Malabsorption: VB12 (Table 1)
Chronic PPI Treatment and Nutrient Malabsorption: Iron (Table 1)
Chronic PPI Treatment and Nutrient Malabsorption: Mg (Table 1)
Chronic PPI Treatment and Nutrient Malabsorption: Calcium (Table 1)
6.3.4. Chronic PPI Treatment and Other Reported PPI Side-Effects and Insights from Studies of ZES Patients (Table 1)
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| BE | Barrett’s esophagus |
| CCK2R | Cholecystokinin type 2 receptor (gastrin receptor) |
| CgA | Chromogranin A |
| CGA/PA | Chronic atrophic gastritis/pernicious anemia |
| CRC | Colorectal cancer |
| EAC | Esophageal adenocarcinoma |
| ECL cell | Gastric enterochromaffin-like cells |
| FSG | Fasting gastrin concentration |
| GC | Gastric cancer |
| GER | Gastroesophageal reflux disease |
| Gly-gastrin | Glycine-extended gastrin |
| HP | Helicobacter pylori |
| Mg | Magnesium |
| MEN1/ZES | Multiple Endocrine Neoplasia type 1 with ZES |
| PDAC | Pancreatic ductal adenocarcinoma |
| PPI | Proton pump inhibitor |
| VB12 | Vitamin B12 |
| ZES | Zollinger–Ellison syndrome |
References
- Strand, D.S.; Kim, D.; Peura, D.A. 25 Years of Proton Pump Inhibitors: A Comprehensive Review. Gut Liver 2017, 11, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, Y.; Ishimura, N.; Ishihara, S. Advantages and Disadvantages of Long-term Proton Pump Inhibitor Use. J. Neurogastroenterol. Motil. 2018, 24, 182–196. [Google Scholar] [CrossRef] [PubMed]
- Jaynes, M.; Kumar, A.B. The risks of long-term use of proton pump inhibitors: A critical review. Ther. Adv. Drug Saf. 2019, 10, 2042098618809927. [Google Scholar] [CrossRef] [PubMed]
- Halfdanarson, O.O.; Pottegard, A.; Bjornsson, E.S.; Lund, S.H.; Ogmundsdottir, M.H.; Steingrímsson, E.; Ogmundsdottir, H.M.; Zoega, H. Proton-pump inhibitors among adults: A nationwide drug-utilization study. Ther. Adv. Gastroenterol. 2018, 11, 1756284818777943. [Google Scholar] [CrossRef] [PubMed]
- Pottegard, A.; Broe, A.; Hallas, J.; De Muckadell, O.B.S.; Lassen, A.T.; Lødrup, A.B. Use of proton-pump inhibitors among adults: A Danish nationwide drug utilization study. Ther. Adv. Gastroenterol. 2016, 9, 671–678. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Fan, Q.; Xiao, S.; Chen, K. Changes in proton pump inhibitor prescribing trend over the past decade and pharmacists’ effect on prescribing practice at a tertiary hospital. BMC Health Serv. Res. 2018, 18, 537. [Google Scholar] [CrossRef] [PubMed]
- Scarpignato, C.; Gatta, L.; Zullo, A.; Blandizzi, C. Effective and safe proton pump inhibitor therapy in acid-related diseases—A position paper addressing benefits and potential harms of acid suppression. BMC Med. 2016, 14, 179. [Google Scholar] [CrossRef]
- Moayyedi, P.; Eikelboom, J.W.; Bosch, J.; Connolly, S.J.; Dyal, L.; Shestakovska, O.; Leong, D.; Anand, S.S.; Störk, S.; Branch, K.R.; et al. Safety of Proton Pump Inhibitors Based on a Large, Multi-Year, Randomized Trial of Patients Receiving Rivaroxaban or Aspirin. Gastroenterology 2019, 157, 682–691. [Google Scholar] [CrossRef]
- Corley, D.A. Safety and Complications of Long-Term Proton Pump Inhibitor Therapy: Getting Closer to the Truth. Gastroenterology 2019, 157, 604–607. [Google Scholar] [CrossRef] [Green Version]
- Waldum, H.L.; Fossmark, R.; Bakke, I.; Martinsen, T.C.; Qvigstad, G. Hypergastrinemia in animals and man: Causes and consequences. Scand. J. Gastroenterol. 2004, 39, 505–509. [Google Scholar] [CrossRef]
- Waldum, H.L.; Sordal, O.; Fossmark, R. Proton pump inhibitors (PPIs) may cause gastric cancer-clinical consequences. Scand. J. Gastroenterol. 2018, 53, 639–642. [Google Scholar] [CrossRef] [PubMed]
- Wormsley, K.G. Is chronic long-term inhibition of gastric secretion really dangerous. Scand. J. Gastroenterol. 1988, 23 (Suppl. 146), 166–174. [Google Scholar] [CrossRef] [PubMed]
- Haastrup, P.F.; Thompson, W.; Sondergaard, J.; Jarbøl, D.E. Side Effects of Long-Term Proton Pump Inhibitor Use: A Review. Basic Clin. Pharmacol. Toxicol. 2018, 123, 114–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nehra, A.K.; Alexander, J.A.; Loftus, C.G.; Nehra, V. Proton Pump Inhibitors: Review of Emerging Concerns. Mayo Clin. Proc. 2018, 93, 240–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, Y.; Tang, J.; Sanagapalli, S.; Kim, B.S.; Leong, R.W. Safety of proton pump inhibitors and risk of gastric cancers: Review of literature and pathophysiological mechanisms. Expert Opin. Drug Saf. 2016, 15, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Aronson, J.K. Inhibiting the proton pump: Mechanisms, benefits, harms, and questions. BMC Med. 2016, 14, 172. [Google Scholar] [CrossRef]
- Lundell, L.; Vieth, M.; Gibson, F.; Nagy, P.; Kahrilas, P.J. Systematic review: The effects of long-term proton pump inhibitor use on serum gastrin levels and gastric histology. Aliment. Pharmacol. Ther. 2015, 42, 649–663. [Google Scholar] [CrossRef]
- Ito, T.; Jensen, R.T. Association of long-term proton pump inhibitor therapy with bone fractures and effects on absorption of calcium, vitamin b (12), iron, and magnesium. Curr. Gastroenterol. Rep. 2010, 12, 448–457. [Google Scholar] [CrossRef]
- Jensen, R.T. Consequences of long-term proton pump blockade: Highlighting insights from studies of patients with gastrinomas. Basic Clin. Pharmacol. Toxicol. 2006, 98, 4–19. [Google Scholar] [CrossRef]
- Zollinger, R.M.; Ellison, E.H. Primary peptic ulcerations of the jejunum associated with islet cell tumors of the pancreas. Ann. Surg. 1955, 142, 709–728. [Google Scholar] [CrossRef]
- Jensen, R.T.; Gardner, J.D. Gastrinoma. In The Pancreas: Biology, Pathobiology and Disease, 2nd ed.; Go, V.L.W., DiMagno, E.P., Gardner, J.D., Lebenthal, E., Reber, H.A., Scheele, G.A., Eds.; Raven Press Publishing Co.: New York, NY, USA, 1993; pp. 931–978. [Google Scholar]
- Jensen, R.T. Zollinger-Ellison syndrome. In Yamada’s Textbook of Gastroenterology, 6th ed.; Podolsky, D.K., Camilleri, M., Fitz, J.G., Kalloo, A.N., Shanahan, F., Wang, T.C., Eds.; John Wiley and Sons, Ltd.: West Sussex, UK, 2016; pp. 1078–1102. [Google Scholar]
- Jensen, R.T. Gastrinoma as a model for prolonged hypergastrinemia in man. In Gastrin; Walsh, J.H., Ed.; Raven Press Publishing Co.: New York, NY, USA, 1993. [Google Scholar]
- Berna, M.J.; Hoffmann, K.M.; Serrano, J.; Gibril, F.; Jensen, R.T. Serum gastrin in Zollinger-Ellison syndrome: I. Prospective study of fasting serum gastrin in 309 patients from the National Institutes of Health and comparison with 2229 cases from the literature. Medicine (Baltimore) 2006, 85, 295–330. [Google Scholar] [CrossRef] [PubMed]
- Peghini, P.L.; Annibale, B.; Azzoni, C.; Milione, M.; Corleto, V.D.; Gibril, F.; Venzon, D.J.; Fave, G.D.; Bordi, C.; Jensen, R.T. Effect of chronic hypergastrinemia on human enterochromaffin-like cells: Insights from patients with sporadic gastrinomas. Gastroenterology 2002, 123, 68–85. [Google Scholar] [CrossRef] [PubMed]
- Orbuch, M.; Venzon, D.J.; Lubensky, I.A.; Weber, H.C.; Gibril, F.; Jensen, R.T. Prolonged hypergastrinemia does not increase the frequency of colonic neoplasia in patients with Zollinger-Ellison syndrome. Dig. Dis. Sci. 1996, 41, 604–613. [Google Scholar] [CrossRef] [PubMed]
- Termanini, B.; Gibril, F.; Sutliff, V.E., III; Yu, F.; Venzon, D.J.; Jensen, R.T. Effect of long-term gastric acid suppressive therapy on serum vitamin B12 levels in patients with Zollinger-Ellison syndrome. Am. J. Med. 1998, 104, 422–430. [Google Scholar] [CrossRef]
- Ito, T.; Igarashi, H.; Uehara, H.; Uehara, H.; Jensen, R.T. Pharmacotherapy of Zollinger-Ellison syndrome. Expert Opin. Pharmacother. 2013, 14, 307–321. [Google Scholar] [CrossRef]
- Roy, P.K.; Venzon, D.J.; Feigenbaum, K.M.; Koviack, P.D.; Bashir, S.; Ojeaburu, J.V.; Gibril, F.; Jensen, R.T. Gastric secretion in Zollinger-Ellison syndrome: Correlation with clinical expression, tumor extent and role in diagnosis—A prospective NIH study of 235 patients and review of the literature in 984 cases. Medicine (Baltimore) 2001, 80, 189–222. [Google Scholar] [CrossRef]
- Metz, D.C.; Strader, D.B.; Orbuch, M.; Koviack, P.D.; Feigenbaum, K.M.; Jensen, R.T. Use of omeprazole in Zollinger-Ellison: A prospective nine-year study of efficacy and safety. Aliment. Pharmacol. Ther. 1993, 7, 597–610. [Google Scholar] [CrossRef]
- Hirschowitz, B.I.; Simmons, J.; Mohnen, J. Clinical outcome using lansoprazole in acid hypersecretors with and without Zollinger-Ellison syndrome: A 13-year prospective study. Clin. Gastroenterol. Hepatol. 2005, 3, 39–48. [Google Scholar] [CrossRef]
- Lamers, C.B.H.W.; Lind, T.; Moberg, S.; Jansen, J.B.M.J.; Olbe, L. Omeprazole in Zollinger-Ellison syndrome: Effects of a single dose and of long term treatment in patients resistant to histamine H2-receptor antagonists. N. Engl. J. Med. 1984, 310, 758–761. [Google Scholar] [CrossRef]
- McArthur, K.E.; Collen, M.J.; Maton, P.N.; Cherner, J.A.; Howard, J.M.; Ciarleglio, C.A.; Cornelius, M.J.; Jensen, R.T.; Gardner, J.D. Omeprazole: Effective, convenient therapy for Zollinger-Ellison syndrome. Gastroenterology 1985, 88, 939–944. [Google Scholar] [CrossRef]
- Jensen, R.T. Use of omeprazole and other proton pump inhibitors in the Zollinger-Ellison syndrome. In Milestones in Drug Therapy; Olbe, L., Ed.; Birkhauser Verlag AG Publish. Co.: Basel, Switzerland, 1999; pp. 205–221. [Google Scholar]
- Metz, D.C.; Pisegna, J.R.; Fishbeyn, V.A.; Benya, R.V.; Feigenbaum, K.M.; Koviack, P.D.; Jensen, R.T. Currently used doses of omeprazole in Zollinger-Ellison syndrome are too high. Gastroenterology 1992, 103, 1498–1508. [Google Scholar] [CrossRef]
- Maton, P.N.; Lack, E.E.; Collen, M.J.; Cornelius, M.J.; David, E.; Gardner, J.D.; Jensen, R.T. The effect of Zollinger-Ellison syndrome and omeprazole therapy on gastric oxyntic endocrine cells. Gastroenterology 1990, 99, 943–950. [Google Scholar] [CrossRef]
- Spindel, E.; Harty, R.F.; Leibach, J.R.; McGuigan, J.E. Decision analysis in evaluation of hypergastrinemia. Am. J. Med. 1986, 80, 11–17. [Google Scholar] [CrossRef]
- Ito, T.; Cadiot, G.; Jensen, R.T. Diagnosis of Zollinger-Ellison syndrome: Increasingly difficult. World J. Gastroenterol. 2012, 18, 5495–5503. [Google Scholar] [CrossRef] [PubMed]
- Rehfeld, J.F.; Friis-Hansen, L.; Goetze, J.P.; Hansen, T.V.O. The biology of cholecystokinin and gastrin peptides. Curr. Top. Med. Chem. 2007, 7, 1154–1165. [Google Scholar] [CrossRef]
- Dockray, G.J. Gastrin. Best Pract. Res. Clin. Endocrinol. Metab. 2004, 18, 555–568. [Google Scholar] [CrossRef]
- Haruma, K.; Kamada, T.; Manabe, N.; Suehiro, M.; Kawamoto, H.; Shiotani, A. Old and New Gut Hormone, Gastrin and Acid Suppressive Therapy. Digestion 2018, 97, 340–344. [Google Scholar] [CrossRef]
- Ito, T.; Igarashi, H.; Uehara, H.; Berna, M.J.; Jensen, R.T. Causes of Death and Prognostic Factors in Multiple Endocrine Neoplasia Type 1: A Prospective Study: Comparison of 106 MEN1/Zollinger-Ellison Syndrome Patients with 1613 Literature MEN1 Patients with or Without Pancreatic Endocrine Tumors. Medicine (Baltimore) 2013, 92, 135–181. [Google Scholar] [CrossRef]
- Creutzfeldt, W. The achlorhydria-carcinoid sequence: Role of gastrin. Digestion 1988, 39, 61–79. [Google Scholar] [CrossRef]
- Waldum, H.L.; Hauso, O.; Brenna, E.; Qvigstad, G.; Fossmark, R. Does long-term profound inhibition of gastric acid secretion increase the risk of ECL cell-derived tumors in man? Scand. J. Gastroenterol. 2016, 51, 767–773. [Google Scholar] [CrossRef]
- Hakanson, R.; Sundler, F. Mechanisms for the development of gastric carcinoids. Digestion 1986, 35 (Suppl. 1), 1–151. [Google Scholar]
- Havu, N. Enterochromaffin-like cell carcinoids of gastric mucosa in rats after life-long inhibition of gastric secretion. Digestion 1986, 35 (Suppl. 1), 42–55. [Google Scholar] [CrossRef] [PubMed]
- Bordi, C.; D’Adda, T.; Azzoni, C.; Pilato, F.P.; Caruana, P. Hypergastrinemia and gastric enterochromaffin-like cells. Am. J. Surg. Pathol. 1995, 19, S8–S19. [Google Scholar] [PubMed]
- Fossmark, R.; Qvigstad, G.; Martinsen, T.C.; Hauso, O.; Waldum, H.L. Animal models to study the role of long-term hypergastrinemia in gastric carcinogenesis. J. Biomed. Biotechnol. 2011, 2011, 975479. [Google Scholar] [CrossRef]
- Van der Hoorn, M.M.C.; Tett, S.E.; de Vries, O.J.; Dobson, A.J.; Peeters, G. The effect of dose and type of proton pump inhibitor use on risk of fractures and osteoporosis treatment in older Australian women: A prospective cohort study. Bone 2015, 81, 675–682. [Google Scholar] [CrossRef] [Green Version]
- Calvete, O.; Reyes, J.; Zuniga, S.; Paumard-Hernández, B.; Fernández, V.; Bujanda, L.; Rodriguez-Pinilla, M.S.; Palacios, J.; Heine-Suñer, D.; Banka, S.; et al. Exome sequencing identifies ATP4A gene as responsible of an atypical familial type I gastric neuroendocrine tumour. Hum. Mol. Genet. 2015, 24, 2914–2922. [Google Scholar] [CrossRef]
- Jensen, R.T. Zollinger-Ellison syndrome. In Surgical Endocrinology: Clinical Syndromes; Doherty, G.M., Skogseid, B., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2001; pp. 291–344. [Google Scholar]
- Howard, J.M.; Chremos, A.N.; Collen, M.J.; McArthur, K.E.; Cherner, J.A.; Maton, P.N.; Ciarleglio, C.A.; Cornelius, M.J.; Gardner, J.D.; Jensen, R.T. Famotidine, a new, potent, long-acting histamine H2-receptor antagonist: Comparison with cimetidine and ranitidine in the treatment of Zollinger-Ellison syndrome. Gastroenterology 1985, 88, 1026–1033. [Google Scholar] [CrossRef]
- Jensen, R.T.; Collen, M.J.; McArthur, K.E.; Howard, J.M.; Maton, P.N.; Cherner, J.A.; Gardner, J.D. Comparison of the effectiveness of ranitidine and cimetidine in inhibiting acid secretion in patients with gastric acid hypersecretory states. Am. J. Med. 1984, 77, 90–105. [Google Scholar]
- Raines, D.; Chester, M.; Diebold, A.E.; Mamikunian, P.; Anthony, C.T.; Mamikunian, G.; Woltering, E.A. A prospective evaluation of the effect of chronic proton pump inhibitor use on plasma biomarker levels in humans. Pancreas 2012, 41, 508–511. [Google Scholar] [CrossRef]
- Jansen, J.B.; Klinkenberg-Knol, E.C.; Meuwissen, S.G.; De Bruijne, J.W.; Festen, H.P.; Snel, P.; Lückers, A.E.; Biemond, I.; Lamers, C.B. Effect of long-term treatment with omeprazole on serum gastrin and serum group A and C pepsinogens in patients with reflux esophagitis. Gastroenterology 1990, 99, 621–628. [Google Scholar] [CrossRef]
- Lamberts, R.; Creutzfeldt, W.; Stockmann, F.; Jacubaschke, U.; Brunner, G.; Maas, S. Long term omeprazole treatment in man: Effects on gastric endocrine cell populations. Digestion 1988, 39, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Wangberg, B.; Nilsson, O.; Theodorsson, E.; Modlin, I.M.; Dahlström, A.; Ahlman, H. Are enterochromaffinlike cell tumours reversible? An experimental study on gastric carcinoids induced in Mastomys by histamine2-receptor blockade. Regul. Pept. 1995, 56, 19–33. [Google Scholar] [CrossRef]
- Grozinsky-Glasberg, S.; Alexandraki, K.I.; Angelousi, A.; Chatzellis, E.; Sougioultzis, S.; Kaltsas, G. Gastric Carcinoids. Endocrinol. Metab. Clin. N. Am. 2018, 47, 645–660. [Google Scholar] [CrossRef] [PubMed]
- Jensen, R.T.; Norton, J.A.; Oberg, K. Neuroendocrine Tumors. In Sleisenger and Fordtran’s Gastrointestinal and Liver Diseases, 10th ed.; Feldman, M., Friedman, L.S., Brandt, L.J., Eds.; Elsevier Saunders: Philadelphia, PA, USA, 2016; pp. 501–541. [Google Scholar]
- Scherubl, H.; Cadiot, G.; Jensen, R.T.; Rosch, T.; Stölzel, U.; Klöppel, G. Neuroendocrine tumors of the stomach (gastric carcinoids) are on the rise: Small tumors, small problems? Endoscopy 2010, 42, 664–671. [Google Scholar] [CrossRef] [PubMed]
- Berna, M.J.; Annibale, B.; Marignani, M.; Luong, T.V.; Corleto, V.; Pace, A.; Ito, T.; Liewehr, D.J.; Venzon, D.J.; Delle Fave, G.; et al. A prospective study of gastric carcinoids and enterochromaffin-like cells changes in Multple Endocrine Neoplaisa Type 1 and Zollinger-Ellison syndrome: Identification of risk factors. J. Clin. Endocrinol. Metab. 2008, 93, 1582–1591. [Google Scholar] [CrossRef] [PubMed]
- Delle Fave, G.; Marignani, M.; Moretti, A.; D’Ambra, G.; Martino, G.; Annibale, B. Hypergastrinemia and enterochromaffin-like cell hyperplasia. Yale J. Biol. Med. 1998, 71, 291–301. [Google Scholar] [PubMed]
- Rindi, G.; Azzoni, C.; La Rosa, S.; Klersy, C.; Paolotti, D.; Rappel, S.; Stolte, M.; Capella, C.; Bordi, C.; Solcia, E. ECL cell tumor and poorly differentiated endocrine carcinoma of the stomach: Prognostic evaluation by pathological analysis. Gastroenterology 1999, 116, 532–542. [Google Scholar] [CrossRef]
- Fossmark, R.; Sordal, O.; Jianu, C.S.; Qvigstad, G.; Nordrum, I.S.; Boyce, M.; Waldum, H.L. Treatment of gastric carcinoids type 1 with the gastrin receptor antagonist netazepide (YF476) results in regression of tumours and normalisation of serum chromogranin A. Aliment. Pharmacol. Ther. 2012, 36, 1067–1075. [Google Scholar] [CrossRef]
- McCarthy, D.M. Commentary: A gastrin antagonist against carcinoids—Implications for PPI-induced hypergastrinaemia. Aliment. Pharmacol. Ther. 2013, 37, 276–277. [Google Scholar] [CrossRef]
- Dawson, R.; Manson, J.M. Omeprazole in oesophageal reflux disease. Lancet 2000, 356, 1770–1771. [Google Scholar] [CrossRef]
- Haga, Y.; Nakatsura, T.; Shibata, Y.; Sameshima, H.; Nakamura, Y.; Tanimura, M.; Ogawa, M. Human gastric carcinoid detected during long-term antiulcer therapy of H2 receptor antagonist and proton pump inhibitor. Dig. Dis. Sci. 1998, 43, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Nandy, N.; Hanson, J.A.; Strickland, R.G.; Mc Carthy, D.M. Solitary Gastric Carcinoid Tumor Associated with Long-Term Use of Omeprazole: A Case Report and Review of the Literature. Dig. Dis. Sci. 2016, 61, 708–712. [Google Scholar] [CrossRef] [PubMed]
- Jianu, C.S.; Lange, O.J.; Viset, T.; Qvigstad, G.; Martinsen, T.C.; Fougner, R.; Kleveland, P.M.; Fossmark, R.; Hauso, O.; Waldum, H.L. Gastric neuroendocrine carcinoma after long-term use of proton pump inhibitor. Scand. J. Gastroenterol. 2012, 47, 64–67. [Google Scholar] [CrossRef] [PubMed]
- Jianu, C.S.; Fossmark, R.; Viset, T.; Qvigstad, G.; Sørdal, Ø.; Mårvik, R.; Waldum, H.L. Gastric carcinoids after long-term use of a proton pump inhibitor. Aliment. Pharmacol. Ther. 2012, 36, 644–649. [Google Scholar] [CrossRef] [PubMed]
- Cadiot, G.; Vissuzaine, C.; Potet, F.; Mignon, M.; Cadiot, D.G. Fundic argyrophil carcinoid tumor in a patient with sporadic-type Zollinger-Ellison syndrome. Dig. Dis. Sci. 1995, 40, 1275–1278. [Google Scholar] [CrossRef] [PubMed]
- Feurle, G.E. Argyrophil cell hyperplasia and a carcinoid tumour in the stomach of a patient with sporadic Zollinger-Ellison syndrome. Gut 1994, 35, 275–277. [Google Scholar] [CrossRef] [PubMed]
- Lamberts, R.; Creutzfeldt, W.; Struber, H.G.; Brunner, G.; Solcia, E. Long-term omeprazole therapy in peptic ulcer disease: Gastrin, endocrine cell growth, and gastritis. Gastroenterology 1993, 104, 1356–1370. [Google Scholar] [CrossRef]
- Solcia, E.; Rindi, G.; Silini, E.; Villani, L. Enterochromaffin-like (ECL) cells and their growths: Relationships to gastrin, reduced acid secretion and gastritis. Bailliere’s Clin. Gastroenterol. 1993, 7, 149–165. [Google Scholar] [CrossRef]
- Campana, D.; Ravizza, D.; Ferolla, P.; Faggiano, A.; Grimaldi, F.; Albertelli, M.; Ricci, C.; Santini, D.; Brighi, N.; Fazio, N.; et al. Risk factors of type 1 gastric neuroendocrine neoplasia in patients with chronic atrophic gastritis. A retrospective, multicentre study. Endocrine 2017, 56, 633–638. [Google Scholar] [CrossRef]
- Vannella, L.; Lahner, E.; Osborn, J.; Annibale, B. Systematic review: Gastric cancer incidence in pernicious anaemia. Aliment. Pharmacol. Ther. 2013, 37, 375–382. [Google Scholar] [CrossRef]
- Solcia, E.; Capella, C.; Sessa, F.; Rindi, G.; Cornaggia, M.; Riva, C.; Villani, L. Gastric carcinoids and related endocrine growths. Digestion 1986, 35 (Suppl. 1), 3–22. [Google Scholar] [CrossRef]
- Aly, A.; Shulkes, A.; Baldwin, G.S. Gastrins, cholecystokinins and gastrointestinal cancer. Biochim. Biophys. Acta 2004, 1704, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Waldum, H.L.; Sagatun, L.; Mjones, P. Gastrin and Gastric Cancer. Front. Endocrinol. (Lausanne) 2017, 8, 1. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.P.; Fantaskey, A.P.; Liu, G.; Zagon, I.S. Identification of gastrin as a growth peptide in human pancreatic cancer. Am. J. Physiol. 1995, 268, R135–R141. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.P.; Shih, A.; Wu, Y.; McLaughlin, P.J.; Zagon, I.S. Gastrin regulates growth of human pancreatic cancer in a tonic and autocrine fashion. Am. J. Physiol. 1996, 270, R1078–R1084. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.P.; Fonkoua, L.K.; Moody, T.W. The Role of Gastrin and CCK Receptors in Pancreatic Cancer and other Malignancies. Int. J. Biol. Sci. 2016, 12, 283–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, J.P.; Nadella, S.; Osborne, N. Gastrin and Gastric Cancer. Cell. Mol. Gastroenterol. Hepatol. 2017, 4, 75–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boursi, B.; Mamtani, R.; Haynes, K.; Yang, Y.-X. Pernicious anemia and colorectal cancer risk—A nested case-control study. Dig. Liver Dis. 2016, 48, 1386–1390. [Google Scholar] [CrossRef]
- Brusselaers, N.; Lagergren, J.; Engstrand, L. Duration of use of proton pump inhibitors and the risk of gastric and oesophageal cancer. Cancer Epidemiol. 2019, 62, 101585. [Google Scholar] [CrossRef]
- Abbas, M.K.; Zaidi, A.R.Z.; Robert, C.A.; Thiha, S.; Malik, B.H. The Safety of Long-term Daily Usage of a Proton Pump Inhibitor: A Literature Review. Cureus 2019, 11, e5563. [Google Scholar] [CrossRef] [Green Version]
- Waldum, H.L.; Rehfeld, J.F. Gastric cancer and gastrin: On the interaction of Helicobacter pylori gastritis and acid inhibitory induced hypergastrinemia. Scand. J. Gastroenterol. 2019, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Thong, B.K.S.; Ima-Nirwana, S.; Chin, K.Y. Proton Pump Inhibitors and Fracture Risk: A Review of Current Evidence and Mechanisms Involved. Int. J. Environ. Res. Public Health 2019, 16, 1571. [Google Scholar] [CrossRef] [PubMed]
- Jensen, R.T. Gastrointestinal endocrine tumors. Gastrinoma. Bailliere’s Clin. Gastroenterol. 1996, 10, 555–766. [Google Scholar]
- Baldwin, G.S. The role of gastrin and cholecystokinin in normal and neoplastic gastrointestinal growth. J. Gastroenterol. Hepatol. 1995, 10, 215–232. [Google Scholar] [CrossRef] [PubMed]
- Shulkes, A.; Baldwin, G. Biology and pathology of non-amidated gastrins. Scand. J. Clin. Lab. Investig. 2001, 61 (Suppl. 234), 123–128. [Google Scholar] [CrossRef]
- Norton, J.A.; Fraker, D.L.; Alexander, H.R.; Venzon, D.J.; Doppman, J.L.; Serrano, J.; Goebel, S.U.; Peghini, P.L.; Roy, P.K.; Gibril, F.; et al. Surgery to cure the Zollinger-Ellison syndrome. N. Engl. J. Med. 1999, 341, 635–644. [Google Scholar] [CrossRef]
- Norton, J.A.; Alexander, H.R.; Fraker, D.L.; Venzon, D.J.; Gibril, F.; Jensen, R.T. Comparison of surgical results in patients with advanced and limited disease with multiple endocrine neoplasia type 1 and Zollinger-Ellison syndrome. Ann. Surg. 2001, 234, 495–506. [Google Scholar] [CrossRef]
- Norton, J.A.; Alexander, H.R.; Fraker, D.L.; Venzon, D.J.; Jensen, R.T. Does the use of routine duodenotomy (DUODX) affect rate of cure, development of liver metastases or survival in patients with Zollinger-Ellison syndrome (ZES)? Ann. Surg. 2004, 239, 617–626. [Google Scholar] [CrossRef]
- Fishbeyn, V.A.; Norton, J.A.; Benya, R.V.; Pisegna, J.R.; Venzon, D.J.; Metz, D.C.; Jensen, R.T. Assessment and prediction of long-term cure in patients with Zollinger-Ellison syndrome: The best approach. Ann. Intern. Med. 1993, 119, 199–206. [Google Scholar] [CrossRef]
- Frucht, H.; Doppman, J.L.; Norton, J.A.; Miller, D.L.; Dwyer, A.J.; Frank, J.A.; Vinayek, R.; Maton, P.N.; Jensen, R.T. Gastrinomas: Comparison of MR Imaging with CT, angiography and US. Radiology 1989, 171, 713–717. [Google Scholar] [CrossRef]
- Doppman, J.L.; Miller, D.L.; Chang, R.; Maton, P.N.; London, J.F.; Gardner, J.D.; Jensen, R.T.; Norton, J.A. Gastrinomas: Localization by means of selective intraarterial injection of secretin. Radiology 1990, 174, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Krudy, A.G.; Doppman, J.L.; Jensen, R.T.; Norton, J.; Collen, M.; Shawker, T.; Gardner, J.; McArthur, K.; Gorden, P. Localization of islet cell tumors by dynamic CT: Comparison with plain CT, arteriography, sonography and venous sampling. Am. J. Roentgenol. 1984, 143, 585–589. [Google Scholar] [CrossRef] [PubMed]
- Cherner, J.A.; Doppman, J.L.; Norton, J.A.; Miller, D.L.; Krudy, A.G.; Raufman, J.-P.; Collen, M.J.; Maton, P.N.; Gardner, J.D.; Jensen, R.T. Selective venous sampling for gastrin to localize gastrinomas. A prospective study. Ann. Intern. Med. 1986, 105, 841–847. [Google Scholar] [CrossRef] [PubMed]
- Gibril, F.; Doppman, J.L.; Reynolds, J.C.; Chen, C.C.; Sutliff, V.E.; Yu, F.; Serrano, J.; Venzon, D.J.; Jensen, R.T. Bone metastases in patients with gastrinomas: A prospective study of bone scanning, somatostatin receptor scanning, and MRI in their detection, their frequency, location and effect of their detection on management. J. Clin. Oncol. 1998, 16, 1040–1053. [Google Scholar] [CrossRef]
- Ito, T.; Jensen, R.T. Molecular imaging in neuroendocrine tumors: Recent advances, controversies, unresolved issues, and roles in management. Curr. Opin. Endocrinol. Diabetes Obes. 2017, 24, 15–24. [Google Scholar] [CrossRef]
- Gibril, F.; Reynolds, J.C.; Doppman, J.L.; Chen, C.C.; Venzon, D.J.; Termanini, B.; Weber, H.C.; Stewart, C.A.; Jensen, R.T. Somatostatin receptor scintigraphy: Its sensitivity compared with that of other imaging methods in detecting primary and metastatic gastrinomas: A prospective study. Ann. Intern. Med. 1996, 125, 26–34. [Google Scholar] [CrossRef]
- Wank, S.A.; Doppman, J.L.; Miller, D.L.; Collen, M.J.; Maton, P.N.; Vinayek, R.; Slaff, J.I.; Norton, J.A.; Gardner, J.D.; Jensen, R.T. Prospective study of the ability of computerized axial tomography to localize gastrinomas in patients with Zollinger-Ellison syndrome. Gastroenterology 1987, 92, 905–912. [Google Scholar] [CrossRef]
- Maton, P.N.; Miller, D.L.; Doppman, J.L.; Collen, M.J.; Norton, J.A.; Vinayek, R.; Slaff, J.I.; Wank, S.A.; Gardner, J.D.; Jensen, R.T. Role of selective angiography in the management of Zollinger- Ellison syndrome. Gastroenterology 1987, 92, 913–918. [Google Scholar] [CrossRef]
- Frucht, H.; Norton, J.A.; London, J.F.; Vinayek, R.; Doppman, J.L.; Gardner, J.D.; Jensen, R.T.; Maton, P.N. Detection of duodenal gastrinomas by operative endoscopic transillumination: A prospective study. Gastroenterology 1990, 99, 1622–1627. [Google Scholar] [CrossRef]
- Sugg, S.L.; Norton, J.A.; Fraker, D.L.; Metz, D.C.; Pisegna, J.R.; Fishbeyn, V.; Benya, R.V.; Shawker, T.H.; Doppman, J.L.; Jensen, R.T. A prospective study of intraoperative methods to diagnose and resect duodenal gastrinomas. Ann. Surg. 1993, 218, 138–144. [Google Scholar] [CrossRef]
- Norton, J.A.; Krampitz, G.W.; Poultsides, G.A.; Visser, B.C.; Fraker, D.L.; Alexander, H.R.; Jensen, R.T. Prospective Evaluation of Results of Reoperation in Zollinger-Ellison Syndrome. Ann. Surg. 2018, 267, 782–788. [Google Scholar] [CrossRef] [PubMed]
- MacFarlane, M.P.; Fraker, D.L.; Alexander, H.R.; Norton, J.A.; Jensen, R.T. A prospective study of surgical resection of duodenal and pancreatic gastrinomas in multiple endocrine neoplasia-Type 1. Surgery 1995, 118, 973–980. [Google Scholar] [CrossRef]
- Pipeleers-Marichal, M.; Somers, G.; Willems, G.; Foulis, A.; Imrie, C.; Bishop, A.E.; Polak, J.M.; Häcki, W.H.; Stamm, B.; Heitz, P.U.; et al. Gastrinomas in the duodenums of patients with multiple endocrine neoplasia type 1 and the Zollinger-Ellison syndrome. N. Engl. J. Med. 1990, 322, 723–727. [Google Scholar] [CrossRef] [PubMed]
- Weber, H.C.; Venzon, D.J.; Lin, J.T.; Fishbein, V.A.; Orbuch, M.; Strader, D.B.; Gibril, F.; Metz, D.C.; Fraker, D.L.; Norton, J.A.; et al. Determinants of metastatic rate and survival in patients with Zollinger-Ellison syndrome: A prospective long-term study. Gastroenterology 1995, 108, 1637–1649. [Google Scholar] [CrossRef]
- Thom, A.K.; Norton, J.A.; Axiotis, C.A.; Jensen, R.T. Location, incidence and malignant potential of duodenal gastrinomas. Surgery 1991, 110, 1086–1093. [Google Scholar]
- Hoffmann, K.M.; Furukawa, M.; Jensen, R.T. Duodenal neuroendocrine tumors: Classification, functional syndromes, diagnosis and medical treatment. Best Pract. Res. Clin. Gastroenterol. 2005, 19, 675–697. [Google Scholar] [CrossRef]
- Donow, C.; Pipeleers-Marichal, M.; Schroder, S.; Stamm, B.; Heitz, P.U.; Klöppel, G. Surgical pathology of gastrinoma: Site, size, multicentricity, association with multiple endocrine neoplasia type 1, and malignancy. Cancer 1991, 68, 1329–1334. [Google Scholar] [CrossRef]
- Norton, J.A.; Foster, D.S.; Ito, T.; Jensen, R.T. Gastrinomas: Medical and SurgicalTreatment. Endocrinol. Metab. Clin. N. Am. 2018, 47, 577–601. [Google Scholar] [CrossRef]
- Jensen, R.T.; Berna, M.J.; Bingham, D.B.; Norton, J.A. Inherited pancreatic endocrine tumor syndromes: Advances in molecular pathogenesis, diagnosis, management and controversies. Cancer 2008, 113 (Suppl. 7), 1807–1843. [Google Scholar] [CrossRef]
- Gibril, F.; Schumann, M.; Pace, A.; Jensen, R.T. Multiple endocrine neoplasia type 1 and Zollinger-Ellison syndrome. A prospective study of 107 cases and comparison with 1009 patients from the literature. Medicine (Baltimore) 2004, 83, 43–83. [Google Scholar] [CrossRef]
- Anlauf, M.; Garbrecht, N.; Henopp, T.; Schmitt, A.; Schlenger, R.; Raffel, A.; Krausch, M.; Gimm, O.; Eisenberger, C.F.; Knoefel, W.T.; et al. Sporadic versus hereditary gastrinomas of the duodenum and pancreas: Distinct clinico-pathological and epidemiological features. World J. Gastroenterol. 2006, 12, 5440–5446. [Google Scholar] [CrossRef] [PubMed]
- Jensen, R.T.; Norton, J.A. Treatment of Pancreatic Neuroendocrine Tumors in Multiple Endocrine Neoplasia Type 1: Some Clarity but Continued Controversy. Pancreas 2017, 46, 589–594. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Jensen, R.T. Imaging in multiple endocrine neoplasia type 1: Recent studies show enhanced sensitivities but increased controversies. Int. J. Endocr. Oncol. 2016, 3, 53–66. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Venzon, D.J.; Serrano, J.; Goebel, S.U.; Doppman, J.L.; Gibril, F.; Jensen, R.T. Prospective study of the clinical course, prognostic factors and survival in patients with longstanding Zollinger-Ellison syndrome. J. Clin. Oncol. 1999, 17, 615–630. [Google Scholar] [CrossRef] [PubMed]
- Norton, J.A.; Krampitz, G.; Jensen, R.T. Multiple Endocrine Neoplasia: Genetics and Clinical Management. Surg. Oncol. Clin. N. Am. 2015, 24, 795–832. [Google Scholar] [CrossRef] [PubMed]
- Jensen, R.T.; Niederle, B.; Mitry, E.; Ramage, J.K.; Steinmüller, T.; Lewington, V.; Scarpa, A.; Sundin, A.; Perren, A.; Gross, D.; et al. Gastrinoma (duodenal and pancreatic). Neuroendocrinology 2006, 84, 173–182. [Google Scholar] [CrossRef]
- Falconi, M.; Eriksson, B.; Kaltsas, G.; Bartsch, D.; Capdevila, J.; Caplin, M.; Kos-Kudla, B.; Kwekkeboom, D.; Rindi, G.; Kloppel, G.; et al. ENETS Consensus Guidelines Update for the Management of Patients with Functional Pancreatic Neuroendocrine Tumors and Non-Functional Pancreatic Neuroendocrine Tumors. Neuroendocrinology 2016, 103, 153–171. [Google Scholar] [CrossRef]
- Jensen, R.T.; Cadiot, G.; Brandi, M.L.; De Herder, W.W.; Kaltsas, G.; Komminoth, P.; Scoazec, J.-Y.; Salazar, R.; Sauvanet, A.; Kianmanesh, R.; et al. ENETS Consensus Guidelines for the Management of Patients with Digestive Neuroendocrine Neoplasms: Functional Pancreatic Endocrine Tumor Syndromes. Neuroendocrinology 2012, 95, 98–119. [Google Scholar] [CrossRef] [Green Version]
- Jensen, R.T. Management of the Zollinger-Ellison syndrome in patients with multiple endocrine neoplasia type 1. J. Intern. Med. 1998, 243, 477–488. [Google Scholar] [CrossRef]
- Roy, P.K.; Venzon, D.J.; Shojamanesh, H.; Abou-Saif, A.; Peghini, P.; Doppman, J.L.; Gibril, F.; Jensen, R.T. Zollinger-Ellison syndrome: Clinical presentation in 261 patients. Medicine (Baltimore) 2000, 79, 379–411. [Google Scholar] [CrossRef]
- Ito, T.; Igarashi, H.; Jensen, R.T. Pancreatic neuroendocrine tumors: Clinical features, diagnosis and medical treatment: Advances. Best Pract. Res. Clin. Gastroenterol. 2012, 26, 737–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shojamanesh, H.; Gibril, F.; Louie, A.; Ojeaburu, J.V.; Bashir, S.; Abou-Saif, A.; Jensen, R.T. Prospective study of the anti-tumor efficacy of long-term octreotide treatment in patients with progressive metastatic gastrinomas. Cancer 2002, 94, 331–343. [Google Scholar] [CrossRef] [PubMed]
- Sutliff, V.E.; Doppman, J.L.; Gibril, F.; Venzon, D.J.; Serrano, J.; Jensen, R.T. Growth of newly diagnosed, untreated metastatic gastrinomas and predictors of growth patterns. J. Clin. Oncol. 1997, 15, 2420–2431. [Google Scholar] [CrossRef] [PubMed]
- Norton, J.A.; Fraker, D.L.; Alexander, H.R.; Gibril, F.; Liewehr, D.J.; Venzon, D.J.; Jensen, R.T. Surgery increases survival in patients with gastrinoma. Ann. Surg. 2006, 244, 410–419. [Google Scholar] [CrossRef]
- Norton, J.A.; Harris, E.J.; Chen, Y.; Visser, B.C.; Poultsides, G.A.; Kunz, P.C.; Fisher, G.A.; Jensen, R.T.; Harris, E.J. Pancreatic endocrine tumors with major vascular abutment, involvement, or encasement and indication for resection. Arch. Surg. 2011, 146, 724–732. [Google Scholar] [CrossRef]
- Fraker, D.L.; Norton, J.A.; Alexander, H.R.; Venzon, D.J.; Jensen, R.T. Surgery in Zollinger-Ellison syndrome alters the natural history of gastrinoma. Ann. Surg. 1994, 220, 320–330. [Google Scholar] [CrossRef]
- Cherner, J.A.; Jensen, R.T.; Dubois, A.; O’Dorisio, T.M.; Gardner, J.D.; Metcalfe, D.D. Gastrointestinal dysfunction in systemic mastocytosis: A prospective study. Gastroenterology 1988, 95, 657–667. [Google Scholar] [CrossRef]
- Lee, L.; Ito, T.; Jensen, R.T. Everolimus in the treatment of neuroendocrine tumors: Efficacy, side-effects, resistance, and factors affecting its place in the treatment sequence. Expert Opin. Pharmacother. 2018, 19, 909–928. [Google Scholar] [CrossRef]
- Ito, T.; Igarashi, H.; Jensen, R.T. Therapy of metastatic pancreatic neuroendocrine tumors (pNETs): Recent insights and advances. J. Gastroenterol. 2012, 47, 941–960. [Google Scholar] [CrossRef]
- Kwekkeboom, D.J.; de Herder, W.W.; Kam, B.L.; Van Eijck, C.H.; Van Essen, M.; Kooij, P.P.; Feelders, R.A.; Van Aken, M.O.; Krenning, E.P. Treatment with the radiolabeled somatostatin analog [177 Lu-DOTA 0, Tyr3] octreotate: Toxicity, efficacy, and survival. J. Clin. Oncol. 2008, 26, 2124–2130. [Google Scholar] [CrossRef]
- Solcia, E.; Fiocca, R.; Villani, L.; Gianatti, A.; Cornaggia, M.; Chiaravalli, A.M.; Curzio, M.; Capella, C. Morphology and pathogenesis of endocrine hyperplasias, precarcinoid lesions, and carcinoids arising in chronic atrophic gastritis. Scand. J. Gastroenterol. 1991, 26, 146–159. [Google Scholar] [CrossRef] [PubMed]
- Borch, K.; Renvall, H.; Liedberg, G. Gastric endocrine cell hyperplasia and carcinoid tumors in pernicious anemia. Gastroenterology 1985, 88, 638–648. [Google Scholar] [CrossRef]
- Kuipers, E.J.; Lundell, L.; Klinkenberg-Knol, E.C.; Havu, N.; Festen, H.P.; Liedman, B.; Lamers, C.B.; Jansen, J.B.; Dalenbäck, J.; Snel, P.; et al. Atrophic gastritis and Helicobacter pylori infection in patients with reflux esophagitis treated with omeprazole or fundoplication. N. Engl. J. Med. 1996, 334, 1018–1022. [Google Scholar] [CrossRef] [PubMed]
- Weber, H.C.; Venzon, D.J.; Jensen, R.T.; Metz, D.C. Studies on the interrelation between Zollinger-Ellison syndrome, Helicobacter pylori and proton pump inhibitor therapy. Gastroenterology 1997, 112, 84–91. [Google Scholar] [CrossRef]
- Saeed, Z.A.; Evans, D.J., Jr.; Evans, D.G.; Cornelius, M.J.; Maton, P.N.; Jensen, R.T. Helicobacter pylori and the Zollinger-Ellison syndrome. Dig. Dis. Sci. 1991, 36, 15–18. [Google Scholar] [CrossRef]
- McColl, K.E. Helicobacter pylori-negative nonsteroidal anti-inflammatory drug-negative ulcer. Gastroenterol. Clin. N. Am. 2009, 38, 353–361. [Google Scholar] [CrossRef]
- Ojeaburu, J.V.; Ito, T.; Crafa, P.; Bordi, C.; Jensen, R.T. Mechanism of Acid hypersecretion post curative gastrinoma resection. Dig. Dis. Sci. 2011, 56, 139–154. [Google Scholar] [CrossRef]
- Pisegna, J.R.; Norton, J.A.; Slimak, G.G.; Metz, D.C.; Maton, P.N.; Jensen, R.T. Effects of curative resection on gastric secretory function and antisecretory drug requirement in the Zollinger-Ellison syndrome. Gastroenterology 1992, 102, 767–778. [Google Scholar] [CrossRef]
- Benya, R.V.; Metz, D.C.; Venzon, D.J.; Fishbeyn, V.A.; Strader, D.B.; Orbuch, M.; Jensen, R.T. Zollinger-Ellison syndrome can be the initial endocrine manifestation in patients with multiple endocrine neoplasia-type 1. Am. J. Med. 1994, 97, 436–444. [Google Scholar] [CrossRef]
- Gibril, F.; Chen, Y.-J.; Schrump, D.S.; Vortmeyer, A.; Zhuang, Z.; Lubensky, I.A.; Reynolds, J.C.; Louie, A.; Entsuah, L.K.; Huang, K.; et al. Prospective study of thymic carcinoids in patients with Multiple Endocrine Neoplasia Type 1. J. Clin. Endocrinol. Metab. 2003, 88, 1066–1081. [Google Scholar] [CrossRef]
- Miller, L.S.; Vinayek, R.; Frucht, H.; Gardner, J.; Jensen, R.T.; Maton, P. Reflux esophagitis in patients with Zollinger-Ellison syndrome. Gastroenterology 1990, 98, 341–346. [Google Scholar] [CrossRef]
- Maton, P.N.; Frucht, H.; Vinayek, R.; Wank, S.; Gardner, J.; Jensen, R.T. Medical management of patients with Zollinger-Ellison syndrome who have had previous gastric surgery: A prospective study. Gastroenterology 1988, 94, 294–299. [Google Scholar] [CrossRef]
- Jensen, R.T.; Gardner, J.D.; Raufman, J.P.; Pandol, S.J.; Doppman, J.L.; Collen, M.J. Zollinger-Ellison syndrome: Current concepts and management. Ann. Intern. Med. 1983, 98, 59–75. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, D.M.; Olinger, E.J.; May, R.J.; Long, B.W.; Gardner, J.D. H2-histamine receptor blocking agents in the Zollinger-Ellison syndrome. Experience in seven cases and implications for long-term therapy. Ann. Intern. Med. 1977, 87, 668–675. [Google Scholar] [CrossRef]
- Neuburger, P.; Lewin, M.; De Recherche, C.; Bonfils, S. Parietal and chief cell population in four cases of the Zollinger-Ellison syndrome. Gastroenterology 1972, 63, 937–942. [Google Scholar] [CrossRef]
- Castrup, H.J.; Fuchs, K.; Peiper, H.J. Cell renewal of gastric mucosa in Zollinger-Ellison syndrome. Acta Hepatogastroenterol. (Stuttg) 1975, 22, 40–43. [Google Scholar]
- Polacek, M.A.; Ellison, E.H. Parietal cell mass and gastric acid secretion in the Zollinger-Ellison syndrome. Surgery 1966, 60, 606–614. [Google Scholar]
- Polacek, M.A.; Ellison, E.H. A Comparative study of the Parietal Cell Mass and Distribution in Normal Stomachs, in Stomachs with Duodenal Ulcer, and in Stomachs of patients with pancreatic adenoma. Surg. Forum 1963, 14, 313–315. [Google Scholar]
- Helander, H.F. Oxyntic mucosa histology in omeprazole treated patients suffering from duodenal ulcer or Zollinger-Ellison syndrome. Digestion 1986, 35 (Suppl. 1), 123–129. [Google Scholar] [CrossRef]
- Rosenlund, M.L. The Zollinger-Ellison syndrome in children. A review. Am. J. Med. Sci. 1967, 254, 884–892. [Google Scholar]
- Sum, P.; Perey, B.J. Parietal-cell mass (PCM) in a man with Zollinger-Ellison syndrome. Can. J. Surg. 1969, 12, 285–288. [Google Scholar] [PubMed]
- Helander, H.F.; Rutgersson, K.; Helander, K.G.; Pisegna, J.R.; Gardner, J.D.; Jensen, R.T. Stereologic investigations of human gastric mucosa: II. Oxyntic mucosa from Zollinger-Ellison syndrome. Scand. J. Gastroenterol. 1992, 27, 875–883. [Google Scholar] [CrossRef] [PubMed]
- Mignon, M.; Lehy, T.; Bonnefond, A.; Ruszniewski, P.; Labeille, D.; Bonfils, S. Development of gastric argyrophil carcinoid tumors in a case of Zollinger-Ellison syndrome with primary hyperparathyroidism during long term antisecretory treatment. Cancer 1987, 59, 1959–1962. [Google Scholar] [CrossRef]
- Waldum, H.L.; Brenna, E.; Kleveland, P.M.; Sandvik, A. Gastrin-physiological and pathophysiological role: Clinical consequences. Dig. Dis. 1995, 13, 25–38. [Google Scholar] [CrossRef]
- Lehy, T.; Mignon, M.; Cadiot, G.; Elouaer-Blanc, L.; Ruszniewski, P.; Lewin, M.; Bonfils, S. Gastric endocrine cell behavior in Zollinger-Ellison patients upon long-term potent antisecretory treatment. Gastroenterology 1989, 96, 1029–1040. [Google Scholar] [CrossRef]
- Bordi, C.; Azzoni, C.; Ferraro, G.; Corleto, V.D.; Gibril, F.; Fave, G.D.; Lubensky, I.A.; Venzon, D.J.; Jensen, R.T. Sampling strategies for analysis of enterochromaffin-like cell changes in Zollinger-Ellison syndrome. Am. J. Clin. Pathol. 2000, 114, 419–425. [Google Scholar] [CrossRef]
- Delle Fave, G.; Marignani, M.; Corleto, V.D.; Angeletti, S.; D’Ambra, G.; Ferraro, G.; D’Adda, T.; Azzoni, C.; Jensen, R.; Annibale, B.; et al. Progression of gastric enterochromaffin-like cells growth in Zollinger-Ellison syndrome and atrophic body gastritis patients. Dig. Liver Dis. 2002, 34, 270–278. [Google Scholar] [CrossRef]
- Cadiot, G.; Lehy, T.; Mignon, M. Gastric endocrine cell proliferation and fundic argyrophil carcinoid tumors in patients with the Zollinger-Ellison syndrome. Acta Oncol. 1993, 32, 135–140. [Google Scholar] [CrossRef]
- Lehy, T.; Cadiot, G.; Mignon, M.; Ruszniewski, P.; Bonfils, S. Influence of multiple endocrine neoplasia type 1 on gastric endocrine cells in patients with the Zollinger-Ellison syndrome. Gut 1992, 33, 1275–1279. [Google Scholar] [CrossRef]
- Cadiot, G.; Lehy, T.; Ruszniewski, P.; Bonfils, S.; Mignon, M. Gastric endocrine cell evolution in patients with Zollinger-Ellison syndrome. Influence of gastrinoma growth and long-term omeprazole treatment. Dig. Dis. Sci. 1993, 38, 1307–1317. [Google Scholar] [CrossRef]
- D’Adda, T.; Corleto, V.; Pilato, F.P.; Baggi, M.T.; Robutti, F.; Fave, G.D.; Bordi, C. Quantitative ultrastructure of endocrine cells of oxyntic mucosa in Zollinger-Ellison syndrome. Correspondence with light microscopic findings. Gastroenterology 1990, 99, 17–26. [Google Scholar] [CrossRef]
- Bardram, L.; Thomsen, P.; Stadil, F. Gastric endocrine cells in omeprazole treated and untreated patients with Zollinger-Ellison syndrome. Digestion 1986, 35 (Suppl. 1), 116–122. [Google Scholar] [CrossRef]
- Hirschowitz, B.I.; Haber, M.M. Helicobacter pylori effects on gastritis, gastrin and enterochromaffin-like cells in Zollinger-Ellison syndrome and non-Zollinger-Ellison syndrome acid hypersecretors treated long-term with lansoprazole. Aliment. Pharmacol. Ther. 2001, 15, 87–103. [Google Scholar] [CrossRef] [PubMed]
- Solcia, E.; Capella, C.; Buffa, R.; Frigerio, G.; Fiocca, R. Pathology of the Zollinger-Ellison syndrome. Prog. Surg. Pathol. 1980, I, 119–133. [Google Scholar]
- Hage, E.; Hendel, L.; Gustafsen, J.; Hendel, J. Histopathology of the gastric oxyntic mucosa in two different patient groups during long-term treatment with omeprazole. Eur. J. Gastroenterol. Hepatol. 2003, 15, 781–789. [Google Scholar] [CrossRef] [PubMed]
- Modlin, I.M.; Tang, L.H. The gastric enterochromaffin-like cell: An enigmatic cellular link. Gastroenterology 1996, 111, 783–810. [Google Scholar] [CrossRef] [PubMed]
- Coupe, M.; Rees, H.; Springer, C.J.; Bishop, A.E.; Morris, J.A.; Polak, J.M.; Calam, J. Gastric enterochromaffin-like (ECL) cells in hypergastrinaemic duodenal ulcer disease. Gut 1990, 31, 144–147. [Google Scholar] [CrossRef]
- Borch, K.; Stridsberg, M.; Burman, P.; Rehfeld, J.F. Basal chromogranin A and gastrin concentrations in circulation correlate to endocrine cell proliferation in type-A gastritis. Scand. J. Gastroenterol. 1997, 32, 198–202. [Google Scholar] [CrossRef]
- Jordan, P.H., Jr.; Barroso, A.; Sweeney, J. Gastric carcinoids in patients with hypergastrinemia. J. Am. Coll. Surg. 2004, 199, 552–555. [Google Scholar] [CrossRef]
- Solcia, E.; Bordi, C.; Creutzfeldt, W.; Dayal, Y.; Dayan, A.; Falkmer, S.; Grimelius, L.; Havu, N. Histopathological classification of nonantral gastric endocrine growths in man. Digestion 1988, 41, 185–200. [Google Scholar] [CrossRef]
- Rindi, G.; Luinetti, O.; Cornaggia, M.; Capella, C.; Solcia, E. Three subtypes of gastric argyrophil carcinoid and the gastric neuroendocrine carcinoma: A clinicopathologic study. Gastroenterology 1993, 104, 994–1006. [Google Scholar] [CrossRef]
- Konturek, S.J.; Konturek, P.C.; Bielanski, W.; Lorens, K.; Sito, E.; Konturek, J.W.; Kwiecień, S.; Bobrzyński, A.; Pawlik, T.; Karcz, D.; et al. Case presentation of gastrinoma combined with gastric carcinoid with the longest survival record—Zollinger-Ellison syndrome: Pathophysiology, diagnosis and therapy. Med. Sci. Monit. 2002, 8, CS43–CS59. [Google Scholar] [PubMed]
- Lee, H.W.; Chung, J.W.; Kim, Y.J.; Kwon, K.A.; Kim, E.J.; Kim, K.K.; Lee, W.K.; Sym, S.J. Synchronous Peripancreatic Lymph Node Gastrinoma and Gastric Neuroendocrine Tumor Type 2. Clin. Endosc. 2016, 49, 483–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tariq, H.; Kamal, M.U.; Vootla, V.; ElZaeedi, M.; Niazi, M.; Gilchrist, B.; Ihimoyan, A.; Dev, A.; Chilimuri, S. A Rare Cause of Abdominal Pain and Mass in an 18-Year-Old Patient: A Diagnostic Dilemma. Gastroenterol. Res. 2018, 11, 75–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massironi, S.; Zilli, A.; Elvevi, A.; Invernizzi, P. The changing face of chronic autoimmune atrophic gastritis: An updated comprehensive perspective. Autoimmun. Rev. 2019, 18, 215–222. [Google Scholar] [CrossRef]
- Kokkola, A.; Sjoblom, S.M.; Haapiainen, R.; Sipponen, P.; Puolakkainen, P.; Jarvinen, H. The risk of gastric carcinoma and carcinoid tumours in patients with pernicious anaemia. Scand. J. Gastroenterol. 1998, 33, 88–92. [Google Scholar]
- Annibale, B.; Azzoni, C.; Corleto, V.D.; Di Giulio, E.; Caruana, P.; D’Ambra, G.; Bordi, C.; Fave, G.D. Atrophic body gastritis patients with enterochromaffin-like cell dysplasia are at increased risk for the development of type I gastric carcinoid. Eur. J. Gastroenterol. Hepatol. 2001, 13, 1449–1456. [Google Scholar] [CrossRef]
- Gibril, F.; Reynolds, J.C.; Roy, P.K.; Peghini, P.L.; Doppman, J.L.; Jensen, R.T. Ability of somatostatin receptor scintigraphy to identify patients with localized gastric carcinoids: A prospective study. J. Nucl. Med. 2000, 41, 1646–1656. [Google Scholar]
- Solcia, E.; Rindi, G.; Havu, N.; Elm, G. Qualitative studies of gastric endocrine cells in patients treated long-term with omeprazole. Scand. J. Gastroenterol. 1989, 24, 129–137. [Google Scholar] [CrossRef]
- Fich, A.; Talley, N.J.; Shorter, R.G.; Phillips, S.F. Zollinger-Ellison syndrome. Relation to Helicobacter pylori-associated chronic gastritis and gastric acid secretion. Dig. Dis. Sci. 1991, 36, 10–14. [Google Scholar] [CrossRef]
- Collen, M.J.; Howard, J.M.; McArthur, K.E.; Raufman, J.-P.; Cornelius, M.J.; Ciarleglio, C.A.; Gardner, J.D.; Jensen, R.T. Comparison of ranitidine and cimetidine in the treatment of gastric hypersecretion. Ann. Intern. Med. 1984, 100, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Raufman, J.P.; Collins, S.M.; Pandol, S.J.; Korman, L.Y.; Collen, M.J.; Cornelius, M.J.; Feld, M.K.; McCarthy, D.M.; Gardner, J.D.; Jensen, R.T. Reliability of symptoms in assessing control of gastric acid secretion in patients with Zollinger-Ellison syndrome. Gastroenterology 1983, 84, 108–113. [Google Scholar] [PubMed]
- Metz, D.C.; Pisegna, J.R.; Fishbeyn, V.A.; Benya, R.V.; Jensen, R.T. Control of gastric acid hypersecretion in the management of patients with Zollinger-Ellison syndrome. World J. Surg. 1993, 17, 468–480. [Google Scholar] [CrossRef] [PubMed]
- Dreijerink, K.M.A.; Timmers, H.T.M.; Brown, M. Twenty years of menin: Emerging opportunities for restoration of transcriptional regulation in MEN1. Endocr. Relat Cancer 2017, 24, T135–T145. [Google Scholar] [CrossRef] [PubMed]
- Ruszniewski, P.; Podevin, P.; Cadiot, G.; Marmuse, J.-P.; Mignon, M.; Vissuzaine, C.; Bonfils, S.; Lehy, T. Clinical, anatomical, and evolutive features of patients with the Zollinger-Ellison syndrome combined with type I multiple endocrine neoplasia. Pancreas 1993, 8, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Rindi, G.; Bordi, C.; Rappel, S.; La Rosa, S.; Stolte, M.; Solcia, E. Gastric carcinoids and neuroendocrine carcinomas: Pathogenesis, pathology, and behavior. World J. Surg. 1996, 20, 168–172. [Google Scholar] [CrossRef]
- Tomassetti, P.; Migliori, M.; Caletti, G.C.; Fusaroli, P.; Corinaldesi, R.; Gullo, L. Treatment of type II gastric carcinoid tumors with somatostatin analogues. N. Engl. J. Med. 2000, 343, 551–554. [Google Scholar] [CrossRef]
- Berger, M.W.; Stephens, D.H. Gastric carcinoid tumors associated with chronic hypergastrinemia in a patient with Zollinger-Ellison syndrome. Radiology 1996, 201, 371–373. [Google Scholar] [CrossRef]
- Caplin, M.; Khan, K.; Grimes, S.; Michaeli, D.; Savage, K.; Pounder, R.; Dhillon, A. Effect of gastrin and anti-gastrin antibodies on proliferation of hepatocyte cell lines. Dig. Dis. Sci. 2001, 46, 1356–1366. [Google Scholar] [CrossRef]
- Norton, J.A.; Melcher, M.L.; Gibril, F.; Jensen, R.T. Gastric carcinoid tumors in multiple endocrine neoplasia-1 patients with Zollinger-Ellison syndrome can be symptomatic, demonstrate aggressive growth, and require surgery. Surgery 2004, 136, 1267–1274. [Google Scholar] [CrossRef]
- Richards, M.L.; Gauger, P.; Thompson, N.W.; Giordano, T.J. Regression of type II gastric carcinoids in multiple endocrine neoplasia type 1 patients with Zollinger-Ellison syndrome after surgical excision of all gastrinomas. World J. Surg. 2004, 28, 652–658. [Google Scholar] [CrossRef] [PubMed]
- Bekele, G.; Felicetta, J.V.; Gani, O.; Shah, I.A. Malignant thymic carcinoid in multiple endocrine neoplasia type I syndrome: Case report and literature review. Endocr. Pract. 1998, 4, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Ruszniewski, P.; Delle Fave, G.; Cadiot, G.; Komminoth, P.; Chung, D.; Kos-Kudla, B.; Kianmanesh, R.; Hochhauser, D.; Arnold, R.; Ahlman, H.; et al. Well-differentiated gastric tumors/carcinomas. Neuroendocrinology 2006, 84, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Hirschowitz, B.I.; Mohnen, J.; Shaw, S. Long-term treatment with lansoprazole for patients with Zollinger-Ellison syndrome. Aliment. Pharmacol. Ther. 1996, 10, 507–522. [Google Scholar] [CrossRef] [PubMed]
- Plockinger, U.; Rindi, G.; Arnold, R.; Eriksson, B.; Krenning, E.P.; de Herder, W.W.; Goede, A.; Caplin, M.; Oberg, K.; Reubi, J.C.; et al. Guidelines for the diagnosis and treatment of neuroendocrine gastrointestinal tumours. A consensus statement on behalf of the European Neuroendocrine Tumour Society (ENETS). Neuroendocrinology 2004, 80, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Debelenko, L.V.; Emmert-Buck, M.R.; Zhuang, Z.; Epshteyn, E.; Moskaluk, C.A.; Jensen, R.T.; Liotta, L.A.; Lubensky, I.A. The Multiple Endocrine Neoplasia Type 1 gene locus is involved in the pathogenesis of Type II gastric carcinoids. Gastroenterology 1997, 113, 773–782. [Google Scholar] [CrossRef]
- Ekman, L.; Hansson, E.; Havu, N.; Carlsson, E.; Lundberg, C. Toxicological studies on omeprazole. Scand. J. Gastroenterol. 1985, 20 (Suppl. 108), 53–69. [Google Scholar]
- Bonfils, S.; Ruszniewski, P.; Costil, V.; Laucournet, H.; Vatier, J.; René, E.; Mignon, M. Prolonged treatment of Zollinger-Ellison syndrome by long-acting somatostatin. Lancet 1986, 327, 554–555. [Google Scholar] [CrossRef]
- Poynter, D.; Pick, C.R.; Harcourt, R.A.; Ainge, G.; Harman, I.W.; Spurling, N.W.; Fluck, P.A.; Cook, J.L. Association of long lasting unsurmountable histamine H2 blockade and gastric carcinoid tumours in the rat. Gut 1985, 26, 1284–1295. [Google Scholar] [CrossRef]
- Poynter, D.; Selway, S.A.; Papworth, S.A.; Riches, S.R. Changes in the gastric mucosa of the mouse associated with long lasting unsurmountable histamine H2 blockade. Gut 1986, 27, 1338–1346. [Google Scholar] [CrossRef]
- Cui, G.; Qvigstad, G.; Falkmer, S.; Sandvik, A.K.; Kawase, S.; Waldum, H.L. Spontaneous ECLomas in cotton rats (Sigmodon hispidus): Tumours occurring in hypoacidic/hypergastrinaemic animals with normal parietal cells. Carcinogenesis 2000, 21, 23–27. [Google Scholar] [CrossRef] [PubMed]
- Borch, K.; Renvall, H.; Liedberg, G.; Andersen, B.N. Relations between circulating gastrin and endocrine cell proliferation in the atrophic gastric fundic mucosa. Scand. J. Gastroenterol. 1986, 21, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Cattan, D.; Roucayrol, A.M.; Launay, J.M.; Callebert, J.; Charasz, N.; Nurit, Y.; Belaiche, J.; Kalifat, R. Circulating gastrin, endocrine cells, histamine content, and histidine decarboxylase activity in atrophic gastritis. Gastroenterology 1989, 97, 586–596. [Google Scholar] [CrossRef]
- Borch, K. Atrophic gastritis and gastric carcinoid tumours. Ann. Med. 1989, 21, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Sjoblom, S.M.; Sipponen, P.; Karonen, S.L.; Järvinen, H.J. Mucosal argyrophil endocrine cells in pernicious anaemia and upper gastrointestinal carcinoid tumours. J. Clin. Pathol. 1989, 42, 371–377. [Google Scholar] [CrossRef] [PubMed]
- Bordi, C.; D’Adda, T.; Pilato, F.P.; Ferrari, C. Carcinoid (ECL cell) tumor of the oxyntic mucosa of the stomach: A hormone-dependent neoplasm? In Progress in Surgical Pathology; Fenoglio-Preiser, C., Wolff, M., Rilke, F., Eds.; Field & Wood: Philadelphia, PA, USA, 1988; Volume 8, pp. 177–195. [Google Scholar]
- Green, D.M.; Bishop, A.E.; Rindi, G.; Lee, F.I.; Daly, M.J.; Domin, J.; Bloom, S.R.; Polak, J.M. Enterochromaffin-like cell populations in human fundic mucosa: Quantitative studies of their variations with age, sex, and plasma gastrin levels. J. Pathol. 1989, 157, 235–241. [Google Scholar] [CrossRef]
- Solcia, E.; Fiocca, R.; Havu, N.; Dalväg, A.; Carlssorf, R.; Carlsson, R. Gastric endocrine cells and gastritis in patients receiving long- term omeprazole treatment. Digestion 1992, 51 (Suppl. 1), 82–92. [Google Scholar] [CrossRef]
- Diebold, M.D.; Richardson, S.; Duchteau, A.; Bigard, M.A.; Colin, R.; Cortot, A.; Fauchère, J.L.; Zeitoun, P. Factors influencing corpus argyrophil cell density and hyperplasia in reflux esophagitis patients treated with antisecretory drugs and controls. Dig. Dis. Sci. 1998, 43, 1629–1635. [Google Scholar]
- Cavalcoli, F.; Zilli, A.; Conte, D.; Ciafardini, C.; Massironi, S. Gastric neuroendocrine neoplasms and proton pump inhibitors: Fact or coincidence? Scand. J. Gastroenterol. 2015, 50, 1397–1403. [Google Scholar] [CrossRef]
- Lahner, E.; Pilozzi, E.; Esposito, G.; Galli, G.; Annibale, B. Gastric carcinoid in the absence of atrophic body gastritis and with low Ki67 index: A clinical challenge. Scand. J. Gastroenterol. 2014, 49, 506–510. [Google Scholar] [CrossRef]
- Kearns, M.D.; Boursi, B.; Yang, Y.X. Proton pump inhibitors on pancreatic cancer risk and survival. Cancer Epidemiol. 2017, 46, 80–84. [Google Scholar] [CrossRef] [PubMed]
- Klinkenberg-Knol, E.C.; Festen, H.P.M.; Jansen, J.B.M.J.; Lamers, C.B.H.W.; Nelis, F.; Snel, P.; Luckers, A.; Dekkers, C.P.M.; Havu, N.; Meuwissen, S.G.M. Long-term treatment wih omeprazole for refractory reflux esophagitis: Efficacy and safety. Ann. Intern. Med. 1994, 121, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Freedberg, D.E.; Haynes, K.; Denburg, M.R.; Zemel, B.S.; Leonard, M.B.; Abrams, J.A.; Yang, Y.-X. Use of proton pump inhibitors is associated with fractures in young adults: A population-based study. Osteoporos. Int. 2015, 26, 2501–2507. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.P.; Solomon, T.E. Cholecystokinin and pancreatic cancer: The chicken or the egg? Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 306, G91–G101. [Google Scholar] [CrossRef] [PubMed]
- Lagergren, J.; Lagergren, P. Recent developments in esophageal adenocarcinoma. CA Cancer J. Clin. 2013, 63, 232–248. [Google Scholar] [CrossRef]
- Hayakawa, Y.; Chang, W.; Jin, G.; Wang, T.C. Gastrin and upper GI cancers. Curr. Opin. Pharmacol. 2016, 31, 31–37. [Google Scholar] [CrossRef]
- Chueca, E.; Lanas, A.; Piazuelo, E. Role of gastrin-peptides in Barrett’s and colorectal carcinogenesis. World J. Gastroenterol. 2012, 18, 6560–6570. [Google Scholar] [CrossRef]
- Singh, S.; Garg, S.K.; Singh, P.P.; Iyer, P.G. El-Serag Acid-suppressive medications and risk of oesophageal adenocarcinoma in patients with Barrett’s oesophagus: A systematic review and meta-analysis. Gut 2014, 63, 1229–1237. [Google Scholar] [CrossRef]
- Lee, Y.; Urbanska, A.M.; Hayakawa, Y.; Wang, H.; Au, A.S.; Luna, A.M.; Chang, W.; Jin, G.; Bhagat, G.; Abrams, J.A.; et al. Gastrin stimulates a cholecystokinin-2-receptor-expressing cardia progenitor cell and promotes progression of Barrett’s-like esophagus. Oncotarget 2017, 8, 203–214. [Google Scholar]
- Haigh, C.R.; Attwood, S.E.; Thompson, D.G.; Jankowski, J.A.; Kirton, C.M.; Pritchard, D.M.; Varro, A.; Dimaline, R. Gastrin induces proliferation in Barrett’s metaplasia through activation of the CCK2 receptor. Gastroenterology 2003, 124, 615–625. [Google Scholar] [CrossRef]
- El-Serag, H.B.; Aguirre, T.V.; Davis, S.; Kuebeler, M.; Bhattacharyya, A.; Sampliner, R.E. Proton pump inhibitors are associated with reduced incidence of dysplasia in Barrett’s esophagus. Am. J. Gastroenterol. 2004, 99, 1877–1883. [Google Scholar] [CrossRef] [PubMed]
- Kastelein, F.; Spaander, M.C.; Steyerberg, E.W.; Biermann, K.; Valkhoff, V.E.; Kuipers, E.J.; Bruno, M.J. Proton pump inhibitors reduce the risk of neoplastic progression in patients with Barrett’s esophagus. Clin. Gastroenterol. Hepatol. 2013, 11, 382–388. [Google Scholar] [CrossRef] [PubMed]
- Jankowski, J.A.Z.; de, C.J.; Love, S.B.; Reilly, G.; Watson, P.; Sanders, S.; Ang, Y.; Morris, D.; Bhandari, P.; Brooks, C.; et al. Esomeprazole and aspirin in Barrett’s oesophagus (AspECT): A randomised factorial trial. Lancet 2018, 392, 400–408. [Google Scholar] [CrossRef]
- Hvid-Jensen, F.; Pedersen, L.; Funch-Jensen, P.; Drewes, A.M. Proton pump inhibitor use may not prevent high-grade dysplasia and oesophageal adenocarcinoma in Barrett’s oesophagus: A nationwide study of 9883 patients. Aliment. Pharmacol. Ther. 2014, 39, 984–991. [Google Scholar] [CrossRef]
- Brusselaers, N.; Engstrand, L.; Lagergren, J. Maintenance proton pump inhibition therapy and risk of oesophageal cancer. Cancer Epidemiol. 2018, 53, 172–177. [Google Scholar] [CrossRef]
- Richter, J.E.; Pandol, S.J.; Castell, D.O.; McCarthy, D.M. Gastroesophageal reflux disease in the Zollinger-Ellison syndrome. Ann. Intern. Med. 1981, 95, 37–43. [Google Scholar] [CrossRef]
- Siewert, R.; Jennewein, H.M.; Arnold, R.; Creutzfeld, W. The lower oesophageal sphincter in the Zollinger-Ellison syndrome. Ger. Med. 1973, 3, 101–102. [Google Scholar]
- McCallum, R.W.; Walsh, J.H. Relationship between lower esophageal sphincter and serum gastrin concentration in Zollinger-Ellison syndrome and other clinical settings. Gastroenterology 1979, 76, 76–81. [Google Scholar]
- Hoffmann, K.M.; Gibril, F.; Entsuah, L.K.; Serrano, J.; Jensen, R.T. Patients with multiple endocrine neoplasia type 1 with gastrinomas have an increased risk of severe esophageal disease including stricture and the premalignant condition, Barrett’s esophagus. J. Clin. Endocrinol. Metab. 2006, 91, 204–212. [Google Scholar] [CrossRef]
- Strader, D.B.; Benjamin, S.; Orbuch, M.; Lubensky, T.A.; Gibril, F.; Weber, C.; Fishbeyn, V.Å.; Jensen, R.T.; Metz, D.C. Esophageal function and occurrence of Barrett’s esophagus in Zollinger-Ellison syndrome. Digestion 1995, 56, 347–356. [Google Scholar] [CrossRef]
- Collen, M.J.; Lewis, J.H.; Benjamin, S.B. Gastric acid hypersecretion in refractory gastroesophageal reflux disease. Gastroenterology 1990, 98, 654–661. [Google Scholar] [CrossRef]
- Sarosiek, J.; Jensen, R.T.; Maton, P.N.; Peura, D.A.; Harlow, T.; Feng, T.; McCallum, R.W.; Pisegna, J.R. Salivary and gastric epidermal growth factor in patients with Zollinger-Ellison syndrome. Am. J. Gastroenterol. 2000, 95, 1158–1165. [Google Scholar] [CrossRef] [PubMed]
- Goudet, P.; Murat, A.; Binquet, C.; Cardot-Bauters, C.; Costa, A.; Ruszniewski, P.; Niccoli, P.; Menegaux, F.; Chabrier, G.; Borson-Chazot, F.; et al. Risk factors and causes of death in MEN1 disease. A GTE (Groupe d’Etude des Tumeurs Endocrines) cohort study among 758 patients. World J. Surg. 2010, 34, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Hur, C.; Miller, M.; Kong, C.Y.; Dowling, E.C.; Nattinger, K.J.; Dunn, M.; Feuer, E.J. Trends in esophageal adenocarcinoma incidence and mortality. Cancer 2013, 119, 1149–1158. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.C.; Chiang, T.H.; Chou, C.K.; Tu, Y.K.; Liao, W.C.; Wu, M.S.; Graham, D.Y. Association Between Helicobacter pylori Eradication and Gastric Cancer Incidence: A Systematic Review and Meta-analysis. Gastroenterology 2016, 150, 1113–1124. [Google Scholar] [CrossRef] [PubMed]
- Kuipers, E.J.; Uyterlinde, A.M.; Pena, A.S.; Hazenberg, H.J.; Bloemena, E.; Lindeman, J.; Klinkenberg-Knol, E.C.; Meuwissen, S.G. Increase of Helicobacter pylori-associated corpus gastritis during acid suppressive therapy: Implications for long-term safety. Am. J. Gastroenterol. 1995, 90, 1401–1406. [Google Scholar] [PubMed]
- Akbari, M.; Tabrizi, R.; Kardeh, S.; Lankarani, K.B. Gastric cancer in patients with gastric atrophy and intestinal metaplasia: A systematic review and meta-analysis. PLoS ONE 2019, 14, e0219865. [Google Scholar] [CrossRef]
- Fossmark, R.; Sagatun, L.; Nordrum, I.S.; Sandvik, A.K.; Waldum, H.L. Hypergastrinemia is associated with adenocarcinomas in the gastric corpus and shorter patient survival. Apmis 2015, 123, 509–514. [Google Scholar] [CrossRef]
- Wang, T.C.; Dangler, C.A.; Chen, D.; Goldenring, J.R.; Koh, T.; Raychowdhury, R.; Coffey, R.J.; Ito, S.; Varro, A.; Dockray, G.J.; et al. Synergistic interaction between hypergastrinemia and Helicobacter infection in a mouse model of gastric cancer. Gastroenterology 2000, 118, 36–47. [Google Scholar] [CrossRef]
- Takaishi, S.; Cui, G.; Frederick, D.M.; Carlson, J.E.; Houghton, J.; Varro, A.; Dockray, G.J.; Ge, Z.; Whary, M.T.; Rogers, A.B.; et al. Synergistic inhibitory effects of gastrin and histamine receptor antagonists on Helicobacter-induced gastric cancer. Gastroenterology 2005, 128, 1965–1983. [Google Scholar] [CrossRef]
- Goetze, J.P.; Eiland, S.; Svendsen, L.B.; Vainer, B.; Hannibal, J.; Rehfeld, J.F. Characterization of gastrins and their receptor in solid human gastric adenocarcinomas. Scand. J. Gastroenterol. 2013, 48, 688–695. [Google Scholar] [CrossRef] [PubMed]
- Cheung, K.S.; Chan, E.W.; Wong, A.Y.S.; Chen, L.; Wong, I.C.K.; Leung, W.K. Long-term proton pump inhibitors and risk of gastric cancer development after treatment for Helicobacter pylori: A population-based study. Gut 2018, 67, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Doggui, M.H.; Ben, Y.L.; Hefaiedh, R.; Bouguassas, W.; Mestiri, A.; Dellagi, K. A gastric collision tumor composed of adenocarcinoma and gastrinoma: Case report. Tunis. Med. 2008, 86, 755–757. [Google Scholar] [PubMed]
- De Leval, L.; Hardy, N.; Deprez, M.; Delwaide, J.; Belaiche, J.; Boniver, J. Gastric collision between a papillotubular adenocarcinoma and a gastrinoma in a patient with Zollinger-Ellison syndrome. Virchows Arch. 2002, 441, 462–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schott, M.; Sagert, C.; Willenberg, H.S.; Schinner, S.; Ramp, U.; Varro, A.; Raffel, A.; Eisenberger, C.; Zacharowski, K.; Scherbaum, W.A.; et al. Carcinogenic hypergastrinemia: Signet-ring cell carcinoma in a patient with multiple endocrine neoplasia type 1 with Zollinger-Ellison’s syndrome. J. Clin. Endocrinol. Metab. 2007, 92, 3378–3382. [Google Scholar] [CrossRef] [PubMed]
- Stewart, C.A.; Termanini, B.; Sutliff, V.E.; Gibril, F.; Jensen, R.T. Assessment of the risk of iron malabsorption occurring in patients with Zollinger-Ellison syndrome (ZES) treated with long-term gastric antisecretory therapy. Gastroenterology 1996, 110, A264. [Google Scholar]
- Dimaline, R.; Varro, A. Novel roles of gastrin. J. Physiol. 2014, 592, 2951–2958. [Google Scholar] [CrossRef]
- Koh, T.J. Extragastric effects of gastrin gene knock-out mice. Pharmacol. Toxicol. 2002, 91, 368–374. [Google Scholar] [CrossRef]
- Hollande, F.; Imdahl, A.; Mantamadiotis, T.; Ciccotosto, G.; Shulkes, A.; Baldwin, G.; Baldwin, G. Glycine-extended gastrin acts as an autocrine growth factor in a nontransformed colon cell line. Gastroenterology 1997, 113, 1576–1588. [Google Scholar] [CrossRef]
- Giraud, J.; Failla, L.M.; Pascussi, J.M.; Lagerqvist, E.L.; Ollier, J.; Finetti, P.; Bertucci, F.; Ya, C.; Gasmi, I.; Bourgaux, J.-F.; et al. Autocrine Secretion of Progastrin Promotes the Survival and Self-Renewal of Colon Cancer Stem-like Cells. Cancer Res. 2016, 76, 3618–3628. [Google Scholar] [CrossRef]
- Lahner, E.; Sbrozzi-Vanni, A.; Vannella, L.; Corleto, V.D.; Di Giulio, E.; Fave, G.D.; Annibale, B. No higher risk for colorectal cancer in atrophic gastritis-related hypergastrinemia. Dig. Liver Dis. 2012, 44, 793–797. [Google Scholar] [CrossRef] [PubMed]
- Lahner, E.; Capasso, M.; Carabotti, M.; Annibale, B. Incidence of cancer (other than gastric cancer) in pernicious anaemia: A systematic review with meta-analysis. Dig. Liver Dis. 2018, 50, 780–786. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.X.; Hennessy, S.; Propert, K.; Hwang, W.; Sedarat, A.; Lewis, J.D. Chronic proton pump inhibitor therapy and the risk of colorectal cancer. Gastroenterology 2007, 133, 748–754. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.S.; Park, S.M.; Eom, C.S.; Kim, S.; Myung, S.-K. Use of Proton Pump Inhibitor and Risk of Colorectal Cancer: A Meta-analysis of Observational Studies. Korean J. Fam. Med. 2012, 33, 272–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, J.P.; Wood, J.G.; Solomon, T.E. Elevated gastrin levels in patients with colon cancer or adenomatous polyps. Dig. Dis. Sci. 1989, 34, 171–174. [Google Scholar] [CrossRef]
- Ciccotosto, G.D.; McLeish, A.; Hardy, K.J.; Shulkes, A. Expression, processing, and secretion of gastrin in patients with colorectal carcinoma. Gastroenterology 1995, 109, 1142–1153. [Google Scholar] [CrossRef]
- Siddheshwar, R.K.; Gray, J.C.; Kelly, S.B. Plasma levels of progastrin but not amidated gastrin or glycine extended gastrin are elevated in patients with colorectal carcinoma. Gut 2001, 48, 47–52. [Google Scholar] [CrossRef]
- Del Valle, J.; Sugano, K.; Yamada, T. Progastrin and its glycine-extended posttranslational processing intermediates in human gastrointestinal tissues. Gastroenterology 1987, 92, 1908–1912. [Google Scholar] [CrossRef]
- Goswami, R.S.; Minoo, P.; Baker, K.; Chong, G.; Foulkes, W.D.; Jass, J.R. Hyperplastic polyposis and cancer of the colon with gastrinoma of the duodenum. Nat. Clin. Pract. Oncol. 2006, 3, 281–284. [Google Scholar] [CrossRef]
- Tobi, M.; Maliakkal, B.J.; Maliakkal, R.; Cats, A.; Kinzie, J.L.; Dullaart, R.P.F.; Luk, G.D. Zollinger-Ellison syndrome, acromegaly, and colorectal neoplasia. J. Clin. Gastroenterol. 1997, 24, 21–24. [Google Scholar] [CrossRef]
- Sobhani, I.; Lehy, T.; Laurent-Puig, P.; Cadiot, G.; Ruszniewski, P.; Mignon, M. Chronic endogenous hypergastrinemia in humans: Evidence for a mitogenic effect on the colonic mucosa. Gastroenterology 1993, 105, 22–30. [Google Scholar] [PubMed]
- Renga, M.; Brandi, G.; Paganelli, G.M.; Calabrese, C.; Papa, S.; Tosti, A.; Tomassetti, P.; Miglioli, M.; Biasco, G. Rectal cell proliferation and colon cancer risk in patients with hypergastrinemia. Gut 1997, 41, 330–332. [Google Scholar] [CrossRef] [PubMed]
- Philip, B.; Roland, C.L.; Daniluk, J.; Liu, Y.; Chatterjee, D.; Gomez, S.B.; Ji, B.; Huang, H.; Wang, H.; Fleming, J.B.; et al. A high-fat diet activates oncogenic Kras and COX2 to induce development of pancreatic ductal adenocarcinoma in mice. Gastroenterology 2013, 145, 1449–1458. [Google Scholar] [CrossRef] [PubMed]
- Matters, G.L.; Harms, J.F.; McGovern, C.O.; Jayakumar, C.; Crepin, K.; Smith, Z.P.; Nelson, M.C.; Stock, H.; Fenn, C.W.; Kaiser, J.; et al. Growth of human pancreatic cancer is inhibited by down-regulation of gastrin gene expression. Pancreas 2009, 38, e151–e161. [Google Scholar] [CrossRef] [PubMed]
- Nadella, S.; Burks, J.; Huber, M.; Wang, J.; Cao, H.; Kallakury, B.; Tucker, R.D.; Boca, S.M.; Jermusyck, A.; Collins, I.; et al. Endogenous Gastrin Collaborates with Mutant KRAS in Pancreatic Carcinogenesis. Pancreas 2019, 48, 894–903. [Google Scholar] [CrossRef] [PubMed]
- Osborne, N.; Sundseth, R.; Gay, M.D.; Cao, H.P.; Tucker, R.D.; Nadella, S.; Wang, S.; Liu, X.; Kroemer, A.; Sutton, L.; et al. Vaccine Against Gastrin, Polyclonal Antibody Stimulator, Decreases Pancreatic Cancer Metastases. Am. J. Physiol. Gastrointest. Liver Physiol. 2019. [Google Scholar] [CrossRef]
- Goetze, J.P.; Nielsen, F.C.; Burcharth, F.; Rehfeld, J.F. Closing the gastrin loop in pancreatic carcinoma: Coexpression of gastrin and its receptor in solid human pancreatic adenocarcinoma. Cancer 2000, 88, 2487–2494. [Google Scholar] [CrossRef]
- Huang, S.C.; Zhang, L.; Chiang, H.C.; Wank, S.A.; Maton, P.N.; Gardner, J.D.; Jensen, R.T. Benzodiazepine analogues L-365,260 and L-364,718 as gastrin and pancreatic CCK receptor antagonists. Am. J. Physiol. 1989, 257, G169–G174. [Google Scholar]
- Fino, K.K.; Matters, G.L.; McGovern, C.O.; Gilius, E.L.; Smith, J.P. Downregulation of the CCK-B receptor in pancreatic cancer cells blocks proliferation and promotes apoptosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G1244–G1252. [Google Scholar] [CrossRef] [Green Version]
- Burks, J.; Nadella, S.; Mahmud, A.; Mankongpaisarnrung, C.; Wang, J.; Hahm, J.-I.; Tucker, R.D.; Shivapurkar, N.; Stern, S.T.; Smith, J.P. Cholecystokinin Receptor-Targeted Polyplex Nanoparticle Inhibits Growth and Metastasis of Pancreatic Cancer. Cell. Mol. Gastroenterol. Hepatol. 2018, 6, 17–32. [Google Scholar] [CrossRef] [Green Version]
- Shah, P.; Rhim, A.D.; Haynes, K.; Hwang, W.-T.; Yang, Y.-X. Diagnosis of pernicious anemia and the risk of pancreatic cancer. Pancreas 2014, 43, 422–426. [Google Scholar] [CrossRef] [PubMed]
- Hwang, I.C.; Chang, J.; Park, S.M. Association between proton pump inhibitor use and the risk of pancreatic cancer: A Korean nationwide cohort study. PLoS ONE 2018, 13, e0203918. [Google Scholar] [CrossRef] [PubMed]
- Creutzfeldt, W.; Lamberts, R. Is hypergastrinemia dangerous to man? Scand. J. Gastroenterol. 1991, 26 (Suppl. 180), 179–191. [Google Scholar] [CrossRef] [PubMed]
- Hsing, A.W.; Hansson, L.E.; McLaughlin, J.K.; Nyren, O.; Blot, W.J.; Ekbom, A.; Fraumeni, J.F. Pernicious anemia and subsequent cancer. A population-based cohort study. Cancer 1993, 71, 745–750. [Google Scholar] [CrossRef]
- Laoveeravat, P.; Thavaraputta, S.; Vutthikraivit, W.; Suchartlikitwong, S.; Mingbunjerdsuk, T.; Motes, A.; Nugent, K.; Rakvit, A.; Islam, E.; Islam, S. Proton Pump Inhibitors and Histamine-2 Receptor Antagonists on the Risk of Pancreatic Cancer: A Systematic Review and Meta-analysis. QJM 2019. [Google Scholar] [CrossRef]
- Karpathakis, A.; Pericleous, M.; Luong, T.V.; Khoo, B.; Thirlwell, C.; Toumpanakis, C.; Caplin, M.E. Pancreatic adenocarcinoma in a patient with multiple endocrine neoplasia 1 syndrome. Pancreas 2013, 42, 725–726. [Google Scholar] [CrossRef]
- Jensen, R.T. Natural history of digestive endocrine tumors. In Recent Advances in Pathophysiology and Management of Inflammatory Bowel Diseases and Digestive Endocrine Tumors; Mignon, M., Colombel, J.F., Eds.; John Libbey Eurotext Publishing Co.: Paris, France, 1999; pp. 192–219. [Google Scholar]
- Maton, P.N.; Vinayek, R.; Frucht, H.; McArthur, K.; Miller, L.; Saeed, Z.; Gardner, J.; Jensen, R. Long-term efficacy and safety of omeprazole in patients with Zollinger-Ellison syndrome: A prospective study. Gastroenterology 1989, 97, 827–836. [Google Scholar] [CrossRef]
- Metz, D.C.; Pisegna, J.R.; Ringham, G.L.; Feigenbaum, K.; Koviack, P.D.; Maton, P.N.; Gardner, J.D.; Jensen, R.T.; Maton, D.P.N.; Gardner, D.J.D. Prospective study of efficacy and safety of lansoprazole in Zollinger-Ellison syndrome. Dig. Dis. Sci. 1993, 38, 245–256. [Google Scholar] [CrossRef]
- Osefo, N.; Ito, T.; Jensen, R.T. Gastric Acid hypersecretory States: Recent insights and advances. Curr. Gastroenterol. Rep. 2009, 11, 433–441. [Google Scholar] [CrossRef]
- von Schrenck, T.; Howard, J.M.; Doppman, J.L.; Norton, J.; Maton, P.; Smith, F.; Vinayek, R.; Frucht, H.; Wank, S.; Gardner, J.; et al. Prospective study of chemotherapy in patients with metastatic gastrinoma. Gastroenterology 1988, 94, 1326–1334. [Google Scholar] [CrossRef]
- Lloyd-Davies, K.A.; Rutgersson, K.; Solvell, L. Omeprazole in the treatment of Zollinger-Ellison syndrome: A 4-year international study. Aliment. Pharmacol. Ther. 1988, 2, 13–32. [Google Scholar] [CrossRef] [PubMed]
- Norton, J.A.; Cornelius, M.J.; Doppman, J.L.; Maton, P.N.; Gardner, J.D.; Jensen, R.T. Effect of parathyroidectomy in patients with hyperparathyroidism, Zollinger-Ellison syndrome and multiple endocrine neoplasia Type I: A prospective study. Surgery 1987, 102, 958–966. [Google Scholar] [PubMed]
- Norton, J.A.; Venzon, D.J.; Berna, M.J.; Alexander, H.R.; Fraker, D.L.; Libutti, S.K.; Marx, S.J.; Gibril, F.; Jensen, R.T. Prospective study of surgery for primary hyperaparathyroidism (HPT) in Multiple Endocrine Neoplasia type 1 (MEN1), and Zollinger-Ellison syndrome (ZES): Longterm outcome of a more virulent form of HPT. Ann. Surg. 2008, 247, 501–510. [Google Scholar] [CrossRef] [PubMed]
- Corsonello, A.; Lattanzio, F.; Bustacchini, S.; Garasto, S.; Cozza, A.; Schepisi, R.; Lenci, F.; Luciani, F.; Maggio, M.G.; Ticinesi, A.; et al. Adverse events of proton pump inhibitors: Potential mechanisms. Curr. Drug Metab. 2018, 19, 142–154. [Google Scholar] [CrossRef] [PubMed]
- Abraham, N.S. Proton pump inhibitors: Potential adverse effects. Curr. Opin. Gastroenterol. 2012, 28, 615–620. [Google Scholar] [CrossRef]
- Freedberg, D.E.; Kim, L.S.; Yang, Y.X. The Risks and Benefits of Long-term Use of Proton Pump Inhibitors: Expert Review and Best Practice Advice from the American Gastroenterological Association. Gastroenterology 2017, 152, 706–715. [Google Scholar] [CrossRef]
- Hirschowitz, B.I.; Worthington, J.; Mohnen, J. Vitamin B12 deficiency in hypersecretors during long-term acid suppression with proton pump inhibitors. Aliment. Pharmacol. Ther. 2008, 27, 1110–1121. [Google Scholar] [CrossRef]
- Lam, J.R.; Schneider, J.L.; Zhao, W.; Corley, D.A. Proton pump inhibitor and histamine 2 receptor antagonist use and vitamin B12 deficiency. JAMA 2013, 310, 2435–2442. [Google Scholar] [CrossRef]
- Jung, S.B.; Nagaraja, V.; Kapur, A.; Eslick, G.D.; Jung, S.B. Association between vitamin B12 deficiency and long-term use of acid-lowering agents: A systematic review and meta-analysis. Intern. Med. J. 2015, 45, 409–416. [Google Scholar] [CrossRef]
- den Elzen, W.P.; Groeneveld, Y.; de Ruijter, W.; Souverijn, J.H.M.; Le Cessie, S.; Assendelft, W.J.J.; Gussekloo, J. Long-term use of proton pump inhibitors and vitamin B12 status in elderly individuals. Aliment. Pharmacol. Ther. 2008, 27, 491–497. [Google Scholar] [CrossRef]
- Qorraj-Bytyqi, H.; Hoxha, R.; Sadiku, S.; Bajraktari, I.H.; Sopjani, M.; Thaçi, K.; Thaçi, S.; Bahtiri, E. Proton Pump Inhibitors Intake and Iron and Vitamin B12 Status: A Prospective Comparative Study with a Follow up of 12 Months. Open Access Maced. J. Med. Sci. 2018, 6, 442–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jensen, R.T. Overview of chronic diarrhea caused by functional neuroendocrine neoplasms. Semin. Gastrointest. Dis. 1999, 10, 156–172. [Google Scholar] [PubMed]
- Umeda, R.; Nakamura, Y.; Masugi, Y.; Shinoda, M.; Hosoe, N.; Ono, Y.; Fujimura, T.; Yamagishi, Y.; Higuchi, H.; Ebinuma, H.; et al. Hemobilia due to biliary intraepithelial neoplasia associated with Zollinger-Ellison syndrome. Clin. J. Gastroenterol. 2012, 5, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Stewart, C.A.; Termanini, B.; Sutliff, V.E.; Serrano, J.; Yu, F.; Gibril, F.; Jensen, R.T. Assessment of the risk of iron malabsorption in patients with Zollinger-Ellison syndrome treated with long-term gastric acid antisecretory therapy. Aliment. Pharmacol. Ther. 1998, 12, 83–98. [Google Scholar] [CrossRef] [PubMed]
- Koop, H.; Bachem, M.G. Serum iron, ferritin, and vitamin B12 during prolonged omeprazole therapy. J. Clin. Gastroenterol. 1992, 14, 288–292. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, C.; Geissler, C.A.; Powell, J.J.; Bomford, A. Proton pump inhibitors suppress absorption of dietary non-haem iron in hereditary haemochromatosis. Gut 2007, 56, 1291–1295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shalev, H.; Quider, A.A.; Harosh, M.B.; Kapelushnik, J. Proton pump inhibitors use suppresses iron absorption in congenital dyserythropoietic anemia. Pediatr. Hematol. Oncol. 2016, 33, 457–461. [Google Scholar] [CrossRef]
- Sharma, N.; Chau, W.Y.; Dobruskin, L. Effect of long-term proton pump inhibitor therapy on hemoglobin and serum iron levels after sleeve gastrectomy. Surg. Obes. Relat. Dis. 2019. [Google Scholar] [CrossRef]
- Ajmera, A.V.; Shastri, G.S.; Gajera, M.J.; Judge, T.A. Suboptimal response to ferrous sulfate in iron-deficient patients taking omeprazole. Am. J. Ther. 2012, 19, 185–189. [Google Scholar] [CrossRef]
- Douwes, R.M.; Gomes-Neto, A.W.; Eisenga, M.F.; Vinke, J.S.J.; de Borst, M.H.; van den Berg, E.; Berger, S.P.; Touw, D.J.; Hak, E.; Blokzijl, H.; et al. Chronic Use of Proton-Pump Inhibitors and Iron Status in Renal Transplant Recipients. J. Clin. Med. 2019, 8, 1382. [Google Scholar] [CrossRef]
- Lam, J.R.; Schneider, J.L.; Quesenberry, C.P.; Corley, D.A. Proton Pump Inhibitor and Histamine-2 Receptor Antagonist Use and Iron Deficiency. Gastroenterology 2017, 152, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Tran-Duy, A.; Connell, N.J.; Vanmolkot, F.H.; Souverein, P.C.; de Wit, N.J.; Stehouwer, C.D.A.; Hoes, A.W.; de Vries, F.; de Boer, A. Use of proton pump inhibitors and risk of iron deficiency: A population-based case-control study. J. Intern. Med. 2019, 285, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Park, C.H.; Kim, E.H.; Roh, Y.H.; Kim, H.Y.; Kil Lee, S. The association between the use of proton pump inhibitors and the risk of hypomagnesemia: A systematic review and meta-analysis. PLoS ONE 2014, 9, e112558. [Google Scholar] [CrossRef] [PubMed]
- Cheungpasitporn, W.; Thongprayoon, C.; Kittanamongkolchai, W.; Srivali, N.; Edmonds, P.J.; Ungprasert, P.; O’Corragain, O.A.; Korpaisarn, S.; Erickson, S.B. Proton pump inhibitors linked to hypomagnesemia: A systematic review and meta-analysis of observational studies. Ren. Fail. 2015, 37, 1237–1241. [Google Scholar] [CrossRef]
- Janett, S.; Camozzi, P.; Peeters, G.G.; Lava, S.A.G.; Simonetti, G.D.; Simonetti, B.G.; Bianchetti, M.G.; Milani, G.P. Hypomagnesemia Induced by Long-Term Treatment with Proton-Pump Inhibitors. Gastroenterol. Res. Pract. 2015, 2015, 951768. [Google Scholar] [CrossRef]
- Zipursky, J.; Macdonald, E.M.; Hollands, S.; Gomes, T.; Mamdani, M.M.; Paterson, J.M.; Lathia, N.; Juurlink, D.N. Proton pump inhibitors and hospitalization with hypomagnesemia: A population-based case-control study. PLoS Med. 2014, 11, e1001736. [Google Scholar] [CrossRef]
- Luk, C.P.; Parsons, R.; Lee, Y.P.; Gomes, T.; Mamdani, M.M.; Paterson, J.M.; Lathia, N.; Juurlink, D.N. Proton pump inhibitor-associated hypomagnesemia: What do FDA data tell us? Ann. Pharmacother. 2013, 47, 773–780. [Google Scholar] [CrossRef]
- Kieboom, B.C.; Kiefte-de Jong, J.C.; Eijgelsheim, M.; Franco, O.H.; Kuipers, E.J.; Hofman, A.; Zietse, R.; Stricker, B.H.; Hoorn, E.J. Proton pump inhibitors and hypomagnesemia in the general population: A population-based cohort study. Am. J. Kidney Dis. 2015, 66, 775–782. [Google Scholar] [CrossRef]
- Mackay, J.D.; Bladon, P.T. Hypomagnesaemia due to proton-pump inhibitor therapy: A clinical case series. QJM 2010, 103, 387–395. [Google Scholar] [CrossRef]
- Hess, M.W.; Hoenderop, J.G.; Bindels, R.J.; Drenth, J.P.H. Systematic review: Hypomagnesaemia induced by proton pump inhibition. Aliment. Pharmacol. Ther. 2012, 36, 405–413. [Google Scholar] [CrossRef]
- William, J.H.; Danziger, J. Magnesium Deficiency and Proton-Pump Inhibitor Use: A Clinical Review. J. Clin. Pharmacol. 2016, 56, 660–668. [Google Scholar] [CrossRef] [PubMed]
- William, J.H.; Danziger, J. Proton-pump inhibitor-induced hypomagnesemia: Current research and proposed mechanisms. World J. Nephrol. 2016, 5, 152–157. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.P.; Hausman, E.; Lionberger, R.; Zhang, X. Modeling and simulation of the effect of proton pump inhibitors on magnesium homeostasis. 1. Oral absorption of magnesium. Mol. Pharm. 2012, 9, 3495–3505. [Google Scholar] [CrossRef] [PubMed]
- Lameris, A.L.; Hess, M.W.; van Kruijsbergen, I.; Hoenderop, J.G.; Bindels, R.J. Omeprazole enhances the colonic expression of the Mg2+ transporter TRPM6. Pflug. Arch. 2013, 465, 1613–1620. [Google Scholar] [CrossRef] [PubMed]
- Gommers, L.M.M.; Ederveen, T.H.A.; van der Wijst, J.; Overmars-Bos, C.; Kortman, G.A.M.; Boekhorst, J.; Bindels, R.J.M.; De Baaij, J.H.F.; Hoenderop, J.G.J. Low gut microbiota diversity and dietary magnesium intake are associated with the development of PPI-induced hypomagnesemia. FASEB J. 2019, fj201900839R. [Google Scholar] [CrossRef]
- Metz, D.C.; Sostek, M.B.; Ruszniewski, P.; Forsmark, C.E.; Monyak, J.; Pisegna, J.R. Effects of esomeprazole on Acid output in patients with zollinger-ellison syndrome or idiopathic gastric Acid hypersecretion. Am. J. Gastroenterol. 2007, 102, 2648–2654. [Google Scholar] [CrossRef]
- Stroker, E.; Leone, L.; Vandeput, Y.; Borbath, I.; Lefebvre, C. Severe symptomatic hypomagnesaemia induced by the chronic use of proton pump inhibitors: A case report of a patient with Zollinger-Ellison syndrome. Acta Clin. Belg. 2014, 69, 62–65. [Google Scholar] [CrossRef]
- Yu, E.W.; Blackwell, T.; Ensrud, K.E.; Hillier, T.A.; Lane, N.E.; Orwoll, E.; Bauer, D.C. Acid-suppressive medications and risk of bone loss and fracture in older adults. Calcif. Tissue Int. 2008, 83, 251–259. [Google Scholar] [CrossRef]
- Kitay, A.M.; Geibel, J.P. Stomach and Bone. Adv. Exp. Med. Biol. 2017, 1033, 97–131. [Google Scholar]
- Yang, Y.X.; Lewis, J.D.; Epstein, S.; Metz, D.C. Long-term proton pump inhibitor therapy and risk of hip fracture. JAMA 2006, 296, 2947–2953. [Google Scholar] [CrossRef]
- Yang, Y.X. Proton pump inhibitor therapy and osteoporosis. Curr. Drug Saf. 2008, 3, 204–209. [Google Scholar] [CrossRef] [PubMed]
- Andersen, B.N.; Johansen, P.B.; Abrahamsen, B. Proton pump inhibitors and osteoporosis. Curr. Opin. Rheumatol. 2016, 28, 420–425. [Google Scholar] [CrossRef] [PubMed]
- Wright, M.J.; Sullivan, R.R.; Gaffney-Stomberg, E.; Caseria, D.M.; O’Brien, K.O.; Proctor, D.D.; Simpson, C.A.; Kerstetter, J.E.; Insogna, K.L. Inhibiting gastric acid production does not affect intestinal calcium absorption in young healthy individuals: A randomized, crossover controlled clinical trial. J. Bone Miner. Res. 2010, 25, 2205–2211. [Google Scholar] [CrossRef] [PubMed]
- Ramsubeik, K.; Keuler, N.S.; Davis, L.A.; Hansen, K.E. Factors associated with calcium absorption in postmenopausal women: A post hoc analysis of dual-isotope studies. J. Acad. Nutr. Diet. 2014, 114, 761–767. [Google Scholar] [CrossRef]
- Vestergaard, P.; Rejnmark, L.; Mosekilde, L. Proton pump inhibitors, histamine H2 receptor antagonists, and other antacid medications and the risk of fracture. Calcif. Tissue Int. 2006, 79, 76–83. [Google Scholar] [CrossRef]
- Targownik, L.E.; Lix, L.M.; Leung, S.; Leslie, W.D. Proton-pump inhibitor use is not associated with osteoporosis or accelerated bone mineral density loss. Gastroenterology 2010, 138, 896–904. [Google Scholar] [CrossRef]
- Kaye, J.A.; Jick, H. Proton pump inhibitor use and risk of hip fractures in patients without major risk factors. Pharmacotherapy 2008, 28, 951–959. [Google Scholar] [CrossRef]
- Kumar, S.; Drake, M.T.; Schleck, C.D.; Johnson, M.L.; Alexander, J.A.; Katzka, D.A.; Iyer, P.G. Incidence and predictors of osteoporotic fractures in patients with Barrett’s oesophagus: A population-based nested case-control study. Aliment. Pharmacol. Ther. 2017, 46, 1094–1102. [Google Scholar] [CrossRef]
- Targownik, L.E. Editorial: Let’s take a break from studying the PPI-fracture association. Aliment. Pharmacol. Ther. 2018, 47, 1543–1544. [Google Scholar] [CrossRef]
- Khalili, H.; Huang, E.S.; Jacobson, B.C.; Camargo, C.A.; Feskanich, D.; Chan, A.T. Use of proton pump inhibitors and risk of hip fracture in relation to dietary and lifestyle factors: A prospective cohort study. BMJ 2012, 344, e372. [Google Scholar] [CrossRef]
- Poly, T.N.; Islam, M.M.; Yang, H.C.; Wu, C.C.; Li, Y.J. Proton pump inhibitors and risk of hip fracture: A meta-analysis of observational studies. Osteoporos. Int. 2019, 30, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Huang, Y.; Li, H.; Sun, W.; Liu, J. Proton-pump inhibitors and risk of fractures: An update meta-analysis. Osteoporos. Int. 2016, 27, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.M.; Yang, S.H.; Liang, C.C.; Huang, H.K. Proton pump inhibitor use and the risk of osteoporosis and fracture in stroke patients: A population-based cohort study. Osteoporos. Int. 2018, 29, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.; Siddiqui, A.N.; Habib, A.; Hussain, M.S.; Najmi, A.K. Proton pump inhibitors’ use and risk of hip fracture: A systematic review and meta-analysis. Rheumatol. Int. 2018, 38, 1999–2014. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, T.; Watarai, K.; Oda, T.; Kim, Y.T.; Oda, H. Proton pump inhibitors and fracture: They impair bone quality and increase fall risk? Osteoporos. Int. 2016, 27, 1675–1676. [Google Scholar] [CrossRef] [PubMed]
- Thaler, H.W.; Sterke, C.S.; van der Cammen, T.J. Association of Proton Pump Inhibitor Use with Recurrent Falls and Risk of Fractures in Older Women: A Study of Medication Use in Older Fallers. J. Nutr. Health Aging 2016, 20, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Yu, E.W.; Bauer, S.R.; Bain, P.A.; Bauer, D.C. Proton pump inhibitors and risk of fractures: A meta-analysis of 11 international studies. Am. J. Med. 2011, 124, 519–526. [Google Scholar] [CrossRef]
- Sugiyama, T. Understanding the Current Evidence on Proton Pump Inhibitor Use and Bone Health. Gastroenterology 2019, 157, 585. [Google Scholar] [CrossRef] [Green Version]
- Targownik, L.E.; Leslie, W.D.; Davison, K.S.; Goltzman, D.; Jamal, S.A.; Kreiger, N.; Josse, R.G.; Kaiser, S.M.; Kovacs, C.S.; Prior, J.C.; et al. The relationship between proton pump inhibitor use and longitudinal change in bone mineral density: A population-based study [corrected] from the Canadian Multicentre Osteoporosis Study (CaMos). Am. J. Gastroenterol. 2012, 107, 1361–1369. [Google Scholar] [CrossRef]
- Targownik, L.E.; Goertzen, A.L.; Luo, Y.; Leslie, W.D. Long-Term Proton Pump Inhibitor Use Is Not Associated with Changes in Bone Strength and Structure. Am. J. Gastroenterol. 2017, 112, 95–101. [Google Scholar] [CrossRef]
- King, C.E.; Toskes, P.P. Nutrient malabsorption in the Zollinger-Ellison Syndrome. Arch. Intern. Med. 1983, 143, 349–351. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.H.; Clement, S. Zollinger-Ellison syndrome presenting with osteomalacia. Va. Med. 1981, 108, 619–621. [Google Scholar] [PubMed]
- Coutinho, F.L.; Lourenco, D.M., Jr.; Toledo, R.A.; Montenegro, F.L.M.; Correia-Deur, J.E.M.; Toledo, S.P.A. Bone mineral density analysis in patients with primary hyperparathyroidism associated with multiple endocrine neoplasia type 1 after total parathyroidectomy. Clin. Endocrinol. (Oxf.) 2010, 72, 462–468. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.M.; Vodopivec, D.; Christakis, I.; Lyons, G.; Wei, Q.; Waguespack, S.G.; Petak, S.M.; Grubbs, E.; Lee, J.E.; Perrier, N. Operative intervention for primary hyperparathyroidism offers greater bone recovery in patients with sporadic disease than in those with multiple endocrine neoplasia type 1-related hyperparathyroidism. Surgery 2017, 161, 107–115. [Google Scholar] [CrossRef]
- Eller-Vainicher, C.; Chiodini, I.; Battista, C.; Viti, R.; Mascia, M.L.; Massironi, S.; Peracchi, M.; D’Agruma, L.; Minisola, S.; Corbetta, S.; et al. Sporadic and MEN1-related primary hyperparathyroidism: Differences in clinical expression and severity. J. Bone Miner. Res. 2009, 24, 1404–1410. [Google Scholar] [CrossRef]
- Burgess, J.R.; Shepherd, J.J.; Murton, F.J.; Parameswaran, V.; Greenaway, T.M. Effective control of bone pain by octreotide in a patient with metastatic gastrinoma. Med. J. Aust. 1996, 164, 725–727. [Google Scholar] [CrossRef]
- Gillen, D.; Mc Coll, K.E. Problems related to acid rebound and tachyphylaxis. Best Pract. Res. Clin. Gastroenterol. 2001, 15, 487–495. [Google Scholar] [CrossRef]
- Hunfeld, N.G.; Geus, W.P.; Kuipers, E.J. Systematic review: Rebound acid hypersecretion after therapy with proton pump inhibitors. Aliment. Pharmacol. Ther. 2007, 25, 39–46. [Google Scholar] [CrossRef]
- Waldum, H.L.; Qvigstad, G.; Fossmark, R.; Kleveland, P.M.; Sandvik, A.K. Rebound acid hypersecretion from a physiological, pathophysiological and clinical viewpoint. Scand. J. Gastroenterol. 2010, 45, 389–394. [Google Scholar] [CrossRef]
- Fossmark, R.; Johnsen, G.; Johanessen, E.; Fossmark, R.; Waldum, H.L. Rebound acid hypersecretion after long-term inhibition of gastric acid secretion. Aliment. Pharmacol. Ther. 2005, 21, 149–154. [Google Scholar] [CrossRef]
- Reimer, C.; Sondergaard, B.; Hilsted, L.; Bytzer, P. Proton-pump inhibitor therapy induces acid-related symptoms in healthy volunteers after withdrawal of therapy. Gastroenterology 2009, 137, 80–87. [Google Scholar] [CrossRef] [PubMed]
- McColl, K.E.; Gillen, D. Evidence that proton-pump inhibitor therapy induces the symptoms it is used to treat. Gastroenterology 2009, 137, 20–22. [Google Scholar] [CrossRef] [PubMed]
- Berna, M.J.; Hoffmann, K.M.; Long, S.H.; Serrano, J.; Gibril, F.; Jensen, R.T. Serum gastrin in Zollinger-Ellison syndrome: II. Prospective study of gastrin provocative testing in 293 patients from the National Institutes of Health and comparison with 537 cases from the literature. evaluation of diagnostic criteria, proposal of new criteria, and correlations with clinical and tumoral features. Medicine (Baltimore) 2006, 85, 331–364. [Google Scholar] [PubMed]





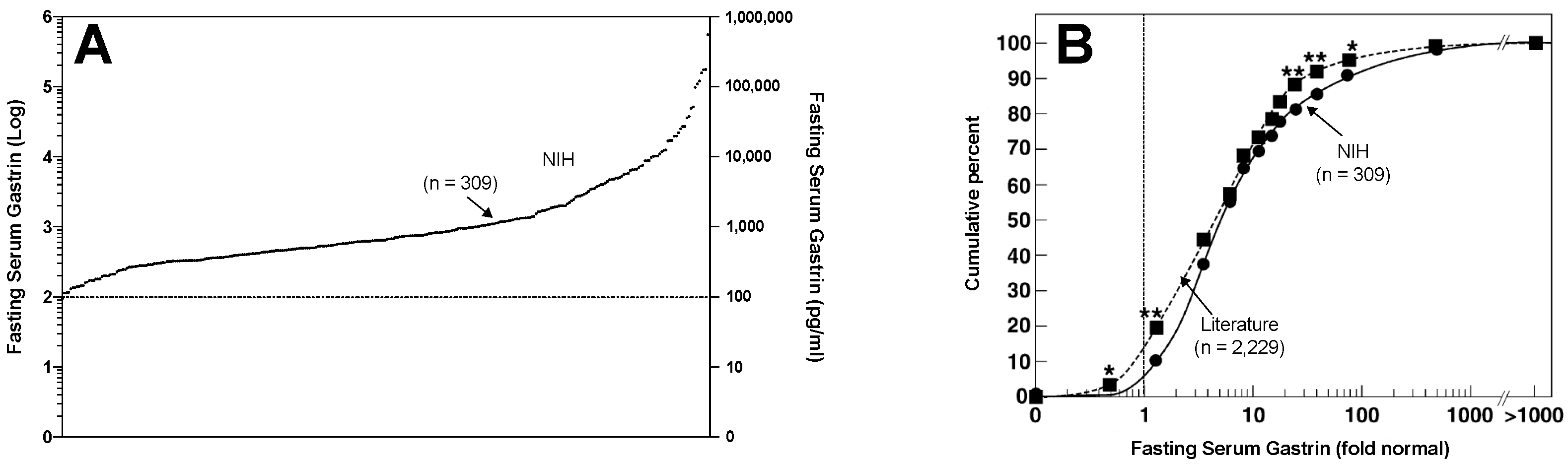{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Potential PPI Side-Effect | Potential Mechanism | Insights from ZES Studies |
|---|---|---|
| General Why ZES useful model of Chr HG | Chronic hypergastrinemia (Chr. HG) |
|
| General Why ZES useful model of long-term PPI use | Lifelong need for potent gastric antisecretory drugs—PPIs drug of choice |
|
| ECL hyperplasia/gastric carcinoids | Chronic hypergastrinemia (Chr. HG) |
|
| Esophageal, gastric, pancreatic adenocarcinomas | Chronic hypergastrinemia (Chr. HG) |
|
| Colorectal cancer | Chronic hypergastrinemia (Chr. HG) |
|
| Nutrient malabsorption (Fe, Ca) | PPI-induced Hypo-/Achlorhydria |
|
| Nutrient malabsorption (VB12) | PPI-induced Hypo-/Achlorhydria |
|
| Hypomagnesemia | Unclear = mechanism |
|
| Bone fractures | Unclear = mechanism |
|
| Rebound hypersecretion after stopping PPI | Unclear = mechanism |
|
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, L.; Ramos-Alvarez, I.; Ito, T.; Jensen, R.T. Insights into Effects/Risks of Chronic Hypergastrinemia and Lifelong PPI Treatment in Man Based on Studies of Patients with Zollinger–Ellison Syndrome. Int. J. Mol. Sci. 2019, 20, 5128. https://doi.org/10.3390/ijms20205128
Lee L, Ramos-Alvarez I, Ito T, Jensen RT. Insights into Effects/Risks of Chronic Hypergastrinemia and Lifelong PPI Treatment in Man Based on Studies of Patients with Zollinger–Ellison Syndrome. International Journal of Molecular Sciences. 2019; 20(20):5128. https://doi.org/10.3390/ijms20205128
Chicago/Turabian StyleLee, Lingaku, Irene Ramos-Alvarez, Tetsuhide Ito, and Robert T. Jensen. 2019. "Insights into Effects/Risks of Chronic Hypergastrinemia and Lifelong PPI Treatment in Man Based on Studies of Patients with Zollinger–Ellison Syndrome" International Journal of Molecular Sciences 20, no. 20: 5128. https://doi.org/10.3390/ijms20205128