Structural and Functional Characterization of Three Novel Fungal Amylases with Enhanced Stability and pH Tolerance

 , , , and
, , , and
Abstract
:1. Introduction
2. Results
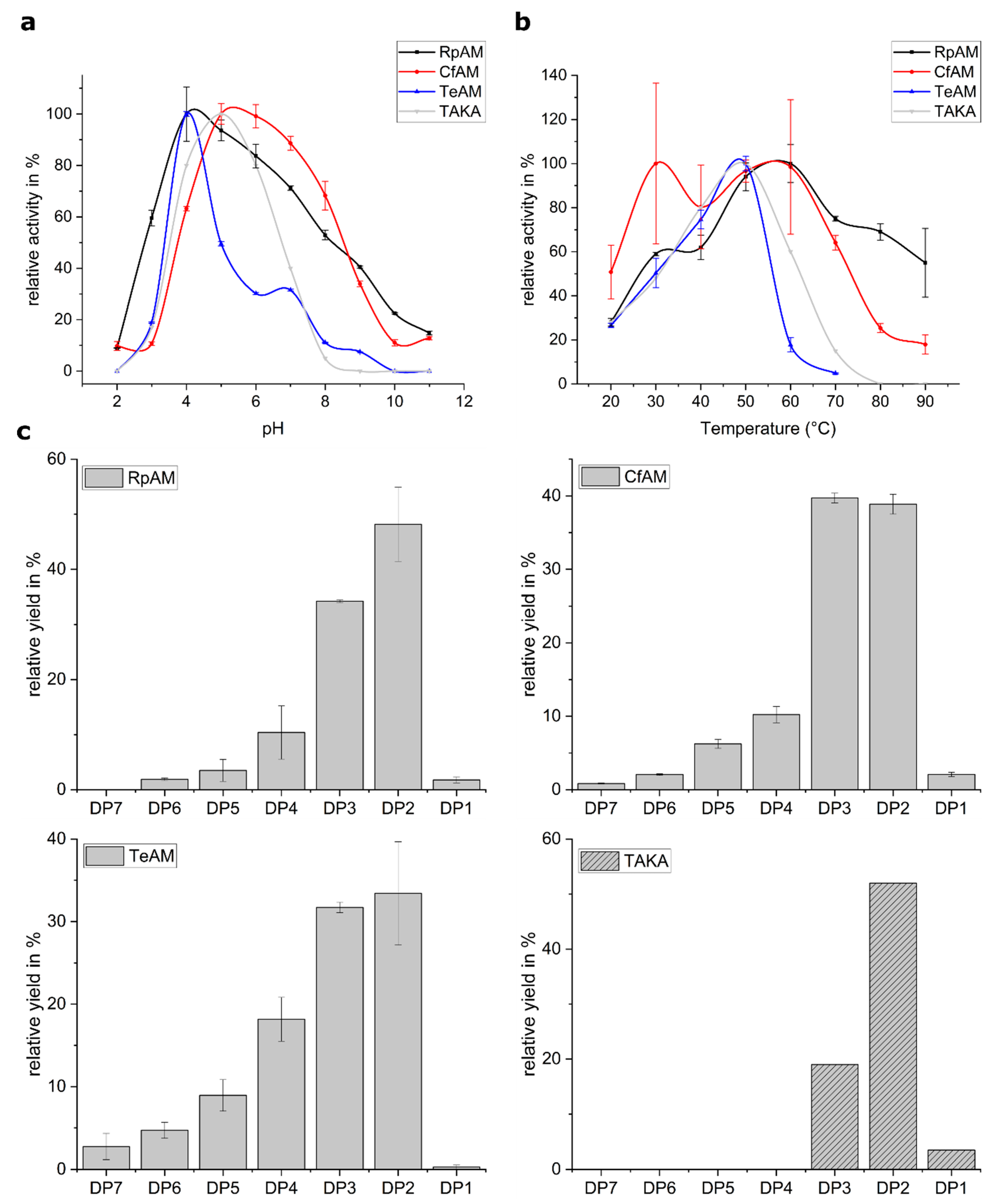
2.1. Biochemical Characterization
2.2. Overall Fold
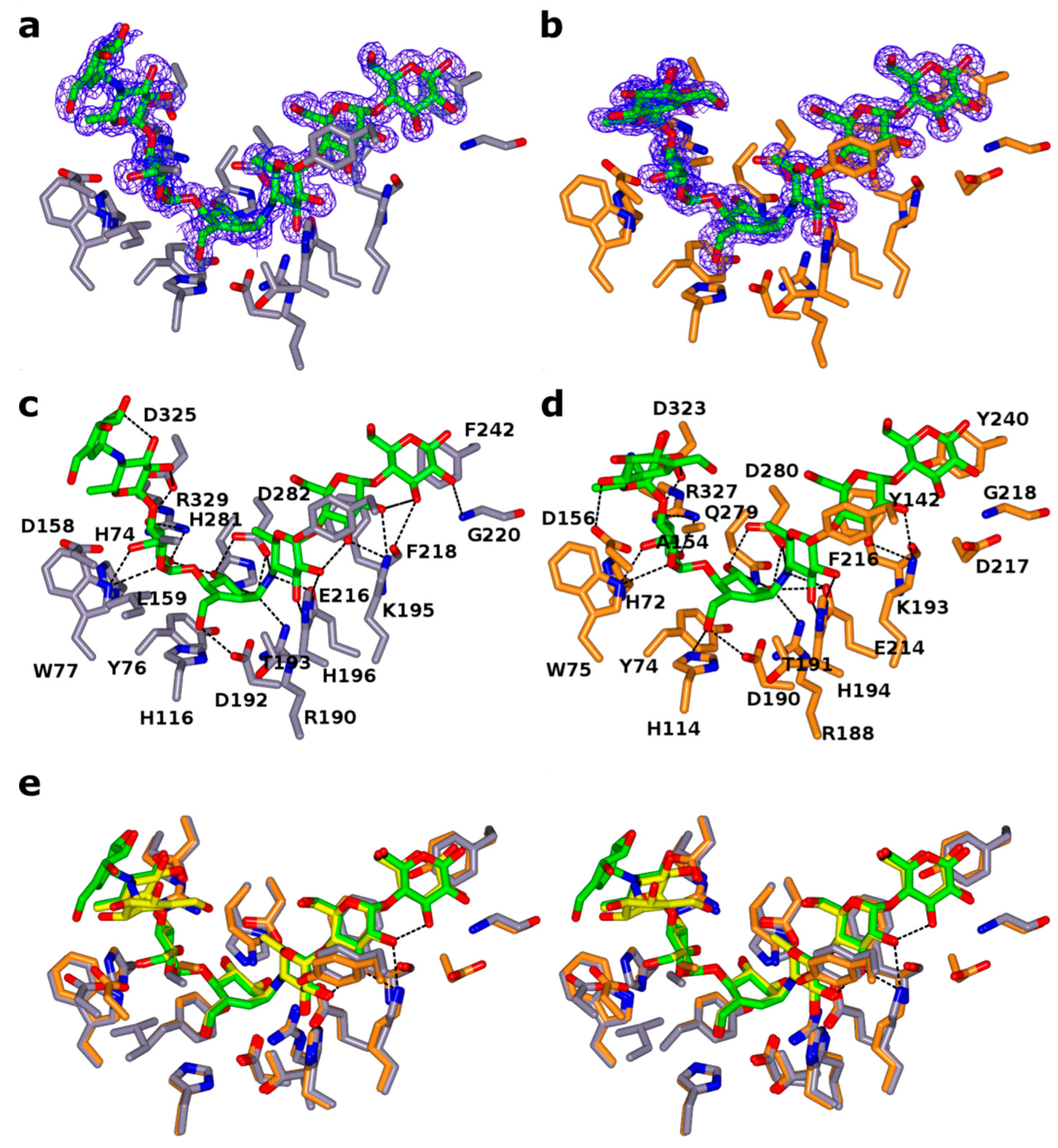
2.3. Ligand Binding Site
2.4. Secondary Glucose Binding Site
2.5. N-Glycosylation
2.6. Isoasparate Formation
3. Discussion
4. Materials and Methods
4.1. Macromolecule Production
4.2. Biochemical Characterisation
4.2.1. pH Optimum
4.2.2. Temperature Optimum
4.2.3. Product Profile
4.3. Crystallisation
4.3.1. RpAM
4.3.2. TeAM
4.3.3. CfAM
4.4. Data Collection and Processing
4.5. Structure Solution and Refinement
5. Conclusions
6. Patents
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CfAM. | Cordyceps farinosa amylase |
| RpAM | Rhizomucor pusillus amylase |
| TeAM | Thamnidium elegans amylase |
| TAKA | Aspergillus oryzae amylase |
| dp | Degree of polymerization |
References
- Payen, A.P.J.F. Memoire sur la diastase, les principaux produits de ses réactions et leurs applications aux arts industriels” (Memoir on diastase, the principal products of its reactions, and their applications to the industrial arts). Annal. Chim. Phys. 1833, 2, 73–92. [Google Scholar]
- Gurung, N.; Ray, S.; Bose, S.; Rai, V. A broader view: Microbial enzymes and their relevance in industries, medicine, and beyond. Biomed Res. Int. 2013, 2013, 329121. [Google Scholar] [CrossRef] [PubMed]
- Roy, J.K.; Manhar, A.K.; Nath, D.; Mandal, M.; Mukherjee, A.K. Cloning and extracellular expression of a raw starch digesting alpha-amylase (Blamy-I) and its application in bioethanol production from a non-conventional source of starch. J. Basic Microbiol. 2015, 55, 1287–1298. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Gigras, P.; Mohapatra, H.; Goswami, V.K.; Chauhan, B. Microbial α-amylases: A biotechnological perspective. Process Biochem. 2003, 38, 1599–1616. [Google Scholar] [CrossRef]
- Niehaus, F.; Bertoldo, C.; Kahler, M.; Antranikian, G. Extremophiles as a source of novel enzymes for industrial application. Appl. Microbiol. Biotechnol. 1999, 51, 711–729. [Google Scholar] [CrossRef]
- Lombard, V.; Golaconda Ramulu, H.; Drula, E.; Coutinho, P.M.; Henrissat, B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014, 42, D490–D495. [Google Scholar] [CrossRef]
- Janecek, S.; Svensson, B.; MacGregor, E.A. Structural and evolutionary aspects of two families of non-catalytic domains present in starch and glycogen binding proteins from microbes, plants and animals. Enzyme Microb. Technol. 2011, 49, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yu, J.; Li, F.; Peng, H.; Zhang, X.; Xiao, Y.; He, C. Crystal structure of a raw-starch-degrading bacterial alpha-amylase belonging to subfamily 37 of the glycoside hydrolase family GH13. Sci. Rep. 2017, 7, 44067. [Google Scholar] [CrossRef]
- Mehta, D.; Satyanarayana, T. Domain C of thermostable alpha-amylase of Geobacillus thermoleovorans mediates raw starch adsorption. Appl. Microbiol. Biotechnol. 2014, 98, 4503–4519. [Google Scholar] [CrossRef]
- Sogaard, M.; Kadziola, A.; Haser, R.; Svensson, B. Site-directed mutagenesis of histidine 93, aspartic acid 180, glutamic acid 205, histidine 290, and aspartic acid 291 at the active site and tryptophan 279 at the raw starch binding site in barley alpha-amylase 1. J. Biol. Chem. 1993, 268, 22480–22484. [Google Scholar]
- Kadziola, A.; Sogaard, M.; Svensson, B.; Haser, R. Molecular structure of a barley alpha-amylase-inhibitor complex: Implications for starch binding and catalysis. J. Mol. Biol. 1998, 278, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Brzozowski, A.M.; Lawson, D.M.; Turkenburg, J.P.; Bisgaard-Frantzen, H.; Svendsen, A.; Borchert, T.V.; Dauter, Z.; Wilson, K.S.; Davies, G.J. Structural analysis of a chimeric bacterial alpha-amylase. High-resolution analysis of native and ligand complexes. Biochemistry 2000, 39, 9099–9107. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, P.E. Studies on the bread-improving mechanism of fungal alpha-amylase. J. Biol. Educ. 1992, 26, 12–18. [Google Scholar] [CrossRef]
- D'Arcy, A.; Bergfors, T.; Cowan-Jacob, S.W.; Marsh, M. Microseed matrix screening for optimization in protein crystallization: What have we learned? Acta Crystallogr F Struct Biol Commun 2014, 70, 1117–1126. [Google Scholar] [CrossRef]
- McNicholas, S.; Agirre, J. Glycoblocks: A schematic three-dimensional representation for glycans and their interactions. Acta Crystallogr. D Struct. Biol. 2017, 73, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Janeček, Š. Does the increased hydrophobicity of the interior and hydrophilicity of the exterior of an enzyme structure reflect its increased thermostability? Int. J. Biol. Macromol. 1993, 15, 317–318. [Google Scholar] [CrossRef]
- Mok, S.C.; Teh, A.H.; Saito, J.A.; Najimudin, N.; Alam, M. Crystal structure of a compact alpha-amylase from Geobacillus thermoleovorans. Enzyme Microb. Technol. 2013, 53, 46–54. [Google Scholar] [CrossRef]
- Mazola, Y.; Guirola, O.; Palomares, S.; Chinea, G.; Menendez, C.; Hernandez, L.; Musacchio, A. A comparative molecular dynamics study of thermophilic and mesophilic beta-fructosidase enzymes. J. Mol. Model. 2015, 21, 228. [Google Scholar] [CrossRef]
- Brzozowski, A.M.; Davies, G.J. Structure of the Aspergillus oryzae alpha-amylase complexed with the inhibitor acarbose at 2.0 A resolution. Biochemistry 1997, 36, 10837–10845. [Google Scholar] [CrossRef]
- Przylas, I.; Terada, Y.; Fujii, K.; Takaha, T.; Saenger, W.; Strater, N. X-ray structure of acarbose bound to amylomaltase from Thermus aquaticus. Implications for the synthesis of large cyclic glucans. Eur. J. Biochem. 2000, 267, 6903–6913. [Google Scholar] [CrossRef]
- Juge, N.; Rodenburg, K.W.; Guo, X.J.; Chaix, J.C.; Svensson, B. Isozyme hybrids within the protruding third loop domain of the barley alpha-amylase (beta/alpha)8-barrel. Implication for BASI sensitivity and substrate affinity. FEBS Lett. 1995, 363, 299–303. [Google Scholar] [CrossRef]
- Rodenburg, K.W.; Juge, N.; Guo, X.J.; Sogaard, M.; Chaix, J.C.; Svensson, B. Domain B protruding at the third beta strand of the alpha/beta barrel in barley alpha-amylase confers distinct isozyme-specific properties. Eur. J. Biochem. 1994, 221, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Penninga, D.; Strokopytov, B.; Rozeboom, H.J.; Lawson, C.L.; Dijkstra, B.W.; Bergsma, J.; Dijkhuizen, L. Site-directed mutations in tyrosine 195 of cyclodextrin glycosyltransferase from Bacillus circulans strain 251 affect activity and product specificity. Biochemistry 1995, 34, 3368–3376. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, A.; Haga, K.; Yamane, K. Four aromatic residues in the active center of cyclodextrin glucanotransferase from alkalophilic Bacillus sp. 1011: Effects of replacements on substrate binding and cyclization characteristics. Biochemistry 1994, 33, 9929–9936. [Google Scholar] [CrossRef]
- Krissinel, E. On the relationship between sequence and structure similarities in proteomics. Bioinformatics 2007, 23, 717–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robert, X.; Haser, R.; Gottschalk, T.E.; Ratajczak, F.; Driguez, H.; Svensson, B.; Aghajari, N. The structure of barley alpha-amylase isozyme 1 reveals a novel role of domain C in substrate recognition and binding: A pair of sugar tongs. Structure 2003, 11, 973–984. [Google Scholar] [CrossRef]
- Sorimachi, K.; Le Gal-Coeffet, M.F.; Williamson, G.; Archer, D.B.; Williamson, M.P. Solution structure of the granular starch binding domain of Aspergillus niger glucoamylase bound to beta-cyclodextrin. Structure 1997, 5, 647–661. [Google Scholar] [CrossRef]
- de Barros, M.C.; do Nascimento Silva, R.; Ramada, M.H.; Galdino, A.S.; de Moraes, L.M.; Torres, F.A.; Ulhoa, C.J. The influence of N-glycosylation on biochemical properties of Amy1, an alpha-amylase from the yeast Cryptococcus flavus. Carbohydr. Res. 2009, 344, 1682–1686. [Google Scholar] [CrossRef]
- Amore, A.; Knott, B.C.; Supekar, N.T.; Shajahan, A.; Azadi, P.; Zhao, P.; Wells, L.; Linger, J.G.; Hobdey, S.E.; Vander Wall, T.A.; et al. Distinct roles of N- and O-glycans in cellulase activity and stability. Proc. Natl. Acad. Sci. USA 2017, 114, 13667–13672. [Google Scholar] [CrossRef] [Green Version]
- Reissner, K.J.; Aswad, D.W. Deamidation and isoaspartate formation in proteins: Unwanted alterations or surreptitious signals? Cell. Mol. Life Sci. 2003, 60, 1281–1295. [Google Scholar] [CrossRef]
- Barends, T.R.; Bultema, J.B.; Kaper, T.; van der Maarel, M.J.; Dijkhuizen, L.; Dijkstra, B.W. Three-way stabilization of the covalent intermediate in amylomaltase, an alpha-amylase-like transglycosylase. J. Biol. Chem. 2007, 282, 17242–17249. [Google Scholar] [CrossRef] [PubMed]
- Roth, C.; Weizenmann, N.; Bexten, N.; Saenger, W.; Zimmermann, W.; Maier, T.; Strater, N. Amylose recognition and ring-size determination of amylomaltase. Sci. Adv. 2017, 3, e1601386. [Google Scholar] [CrossRef] [PubMed]
- Dalboege, H.; Christgau, S.; Andersen, L.N.; Kofod, L.V.; Kauppinen, M.S. DNA encoding an enxyme with endoglucanase activity from Trichoderma harzianum. World Patent WO/1995/002043, 1995. [Google Scholar]
- Sun, Y.; Li, M.; Zhang, Y.; Liu, L.; Liu, Y.; Liu, Z.; Li, X.; Lou, Z. Crystallization and preliminary crystallographic analysis of Gibberella zeae extracellular lipase. Acta Crystallogr. Sect. F Struct. Biol. Cryst Commun. 2008, 64, 813–815. [Google Scholar] [CrossRef] [PubMed]
- Britton, H.T.S.; Robinson, R.A. CXCVIII.—Universal buffer solutions and the dissociation constant of veronal. J. Chem. Soc. 1931, 1456–1462. [Google Scholar] [CrossRef]
- Shaw Stewart, P.D.; Kolek, S.A.; Briggs, R.A.; Chayen, N.E.; Baldock, P.F.M. Random Microseeding: A Theoretical and Practical Exploration of Seed Stability and Seeding Techniques for Successful Protein Crystallization. Cryst. Growth Des. 2011, 11, 3432–3441. [Google Scholar] [CrossRef]
- Shah, A.K.; Liu, Z.-J.; Stewart, P.D.; Schubot, F.D.; Rose, J.P.; Newton, M.G.; Wang, B.-C. On increasing protein-crystallization throughput for X-ray diffraction studies. Acta Crystallogr. Sect. D 2005, 61, 123–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabsch, W. Xds. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Evans, P.R.; Murshudov, G.N. How good are my data and what is the resolution? Acta Crystallogr. D Biol. Crystallogr. 2013, 69, 1204–1214. [Google Scholar] [CrossRef]
- Vagin, A.; Teplyakov, A. Molecular replacement with MOLREP. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 22–25. [Google Scholar] [CrossRef]
- Cowtan, K. The Buccaneer software for automated model building. 1. Tracing protein chains. Acta Crystallogr. D Biol. Crystallogr. 2006, 62, 1002–1011. [Google Scholar] [CrossRef]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [PubMed]
- Murshudov, G.N.; Vagin, A.A.; Dodson, E.J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 1997, 53, 240–255. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CfAM | TeAM | RpAM | |
|---|---|---|---|
| Diffraction source | Diamond I02 | Diamond I02 | ESRF ID29 |
| Wavelength (Å) | 0.9795 | 0.9795 | 1.0004 |
| Temperature (K) | 100 | 100 | 100 |
| Space group | P1 | P212121 | P1 |
| a, b, c (Å) | 56.88, 61.97, 70.40 | 51.02, 56.63, 166.01 | 51.22, 62.60, 66.81 |
| α, β, γ (°) | 79.33, 82.88, 67.99 | 90, 90, 90 | 77.03, 81.04, 89.62 |
| Resolution range (Å) | 33.1–1.35 (1.37–1.35) | 48.76–1.20 (1.22-1.20) | 43.21–1.4 (1.42–1.40) |
| Total No. of reflections | 342,708 | 1,149,540 | 315,876 |
| No. of unique reflections | 163,777 | 150,529 | 146,177 |
| Completeness (%) | 85.3 (38.2) | 99.8 (96.7) | 92.9 (61.9) |
| Redundancy | 2.1 (2.1) | 7.6 (4.6) | 2.2 (2.1) |
| 〈I/σ(I)〉 | 13.7 (10.3) | 14.1(1.7) | 9.6 (2.3) |
| Rr.i.m. | 0.076 (0.129) | 0.021 (0.446) | 0.030 (0.225) |
| CC1/2 | 0.983 (0.970 | 0.999 (0.615) | 0.998 (0.892) |
| Overall B factor from Wilson plot (Å2) | 6.8 | 8.7 | 8.1 |
| CfAM | TeAM | RpAM | |
|---|---|---|---|
| PDB-ID | 6SAV | 6SAO | 6SAU |
| Resolution range (Å) | 33.1–1.35 (1.385–1.35) | 48.76–1.20 (1.22–1.20) | 39.99–1.4 (1.42–1.40) |
| Completeness (%) | 85.3 (39.7) | 99.8 (96.7) | 92.8 (89.2) |
| No. of reflections, working set | 155,488 | 143,033 | 138,848 |
| No. of reflections, test set | 8289 | 7574 | 7328 |
| Final Rcryst | 0.113 (0.09) | 0.110 (0.27) | 0.136 (0.22) |
| Final Rfree | 0.150 (0.17) | 0.134 (0.29) | 0.164 (0.26) |
| Cruickshank DPI | 0.051 | 0.027 | 0.056 |
| No. of subunits in the asymmetric unit | 2 | 1 | 2 |
| No. of non-H atoms | Chain A/B | Chain A | Chain A/B |
| Protein | 3557/3609 | 3570 | 3662/3592 |
| Ion | 1/2 | 1 | 1/1 |
| Ligand | 99/120 | 133 | 14/36 |
| Water | 875 | 568 | 943 |
| Total | 8263 | 4272 | 8306 |
| R.m.s. deviations | |||
| Bonds (Å) | 0.0191 | 0.0163 | 0.0147 |
| Angles (°) | 2.06 | 1.937 | 1.875 |
| Average B factors (Å2) | Chain A/B | Chain A | Chain A/B |
| Protein | 10/8.7 | 12.9 | 12.9/12.3 |
| Ions | |||
| Ca2+ | 6.7/5.8 | 9.5 | 7.39/7.5 |
| Na2+ | N/A/10.9 | ||
| Ligand | 19.6/18.0 | 22.2 | 19.2/21.4 |
| Water | 19.0 | 28.8 | 24.21 |
| Ramachandran plot | |||
| Most favoured (%) | 98.6 | 97.7 | 97.2 |
| Allowed (%) | 1.4 | 2.3 | 2.7 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roth, C.; Moroz, O.V.; Turkenburg, J.P.; Blagova, E.; Waterman, J.; Ariza, A.; Ming, L.; Tianqi, S.; Andersen, C.; Davies, G.J.; et al. Structural and Functional Characterization of Three Novel Fungal Amylases with Enhanced Stability and pH Tolerance. Int. J. Mol. Sci. 2019, 20, 4902. https://doi.org/10.3390/ijms20194902
Roth C, Moroz OV, Turkenburg JP, Blagova E, Waterman J, Ariza A, Ming L, Tianqi S, Andersen C, Davies GJ, et al. Structural and Functional Characterization of Three Novel Fungal Amylases with Enhanced Stability and pH Tolerance. International Journal of Molecular Sciences. 2019; 20(19):4902. https://doi.org/10.3390/ijms20194902
Chicago/Turabian StyleRoth, Christian, Olga V. Moroz, Johan P. Turkenburg, Elena Blagova, Jitka Waterman, Antonio Ariza, Li Ming, Sun Tianqi, Carsten Andersen, Gideon J. Davies, and et al. 2019. "Structural and Functional Characterization of Three Novel Fungal Amylases with Enhanced Stability and pH Tolerance" International Journal of Molecular Sciences 20, no. 19: 4902. https://doi.org/10.3390/ijms20194902