Targeting Tyrosine Kinases in Acute Myeloid Leukemia: Why, Who and How?

 , ,
, ,  and
and
Abstract
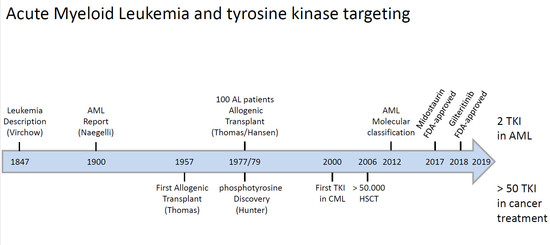
:
1. Introduction
2. Kinome and Tyrosine Kinase
3. Tyrosine Kinase in Hematopoietic Tissue
4. Tyrosine Kinase in Acute Myeloid Leukemia
5. Tyrosine Kinase Inhibitors
5.1. FLT3 Tyrosine Kinase Inhibitors
5.2. KIT Tyrosine Kinase Inhibitors
5.3. TAM Tyrosine Kinase Inhibitors
5.4. SYK Tyrosine Kinase Inhibitors
5.5. SFK Tyrosine Kinase Inhibitors
5.6. MET/RON Tyrosine Kinase Inhibitors
5.7. TEC Family Tyrosine Kinase Inhibitors
6. Concluding Remarks
Funding
Conflicts of Interest
References
- Robinson, D.R.; Wu, Y.M.; Lin, S.F. The protein tyrosine kinase family of the human genome. Oncogene 2000, 19, 5548–5557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The protein kinase complement of the human genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Chevalier, D.; Larue, C.; Ki Cho, S.; Walker, J.C. The protein phosphatases and protein kinases of Arabidopsis thaliana. Arabidopsis Book 2007, 5, e0106. [Google Scholar]
- Eckhart, W.; Hutchinson, M.; Hunter, T. An activity phosphorylating tyrosine in polyoma T antigen immunoprecipitates. Cell 1979, 18, 925–933. [Google Scholar] [CrossRef]
- Weiss, A.; Schlessinger, J. Switching signals on or off by receptor dimerization. Cell 1998, 94, 277–280. [Google Scholar] [CrossRef]
- Duong-Ly, K.C.; Peterson, J.R. The human kinome and kinase inhibition. Curr. Prot. Pharmacol. 2013, 60, 2–9. [Google Scholar]
- Traxler, P.M.; Furet, P.; Mett, H.; Buchdunger, E.; Meyer, T.; Lydon, N. 4-(Phenylamino)pyrrolopyrimidines: Potent and selective, ATP site directed inhibitors of the EGF-receptor protein tyrosine kinase. J. Med. Chem. 1996, 39, 2285–2292. [Google Scholar] [CrossRef]
- Carroll, M. CGP 57148, a tyrosine kinase inhibitor, inhibits the growth of cells expressing BCR-ABL, TEL-ABL, and TEL-PDGFR fusion proteins. Blood 1997, 90, 4947–4952. [Google Scholar]
- Fleuren, E.D.; Zhang, L.; Wu, J.; Daly, R.J. The kinome ‘at large’ in cancer. Nat. Rev. Cancer 2016, 16, 83–98. [Google Scholar] [CrossRef]
- Gross, S.; Rahal, R.; Stransky, N.; Lengauer, C.; Hoeflich, K.P. Targeting cancer with kinase inhibitors. J. Clin. Invest. 2015, 125, 1780–1789. [Google Scholar] [CrossRef]
- Roskoski, R. Classification of small molecule protein kinase inhibitors based upon the structures of their drug-enzyme complexes. Pharmacol. Res. 2016, 103, 26–48. [Google Scholar] [CrossRef] [PubMed]
- Bhullar, K.S.; Lagaron, N.O.; McGowan, E.M.; Parmar, I.; Jha, A.; Hubbard, B.P.; Rupasinghe, H.P.V. Kinase-targeted cancer therapies: Progress, challenges and future directions. Mol. Cancer 2018, 17, 48. [Google Scholar] [CrossRef] [PubMed]
- Paulson, R.F.; Bernstein, A. Receptor tyrosine kinases and the regulation of hematopoiesis. Semin. Immunol. 1995, 7, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Rohrschneider, L.R.; Bourette, R.P.; Lioubin, M.N.; Algate, P.A.; Myles, G.M.; Carlberg, K. Growth and differentiation signals regulated by the M-CSF receptor. Mol. Reprod. Dev. 1997, 46, 96–103. [Google Scholar] [CrossRef]
- Bernstein, A.; Forrester, L.; Reith, A.D.; Dubreuil, P.; Rottapel, R. The murine W/c-kit and Steel loci and the control of hematopoiesis. Semin. Hematol. 1991, 28, 138–142. [Google Scholar]
- Gilliland, D.G.; Griffin, J.D. Role of FLT3 in leukemia. Curr. Opin. Hematol. 2002, 9, 274–281. [Google Scholar] [CrossRef] [PubMed]
- Ihle, J.N. TheJanusprotein tyrosine kinases in hematopoietic cytokine signaling. Semin. Immonol. 1995, 7, 247–254. [Google Scholar] [CrossRef]
- Harrison, D.A.; Binari, R.; Nahreini, T.S.; Gilman, M.; Perrimon, N. Activation of a Drosophila Janus kinase (JAK) causes hematopoietic neoplasia and developmental defects. EMBO J. 1995, 14, 2857–2865. [Google Scholar] [CrossRef]
- Chow, L.M.L.; Veillette, A. The Src and Csk families of tyrosine protein kinases in hemopoietic cells. Semin. Immonol. 1995, 7, 207–226. [Google Scholar] [CrossRef]
- Corey, S.J.; Anderson, S.M. Src-related protein tyrosine kinases in hematopoiesis. Blood 1999, 93, 1–14. [Google Scholar]
- Ingley, E. Src family kinases: Regulation of their activities, levels and identification of new pathways. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2008, 1784, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Klingmüller, U.; Wu, H.; Hsiao, J.G.; Toker, A.; Duckworth, B.C.; Cantley, L.C.; Lodish, H.F. Identification of a novel pathway important for proliferation and differentiation of primary erythroid progenitors. Proc. Natl. Acad. Sci. USA 1997, 94, 3016–3021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lannutti, B.J.; Shim, M.-H.; Blake, N.; Reems, J.A.; Drachman, J.G. Identification and activation of Src family kinases in primary megakaryocytes. Exp. Hematol. 2003, 31, 1268–1274. [Google Scholar] [CrossRef] [PubMed]
- Hibbs, M.L.; Tarlinton, D.M.; Armes, J.; Grail, D.; Hodgson, G.; Maglitto, R.; Stacker, S.A.; Dunn, A.R. Multiple defects in the immune system of Lyn-deficient mice, culminating in autoimmune disease. Cell 1995, 83, 301–311. [Google Scholar] [CrossRef] [Green Version]
- Stein, P.L.; Vogel, H.; Soriano, P. Combined deficiencies of Src, Fyn, and Yes tyrosine kinases in mutant mice. Genes Dev. 1994, 8, 1999–2007. [Google Scholar] [CrossRef] [PubMed]
- Lowell, C.A.; Niwa, M.; Soriano, P.; Varmus, H.E. Deficiency of the Hck and Src tyrosine kinases results in extreme levels of extramedullary hematopoiesis. Blood 1996, 87, 1780–1792. [Google Scholar] [PubMed]
- Roche, S.; Koegl, M.; Barone, M.V.; Roussel, M.F.; Courtneidge, S.A. DNA synthesis induced by some but not all growth factors requires Src family protein tyrosine kinases. Mol. Cell Biol. 1995, 15, 1102–1109. [Google Scholar] [CrossRef] [Green Version]
- Corey, S.J.; Dombrosky-Ferlan, P.M.; Zuo, S.; Krohn, E.; Donnenberg, A.D.; Zorich, P.; Romero, G.; Takata, M.; Kurosaki, T. Requirement of src kinase lyn for induction of DNA synthesis by granulocyte colony-stimulating factor. J. Biol. Chem. 1998, 273, 3230–3235. [Google Scholar] [CrossRef]
- Linnekin, D.; DeBerry, C.S.; Mou, S. Lyn associates with the juxtamembrane region of c-Kit and is activated by stem cell factor in hematopoietic cell lines and normal progenitor cells. J. Biol. Chem. 1997, 272, 27450–27455. [Google Scholar] [CrossRef]
- Mano, H.; Ishikawa, F.; Nishida, J.; Hirai, H.; Takaku, F. A novel protein-tyrosine kinase, tec, is preferentially expressed in liver. Oncogene 1990, 5, 1781–1786. [Google Scholar]
- Yang, W.-C.; Collette, Y.; Nunes, J.A.; Olive, D. Tec kinases: A family with multiple roles in immunity. Immunity 2000, 12, 373–382. [Google Scholar] [CrossRef]
- Vihinen, M.; Brandau, O.; Brandén, L.J.; Kwan, S.P.; Lappalainen, I.; Lester, T.; Noordzij, J.G.; Ochs, H.D.; Ollila, J.; Pienaar, S.M.; et al. BTKbase, mutation database for X-linked agammaglobulinemia (XLA). Nucleic Acids Res. 1998, 26, 242–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Oers, N.S.C.; Weiss, A. The Syk/ZAP-70 protein tyrosine kinase connection to antigen receptor signalling processes. Semin. Immonol. 1995, 7, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Latour, S.; Chow, L.M.L.; Veillette, A. Differential intrinsic enzymatic activity of Syk and Zap-70 protein-tyrosine kinases. J. Biol. Chem. 1996, 271, 22782–22790. [Google Scholar] [CrossRef] [PubMed]
- Efremov, D.G.; Laurenti, L. The Syk kinase as a therapeutic target in leukemia and lymphoma. Expert Opin. Invest. Drugs 2011, 20, 623–636. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.M.; Rowley, B.; Pao, W.; Hayday, A.; Bolen, J.B.; Pawson, T. Syk tyrosine kinase required for mouse viability and B-cell development. Nature 1995, 378, 303–306. [Google Scholar] [CrossRef]
- Turner, M.; Joseph Mee, P.; Costello, P.S.; Williams, O.; Price, A.A.; Duddy, L.P.; Furlong, M.T.; Geahlen, R.L.; Tybulewicz, V.L.J. Perinatal lethality and blocked B-cell development in mice lacking the tyrosine kinase Syk. Nature 1995, 378, 298–302. [Google Scholar] [CrossRef]
- MacDonald, I.; Levy, J.; Pawson, T. Expression of the mammalian c-fes protein in hematopoietic cells and identification of a distinct fes-related protein. Mol. Cell Biol. 1985, 5, 2543–2551. [Google Scholar] [CrossRef]
- Yates, K.E.; Gasson, J.C. Role of c-Fes in normal and neoplastic hematopoiesis. Stem Cell J. 1996, 14, 117–123. [Google Scholar] [CrossRef]
- Craig, A.W. FES/FER kinase signaling in hematopoietic cells and leukemias. Front Biosci. 2012, 17, 861–875. [Google Scholar] [CrossRef]
- Blume-Jensen, P.; Hunter, T. Oncogenic kinase signalling. Nature 2001, 411, 355. [Google Scholar] [CrossRef] [PubMed]
- Scheijen, B.; Griffin, J.D. Tyrosine kinase oncogenes in normal hematopoiesis and hematological disease. Oncogene 2002, 21, 3314–3333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ku, M.; Wall, M.; MacKinnon, R.N.; Walkley, C.R.; Purton, L.E.; Tam, C.; Izon, D.; Campbell, L.; Cheng, H.-C.; Nandurkar, H. Src family kinases and their role in hematological malignancies. Leuk. Lymphoma 2015, 56, 577–586. [Google Scholar] [CrossRef] [PubMed]
- Morris, C.; Kennedy, M.; Heisterkamp, N.; Columbano-Green, L.; Romeril, K.; Groffen, J.; Fitzgerald, P. A complex chromosome rearrangement forms the BCR-ABL fusion gene in leukemic cells with a normal karyotype. Genes Chromosomes Cancer 1991, 3, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Patel, J.P.; Gonen, M.; Figueroa, M.E.; Fernandez, H.; Sun, Z.; Racevskis, J.; Van Vlierberghe, P.; Dolgalev, I.; Thomas, S.; Aminova, O.; et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N. Engl. J. Med. 2012, 366, 1079–1089. [Google Scholar] [CrossRef]
- Kaushansky, K. On the molecular origins of the chronic myeloproliferative disorders: It all makes sense. Blood 2005, 105, 4187–4190. [Google Scholar] [CrossRef]
- James, C.; Ugo, V.; Le Couedic, J.P.; Staerk, J.; Delhommeau, F.; Lacout, C.; Garcon, L.; Raslova, H.; Berger, R.; Bennaceur-Griscelli, A.; et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005, 434, 1144–1148. [Google Scholar] [CrossRef]
- Deschler, B.; Lübbert, M. Acute myeloid leukemia: Epidemiology and etiology. Int. J. Am. Cancer Soc. 2006, 107, 2099–2107. [Google Scholar] [CrossRef]
- Bullinger, L.; Döhner, K.; Döhner, H. Genomics of acute myeloid leukemia diagnosis and pathways. J. Clin. Oncol. 2017, 35, 934–946. [Google Scholar] [CrossRef]
- Hahn, C.K.; Berchuck, J.E.; Ross, K.N.; Kakoza, R.M.; Clauser, K.; Schinzel, A.C.; Ross, L.; Galinsky, I.; Davis, T.N.; Silver, S.J.; et al. Proteomic and genetic approaches identify Syk as an AML target. Cancer Cell 2009, 16, 281–294. [Google Scholar] [CrossRef]
- Puissant, A.; Fenouille, N.; Alexe, G.; Pikman, Y.; Bassil, C.F.; Mehta, S.; Du, J.; Kazi, J.U.; Luciano, F.; Rönnstrand, L.; et al. SYK is a critical regulator of FLT3 in acute myeloid leukemia. Cancer Cell 2014, 25, 226–242. [Google Scholar] [CrossRef] [PubMed]
- Farge, T.; Saland, E.; de Toni, F.; Aroua, N.; Hosseini, M.; Perry, R.; Bosc, C.; Sugita, M.; Stuani, L.; Fraisse, M.; et al. Chemotherapy-resistant human acute myeloid leukemia cells are not enriched for leukemic stem cells but require oxidative metabolism. Cancer Discov. 2017, 7, 716–735. [Google Scholar] [CrossRef] [PubMed]
- Kainz, B.; Heintel, D.; Marculescu, R.; Schwarzinger, I.; Sperr, W.; Le, T.; Weltermann, A.; Fonatsch, C.; Haas, O.A.; Mannhalter, C.; et al. Variable prognostic value of FLT3 internal tandem duplications in patients with de novo AML and a normal karyotype, t(15;17), t(8;21) or inv(16). Hematol. J. 2002, 3, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Stirewalt, D.L.; Radich, J.P. The role of FLT3 in haematopoietic malignancies. Nat. Rev. Cancer 2003, 3, 650–665. [Google Scholar] [CrossRef] [PubMed]
- Toffalini, F.; Demoulin, J.-B. New insights into the mechanisms of hematopoietic cell transformation by activated receptor tyrosine kinases. Blood 2010, 116, 2429–2437. [Google Scholar] [CrossRef] [Green Version]
- Whitman, S.P.; Maharry, K.; Radmacher, M.D.; Becker, H.; Mrozek, K.; Margeson, D.; Holland, K.B.; Wu, Y.Z.; Schwind, S.; Metzeler, K.H.; et al. FLT3 internal tandem duplication associates with adverse outcome and gene- and microRNA-expression signatures in patients 60 years of age or older with primary cytogenetically normal acute myeloid leukemia: A cancer and leukemia group B study. Blood 2010, 116, 3622–3626. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Ravandi, F.; Bayraktar, U.D.; Chiattone, A.; Bashir, Q.; Giralt, S.; Chen, J.; Qazilbash, M.; Kebriaei, P.; Konopleva, M.; et al. Treatment of FLT3-ITD-positive acute myeloid leukemia relapsing after allogeneic stem cell transplantation with Sorafenib. Biol. Blood Marrow Transplant. 2011, 17, 1874–1877. [Google Scholar] [CrossRef]
- Kindler, T.; Lipka, D.B.; Fischer, T. FLT3 as a therapeutic target in AML: Still challenging after all these years. Blood 2010, 116, 5089–5102. [Google Scholar] [CrossRef]
- Grunwald, M.R.; Levis, M.J. FLT3 inhibitors for acute myeloid leukemia: A review of their efficacy and mechanisms of resistance. Int. J. Hematol. 2013, 97, 683–694. [Google Scholar] [CrossRef]
- Levis, M.; Allebach, J.; Tse, K.F.; Zheng, R.; Baldwin, B.R.; Smith, B.D.; Jones-Bolin, S.; Ruggeri, B.; Dionne, C.; Small, D. A FLT3-targeted tyrosine kinase inhibitor is cytotoxic to leukemia cells in vitro and in vivo. Blood 2002, 99, 3885–3891. [Google Scholar] [CrossRef] [Green Version]
- Levis, M.; Ravandi, F.; Wang, E.S.; Baer, M.R.; Perl, A.; Coutre, S.; Erba, H.; Stuart, R.K.; Baccarani, M.; Cripe, L.D.; et al. Results from a randomized trial of salvage chemotherapy followed by lestaurtinib for patients with FLT3 mutant AML in first relapse. Blood 2011, 117, 3294–3301. [Google Scholar] [CrossRef] [PubMed]
- Rahmani, M.; Davis, E.M.; Bauer, C.; Dent, P.; Grant, S. Apoptosis induced by the kinase inhibitor BAY 43-9006 in human leukemia cells involves down-regulation of Mcl-1 through inhibition of translation. J. Biol. Chem. 2005, 280, 35217–35227. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Konopleva, M.; Ruvolo, V.R.; McQueen, T.; Evans, R.L.; Bornmann, W.G.; McCubrey, J.; Cortes, J.; Andreeff, M. Sorafenib induces apoptosis of AML cells via Bim-mediated activation of the intrinsic apoptotic pathway. Leukemia 2008, 22, 808–818. [Google Scholar] [CrossRef] [PubMed]
- Burchert, A.; Bug, G.; Finke, J.; Stelljes, M.; Rollig, C.; Wäsch, R.; Bornhauser, M.; Berg, T.; Lang, F.; Ehninger, G.; et al. Sorafenib as maintenance therapy post allogeneic stem cell transplantation for FLT3-ITD positive AML: Results from the randomized, double-blind, placebo-controlled multicentre sormain trial. Blood 2018, 132, 661. [Google Scholar]
- Mathew, N.R.; Baumgartner, F.; Braun, L.; O’Sullivan, D.; Thomas, S.; Waterhouse, M.; Muller, T.A.; Hanke, K.; Taromi, S.; Apostolova, P.; et al. Sorafenib promotes graft-versus-leukemia activity in mice and humans through IL-15 production in FLT3-ITD-mutant leukemia cells. Nat. Med. 2018, 24, 282. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, E.; Boulton, C.; Kelly, L.M.; Manley, P.; Fabbro, D.; Meyer, T.; Gilliland, D.G.; Griffin, J.D. Inhibition of mutant FLT3 receptors in leukemia cells by the small molecule tyrosine kinase inhibitor PKC412. Cancer Cell 2002, 1, 433–443. [Google Scholar] [CrossRef] [Green Version]
- Stone, R.M.; Manley, P.W.; Larson, R.A.; Capdeville, R. Midostaurin: Its odyssey from discovery to approval for treating acute myeloid leukemia and advanced systemic mastocytosis. Blood Adv. 2018, 2, 444–453. [Google Scholar] [CrossRef] [PubMed]
- O’Farrell, A.-M.; Abrams, T.J.; Yuen, H.A.; Ngai, T.J.; Louie, S.G.; Yee, K.W.H.; Wong, L.M.; Hong, W.; Lee, L.B.; Town, A.; et al. SU11248 is a novel FLT3 tyrosine kinase inhibitor with potent activity in vitro and in vivo. Blood 2003, 101, 3597–3605. [Google Scholar]
- Fiedler, W.; Serve, H.; Dohner, H.; Schwittay, M.; Ottmann, O.G.; O’Farrell, A.-M.; Bello, C.L.; Allred, R.; Manning, W.C.; Cherrington, J.M.; et al. A phase 1 study of SU11248 in the treatment of patients with refractory or resistant acute myeloid leukemia (AML) or not amenable to conventional therapy for the disease. Blood 2005, 105, 986–993. [Google Scholar] [CrossRef]
- Fiedler, W.; Kayser, S.; Kebenko, M.; Janning, M.; Krauter, J.; Schittenhelm, M.; Gotze, K.; Weber, D.; Gohring, G.; Teleanu, V.; et al. A phase I/II study of sunitinib and intensive chemotherapy in patients over 60 years of age with acute myeloid leukaemia and activating FLT3 mutations. Br. J. Haematol. 2015, 169, 694–700. [Google Scholar] [CrossRef]
- Zarrinkar, P.P.; Gunawardane, R.N.; Cramer, M.D.; Gardner, M.F.; Brigham, D.; Belli, B.; Karaman, M.W.; Pratz, K.W.; Pallares, G.; Chao, Q.; et al. AC220 is a uniquely potent and selective inhibitor of FLT3 for the treatment of acute myeloid leukemia (AML). Blood 2009, 114, 2984–2992. [Google Scholar] [CrossRef] [PubMed]
- Levis, M. Quizartinib for the treatment of FLT3/ITD acute myeloid leukemia. Future Oncol. 2014, 10, 1571–1579. [Google Scholar] [CrossRef] [PubMed]
- Altman, J.K.; Foran, J.M.; Pratz, K.W.; Trone, D.; Cortes, J.E.; Tallman, M.S. Phase 1 study of quizartinib in combination with induction and consolidation chemotherapy in patients with newly diagnosed acute myeloid leukemia. Am. J. Hematol. 2013, 93, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Hills, R.K.; Gammon, G.; Trone, D.; Burnett, A.K. Quizartinib significantly improves overall survival in FLT3-ITD positive AML patients relapsed after stem cell transplantation or after failure of salvage chemotherapy: A comparison with historical AML database (UK NCRI data). Blood 2015, 126, 2557. [Google Scholar]
- Cortes, J.; Perl, A.E.; Dohner, H.; Kantarjian, H.; Martinelli, G.; Kovacsovics, T.; Rousselot, P.; Steffen, B.; Dombret, H.; Estey, E.; et al. Quizartinib, an FLT3 inhibitor, as monotherapy in patients with relapsed or refractory acute myeloid leukaemia: An open-label, multicentre, single-arm, phase 2 trial. Lancet 2018, 19, 889–903. [Google Scholar] [CrossRef]
- Cortes, J.E.; Khaled, S.K.; Martinelli, G.; Perl, A.E.; Ganguly, S.; Russell, N.H.; Kramer, A.; Dombret, H.; Hogge, D.; Jonas, B.A.; et al. Efficacy and safety of single-agent Quizartinib (Q), a potent and selective FLT3 inhibitor (FLT3i), in patients (pts) with FLT3-internal tandem duplication (FLT3-ITD)-mutated relapsed/refractory (R/R) acute myeloid leukemia (AML) enrolled in the global, phase 3, randomized controlled quantum-R trial. Blood 2018, 132, 563. [Google Scholar]
- Zimmerman, E.I.; Turner, D.C.; Buaboonnam, J.; Hu, S.; Orwick, S.; Roberts, M.S.; Janke, L.J.; Ramachandran, A.; Stewart, C.F.; Inaba, H.; et al. Crenolanib is active against models of drug-resistant FLT3-ITD−positive acute myeloid leukemia. Blood 2013, 122, 3607–3615. [Google Scholar] [CrossRef] [PubMed]
- Galanis, A.; Ma, H.; Rajkhowa, T.; Ramachandran, A.; Small, D.; Cortes, J.; Levis, M. Crenolanib is a potent inhibitor of FLT3 with activity against resistance-conferring point mutants. Blood 2014, 123, 94–100. [Google Scholar] [CrossRef] [Green Version]
- Randhawa, J.K.; Kantarjian, H.M.; Borthakur, G.; Thompson, P.A.; Konopleva, M.; Daver, N.; Pemmaraju, N.; Jabbour, E.; Kadia, T.M.; Estrov, Z.; et al. Results of a phase II study of crenolanib in relapsed/refractory acute myeloid leukemia patients (Pts) with activating FLT3 mutations. Blood 2014, 124, 389. [Google Scholar]
- Jorge, E.C.; Hagop, M.K.; Tapan, M.K.; Gautam, B.; Marina, K.; Guillermo, G.-M.; Naval Guastad, D.; Naveen, P.; Elias, J.; Zeev, E.; et al. Crenolanib besylate, a type I pan-FLT3 inhibitor, to demonstrate clinical activity in multiply relapsed FLT3-ITD and D835 AML. J. Clin. Oncol. 2016, 34, 7008. [Google Scholar]
- Zhang, H.; Savage, S.; Schultz, A.R.; Bottomly, D.; White, L.; Segerdell, E.; Wilmot, B.; McWeeney, S.K.; Eide, C.A.; Nechiporuk, T.; et al. Clinical resistance to crenolanib in acute myeloid leukemia due to diverse molecular mechanisms. Nat. Commun. 2019, 10, 244. [Google Scholar] [CrossRef] [PubMed]
- Levis, M.; Alexander, E.P.; Jessica, K.A.; Jorge, E.C.; Ellen, K.R.; Richard, A.L.; Catherine Choy, S.; Eunice, S.W.; Stephen Anthony, S.; Maria, R.B.; et al. Results of a first-in-human, phase I/II trial of ASP2215, a selective, potent inhibitor of FLT3/Axl in patients with relapsed or refractory (R/R) acute myeloid leukemia (AML). J. Clin. Oncol. 2015, 33, 7003. [Google Scholar] [CrossRef]
- Perl, A.E.; Altman, J.K.; Cortes, J.; Smith, C.; Litzow, M.; Baer, M.R.; Claxton, D.; Erba, H.P.; Gill, S.; Goldberg, S.; et al. Selective inhibition of FLT3 by gilteritinib in relapsed or refractory acute myeloid leukaemia: A multicentre, first-in-human, open-label, phase 1–2 study. Lancet Oncol. 2017, 18, 1061–1075. [Google Scholar] [CrossRef]
- McMahon, C.M.; Ferng, T.; Canaani, J.; Wang, E.S.; Morrissette, J.J.D.; Eastburn, D.J.; Pellegrino, M.; Durruthy-Durruthy, R.; Watt, C.D.; Asthana, S.; et al. Clonal selection with Ras pathway activation mediates secondary clinical resistance to selective FLT3 inhibition in acute myeloid leukemia. Cancer Discov. 2019. [Google Scholar] [CrossRef]
- Perl, A.; Martinelli, G.; Cortes, J. Gilteritinib significantly prolongs overall survival in patients with FLT3-mutated (FLT3mut+) relapsed/refractory (R/R) acute myeloid leukemia (AML): Results from the phase III ADMIRAL trial. In Proceedings of the AACR Annual Meeting, Atlanta, GA, USA, 29 March–3 April 2019. [Google Scholar]
- Nakatani, T.; Uda, K.; Yamaura, T.; Takasaki, M.; Akashi, A.; Chen, F.; Ishikawa, Y.; Hayakawa, F.; Hagiwara, S.; Kiyoi, H.; et al. Development of FF-10101, a novel irreversible FLT3 inhibitor, which overcomes drug resistance mutations. Blood 2015, 126, 1353. [Google Scholar]
- Yamaura, T.; Nakatani, T.; Uda, K.; Ogura, H.; Shin, W.; Kurokawa, N.; Saito, K.; Fujikawa, N.; Date, T.; Takasaki, M.; et al. A novel irreversible FLT3 inhibitor, FF-10101, shows excellent efficacy against AML cells with FLT3 mutations. Blood 2018, 131, 426–438. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, H.; Kanakura, Y.; Tamaki, T.; Kuriu, A.; Kitayama, H.; Ishikawa, J.; Kanayama, Y.; Yonezawa, T.; Tarui, S.; Griffin, J.D. Expression and functional role of the proto-oncogene c-kit in acute myeloblastic leukemia cells. Blood 1991, 78, 2962–2968. [Google Scholar] [PubMed]
- Cairoli, R.; Beghini, A.; Grillo, G.; Nadali, G.; Elice, F.; Ripamonti, C.B.; Colapietro, P.; Nichelatti, M.; Pezzetti, L.; Lunghi, M.; et al. Prognostic impact of c-KIT mutations in core binding factor leukemias: An Italian retrospective study. Blood 2006, 107, 3463–3468. [Google Scholar] [CrossRef]
- Malaise, M.; Steinbach, D.; Corbacioglu, S. Clinical implications of c-Kit mutations in acute myelogenous leukemia. Curr. Hematol. Malig. Rep. 2009, 4, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Ashman, L.K.; Griffith, R. Therapeutic targeting of c-KIT in cancer. Expert Opin. Invest. Drugs 2013, 22, 103–115. [Google Scholar] [CrossRef]
- Lennartsson, J.; Jelacic, T.; Linnekin, D.; Shivakrupa, R. Normal and oncogenic forms of the receptor tyrosine kinase kit. Stem Cells 2005, 23, 16–43. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Xie, H.; Wang, H.; Chen, L.; Sun, Y.; Chen, Z.; Li, Q. Prognostic significance of KIT mutations in core-binding factor acute myeloid leukemia: A systematic review and meta-analysis. PLoS ONE 2016, 11, e0146614. [Google Scholar] [CrossRef] [PubMed]
- Ayatollahi, H.; Shajiei, A.; Sadeghian, M.H.; Sheikhi, M.; Yazdandoust, E.; Ghazanfarpour, M.; Shams, S.F.; Shakeri, S. Prognostic importance of C-KIT mutations in core binding factor acute myeloid leukemia: A systematic review. Hematol. Oncol. Stem Cell Ther. 2017, 10, 1–7. [Google Scholar] [CrossRef]
- Dos Santos, C.; McDonald, T.; Ho, Y.W.; Liu, H.; Lin, A.; Forman, S.J.; Kuo, Y.-H.; Bhatia, R. The Src and c-Kit kinase inhibitor dasatinib enhances p53-mediated targeting of human acute myeloid leukemia stem cells by chemotherapeutic agents. Blood 2013, 122, 1900–1913. [Google Scholar] [CrossRef] [PubMed]
- Heo, S.-K.; Noh, E.-K.; Kim, J.Y.; Jeong, Y.K.; Jo, J.-C.; Choi, Y.; Koh, S.; Baek, J.H.; Min, Y.J.; Kim, H. Targeting c-KIT (CD117) by dasatinib and radotinib promotes acute myeloid leukemia cell death. Sci. Rep. 2017, 7, 15278. [Google Scholar] [CrossRef]
- Fiedler, W.; Mesters, R.; Tinnefeld, H.; Loges, S.; Staib, P.; Dührsen, U.; Flasshove, M.; Ottmann, O.G.; Jung, W.; Cavalli, F.; et al. A phase 2 clinical study of SU5416 in patients with refractory acute myeloid leukemia. Blood 2003, 102, 2763–2767. [Google Scholar] [CrossRef] [PubMed]
- Smolich, B.D.; Yuen, H.A.; West, K.A.; Giles, F.J.; Albitar, M.; Cherrington, J.M. The antiangiogenic protein kinase inhibitors SU5416 and SU6668 inhibit the SCF receptor (c-kit) in a human myeloid leukemia cell line and in acute myeloid leukemia blasts. Blood 2001, 97, 1413–1421. [Google Scholar] [CrossRef] [Green Version]
- Linger, R.M.A.; Keating, A.K.; Earp, H.S.; Graham, D.K. TAM receptor tyrosine kinases: Biologic functions, signaling, and potential therapeutic targeting in human cancer. Adv. Cancer Res. 2008, 100, 35–83. [Google Scholar]
- Neubauer, A.; O’Bryan, J.P.; Fiebeler, A.; Schmidt, C.; Huhn, D.; Liu, E.T. Axl, a novel receptor tyrosine kinase isolated from chronic myelogenous leukemia. Semin. Hematol. 1993, 30, 34. [Google Scholar]
- Caberoy, N.B.; Zhou, Y.; Li, W. Tubby and tubby-like protein 1 are new MerTK ligands for phagocytosis. EMBO J. 2010, 29, 3898–3910. [Google Scholar] [CrossRef] [Green Version]
- Schoumacher, M.; Burbridge, M. Key roles of AXL and MER receptor tyrosine kinases in resistance to multiple anticancer therapies. Curr. Oncol. Rep. 2017, 19, 19. [Google Scholar] [CrossRef] [PubMed]
- Graham, D.K.; DeRyckere, D.; Davies, K.D.; Earp, H.S. The TAM family: Phosphatidylserine-sensing receptor tyrosine kinases gone awry in cancer. Nat. Rev. Cancer 2014, 14, 769–785. [Google Scholar] [CrossRef] [PubMed]
- Mollard, A.; Warner, S.L.; Call, L.T.; Wade, M.L.; Bearss, J.J.; Verma, A.; Sharma, S.; Vankayalapati, H.; Bearss, D.J. Design, synthesis and biological evaluation of a series of novel Axl kinase inhibitors. ACS Med. Chem. Lett. 2011, 2, 907–912. [Google Scholar] [CrossRef] [PubMed]
- Rochlitz, C.; Lohri, A.; Bacchi, M.; Schmidt, M.; Nagel, S.; Fopp, M.; Fey, M.F.; Herrmann, R.; Neubauer, A. Axl expression is associated with adverse prognosis and with expression of Bcl-2 and CD34 in de novo acute myeloid leukemia (AML): Results from a multicenter trial of the Swiss group for clinical cancer research (SAKK). Leukemia 1999, 13, 1352–1358. [Google Scholar] [CrossRef] [PubMed]
- Whitman, S.P.; Kohlschmidt, J.; Maharry, K.; Volinia, S.; Mrozek, K.; Nicolet, D.; Schwind, S.; Becker, H.; Metzeler, K.H.; Mendler, J.H.; et al. GAS6 expression identifies high-risk adult AML patients: Potential implications for therapy. Leukemia 2014, 28, 1252–1258. [Google Scholar] [CrossRef]
- Dufies, M.; Jacquel, A.; Belhacene, N.; Robert, G.; Cluzeau, T.; Luciano, F.; Cassuto, J.P.; Raynaud, S.; Auberger, P. Mechanisms of AXL overexpression and function in Imatinib-resistant chronic myeloid leukemia cells. Oncotarget 2011, 2, 874–885. [Google Scholar] [CrossRef] [PubMed]
- Gioia, R.; Leroy, C.; Drullion, C.; Lagarde, V.; Etienne, G.; Dulucq, S.; Lippert, E.; Roche, S.; Mahon, F.X.; Pasquet, J.M. Quantitative phosphoproteomics revealed interplay between Syk and Lyn in the resistance to nilotinib in chronic myeloid leukemia cells. Blood 2011, 118, 2211–2221. [Google Scholar] [CrossRef]
- Gioia, R.; Tregoat, C.; Dumas, P.Y.; Lagarde, V.; Prouzet-Mauleon, V.; Desplat, V.; Sirvent, A.; Praloran, V.; Lippert, E.; Villacreces, A.; et al. CBL controls a tyrosine kinase network involving AXL, SYK and LYN in nilotinib-resistant chronic myeloid leukaemia. J. Pathol. 2015, 237, 14–24. [Google Scholar] [CrossRef]
- Hong, C.C.; Lay, J.D.; Huang, J.S.; Cheng, A.L.; Tang, J.L.; Lin, M.T.; Lai, G.M.; Chuang, S.E. Receptor tyrosine kinase AXL is induced by chemotherapy drugs and overexpression of AXL confers drug resistance in acute myeloid leukemia. Cancer Lett. 2008, 268, 314–324. [Google Scholar] [CrossRef]
- Ben-Batalla, I.; Schultze, A.; Wroblewski, M.; Erdmann, R.; Heuser, M.; Waizenegger, J.S.; Riecken, K.; Binder, M.; Schewe, D.; Sawall, S.; et al. Axl, a prognostic and therapeutic target in acute myeloid leukemia mediates paracrine cross-talk of leukemia cells with bone marrow stroma. Blood 2013, 122, 2443–2452. [Google Scholar] [CrossRef]
- Park, I.-K.; Mishra, A.; Chandler, J.; Whitman, S.P.; Marcucci, G.; Caligiuri, M.A. Inhibition of the receptor tyrosine kinase Axl impedes activation of the FLT3 internal tandem duplication in human acute myeloid leukemia: Implications for Axl as a potential therapeutic target. Blood 2013, 121, 2064–2073. [Google Scholar] [CrossRef] [PubMed]
- Park, I.K.; Mundy-Bosse, B.; Whitman, S.P.; Zhang, X.; Warner, S.L.; Bearss, D.J.; Blum, W.; Marcucci, G.; Caligiuri, M.A. Receptor tyrosine kinase Axl is required for resistance of leukemic cells to FLT3-targeted therapy in acute myeloid leukemia. Leukemia 2015, 29, 2382–2389. [Google Scholar] [CrossRef] [PubMed]
- Dumas, P.-Y.; Naudin, C.c.; Martin-Lanner2e, S.V.; Izac, B.; Casetti, L.; Mansier, O.; Rousseau, B.T.; Artus, A.; Dufossée, M.L.; Giese, A.; et al. Hematopoietic niche drives FLT3-ITD acute myeloid leukemia resistance to quizartinib via STAT5- and hypoxia- dependent up-regulation of AXL. Haematologica 2019, 104. [Google Scholar] [CrossRef] [PubMed]
- Huey, M.G.; Minson, K.A.; Earp, H.S.; DeRyckere, D.; Graham, D.K. Targeting the TAM receptors in leukemia. Cancers 2016, 8, 101. [Google Scholar] [CrossRef] [PubMed]
- Gay, C.M.; Balaji, K.; Byers, L.A. Giving AXL the axe: Targeting AXL in human malignancy. Br. J. Cancer 2017, 116, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Chen, X.; He, J.; Liao, D.; Zu, X. Axl inhibitors as novel cancer therapeutic agents. Life Sci. 2018, 198, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Myers, S.H.; Brunton, V.G.; Unciti-Broceta, A. AXL Inhibitors in cancer: A medicinal chemistry perspective. J. Med. Chem. 2016, 59, 3593–3608. [Google Scholar] [CrossRef]
- Holland, S.J.; Pan, A.; Franci, C.; Hu, Y.; Chang, B.; Li, W.; Duan, M.; Torneros, A.; Yu, J.; Heckrodt, T.J.; et al. R428, a selective small molecule inhibitor of Axl kinase, blocks tumor spread and prolongs survival in models of metastatic breast cancer. Cancer Res. 2010, 70, 1544–1554. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Secreto, C.; Boysen, J.; Sassoon, T.; Shanafelt, T.D.; Mukhopadhyay, D.; Kay, N.E. A novel receptor tyrosine kinase Axl is constitutively active in B-cell chronic lymphocytic leukemia and acts as a docking site of non-receptor kinases: Implications for therapy. Blood 2010, 117, 1928–1937. [Google Scholar] [CrossRef]
- Loges, S.; Björn Tore, G.; Michael, H.; Jörg, C.; Carlos Enrique, V.; Peter, P.; Ben-Batalla, I.; Nuray, A.; David, M.; Anthony, B.; et al. The immunomodulatory activity of bemcentinib (BGB324): A first-in-class selective oral AXL inhibitor in patients with relapsed/refractory acute myeloid leukemia or myelodysplastic syndrome. J. Clin. Oncol. 2018, 36, 70. [Google Scholar] [CrossRef]
- Loges, S.; Bjorn Torre, G.; Michael, H.; Ben-Batalla, I.; David, M.; Chromik, J.; Maxim, K.; Walter, M.F.; Jorge, E.C. A first-in-patient phase I study of BGB324, a selective Axl kinase inhibitor in patients with refractory/relapsed AML and high-risk MDS. J. Clin. Oncol. 2016, 34, 2561. [Google Scholar] [CrossRef]
- Sonja, L.; Gjertsen, B.T.; Heuser, M.; Chromik, J.; Batalla, I.B.; Akyüz, N.; Micklem, D.; Brown, A.; Lorens, J.; Kebenko, M.; et al. BGB324, an orally available selective Axl inhibitor exerts anti-leukemic activity in the first-in-patient trial BGBC003 and induces unique changes in biomarker profiles. Blood 2016, 128, 592. [Google Scholar]
- Eryildiz, F.; Tyner, J.W. Abstract 1265: Dysregulated tyrosine kinase Tyro3 signaling in acute myeloid leukemia. Cancer Res. 2016, 76, 1265. [Google Scholar]
- Gilmour, M.; Scholtz, A.; Ottmann, O.G.; Hills, R.K.; Knapper, S.; Zabkiewicz, J. Axl/Mer Inhibitor ONO-9330547 As a novel therapeutic agent in a stromal co-culture model of primary acute myeloid leukaemia (AML). Blood 2016, 128, 2754. [Google Scholar]
- Ruvolo, P.; Ma, H.; Ruvolo, V.; Mu, H.; Schober, W.; Yasuhiro, T.; Yoshizawa, T.; Gallardo, M.; Zhang, X.; Khoury, J.D.; et al. AXL inhibitor ONO-9330547 suppresses PLK1 gene and protein expression and effectively induces growth arrest and apoptosis in FLT3 ITD acute myeloid leukemia cells. Blood 2016, 128, 3939. [Google Scholar]
- Lee-Sherick, A.B.; Eisenman, K.M.; Sather, S.; McGranahan, A.; Armistead, P.M.; McGary, C.S.; Hunsucker, S.A.; Schlegel, J.; Martinson, H.; Cannon, C.; et al. Aberrant Mer receptor tyrosine kinase expression contributes to leukemogenesis in acute myeloid leukemia. Oncogene 2013, 32, 5359. [Google Scholar] [CrossRef] [PubMed]
- Minson, K.A.; Huey, M.G.; Hill, A.A.; Perez, I.; Wang, X.; Frye, S.; Earp, H.S.; DeRyckere, D.; Graham, D.K. Bone marrow stromal cell mediated resistance to mertk inhibition in acute leukemia. Blood 2016, 128, 2819. [Google Scholar]
- Ruvolo, P.P.; Ma, H.; Ruvolo, V.R.; Zhang, X.; Mu, H.; Schober, W.; Hernandez, I.; Gallardo, M.; Khoury, J.D.; Cortes, J.; et al. Anexelekto/MER tyrosine kinase inhibitor ONO-7475 arrests growth and kills FMS-like tyrosine kinase 3-internal tandem duplication mutant acute myeloid leukemia cells by diverse mechanisms. Haematologica 2017, 102, 2048–2057. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, K.; Li, C.; Hirosaki, T.; Kato, H.; Ishikawa, Y.; Oka, M.; Egawa, H.; Kozaki, R.; Yoshizawa, T. Abstract 1883: A novel Axl and Mertk dual inhibitor ONO-7475: A new therapeutic agent for the treatment of FLT3-ITD and -wild-type acute myeloid leukemia (AML) overexpressing. Cancer Res. 2018, 78, 1883. [Google Scholar]
- Lu, J.W.; Wang, A.N.; Liao, H.A.; Chen, C.Y.; Hou, H.A.; Hu, C.Y.; Tien, H.F.; Ou, D.L.; Lin, L.I. Cabozantinib is selectively cytotoxic in acute myeloid leukemia cells with FLT3-internal tandem duplication (FLT3-ITD). Cancer Lett. 2016, 376, 218–225. [Google Scholar] [CrossRef]
- Fathi, A.T.; Blonquist, T.M.; Hernandez, D.; Amrein, P.C.; Ballen, K.K.; McMasters, M.; Avigan, D.E.; Joyce, R.; Logan, E.K.; Hobbs, G.; et al. Cabozantinib is well tolerated in acute myeloid leukemia and effectively inhibits the resistance-conferring FLT3/tyrosine kinase domain/F691 mutation. Cancer 2018, 124, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Jimbo, T.; Taira, T.; Komatsu, T.; Kumazawa, K.; Maeda, N.; Haginoya, N.; Suzuki, T.; Ota, M.; Totoki, Y.; Wada, C.; et al. DS-1205b, a novel, selective, small-molecule inhibitor of AXL, delays the onset of resistance and overcomes acquired resistance to EGFR-TKIs in a human EGFR-mutant NSCLC (T790M-negative) xenograft model. Ann. Oncol. 2017, 28, 395. [Google Scholar] [CrossRef]
- Oellerich, T.; Oellerich, M.F.; Engelke, M.; Munch, S.; Mohr, S.; Nimz, M.; Hsiao, H.-H.; Corso, J.; Zhang, J.; Bohnenberger, H.; et al. b2 integrine derived signals induce cell survival and proliferation of AML blasts by activating a Syk/STAT signaling axis. Blood 2013, 121, 3889–3899. [Google Scholar] [CrossRef] [PubMed]
- Boros, K.; Puissant, A.; Back, M.; Alexe, G.; Bassil, C.F.; Sinha, P.; Tholouli, E.; Stegmaier, K.; Byers, R.J.; Rodig, S.J. Increased SYK activity is associated with unfavorable outcome among patients with acute myeloid leukemia. Oncotarget 2015, 6, 25575–25587. [Google Scholar] [CrossRef] [PubMed]
- Bartaula-Brevik, S.; Lindstad Brattas, M.K.; Tvedt, T.H.A.; Reikvam, H.; Bruserud, O. Splenic tyrosine kinase (SYK) inhibitors and their possible use in acute myeloid leukemia. Expert Opin. Invest. Drugs 2018, 27, 377–387. [Google Scholar] [CrossRef] [PubMed]
- Richine, B.M.; Virts, E.L.; Bowling, J.D.; Ramdas, B.; Mali, R.; Naoye, R.; Liu, Z.; Zhang, Z.Y.; Boswell, H.S.; Kapur, R.; et al. Syk kinase and Shp2 phosphatase inhibition cooperate to reduce FLT3-ITD-induced STAT5 activation and proliferation of acute myeloid leukemia. Leukemia 2016, 30, 2094–2097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, A.R.; Bhatnagar, B.; Marcondes, A.M.Q.; DiPaolo, J.; Vasu, S.; Mims, A.S.; Klisovic, R.B.; Walsh, K.J.; Canning, R.; Behbehani, G.K.; et al. Interim results of a phase 1b/2 study of entospletinib (GS-9973) monotherapy and in combination with chemotherapy in patients with acute myeloid leukemia. Blood 2016, 128, 2831. [Google Scholar]
- Walker, A.R.; Byrd, J.C.; Blum, W.; Lin, T.; Crosswell, H.E.; Zhang, D.; Gao, J.; Rao, A.V.; Minden, M.D.; Stock, W. Abstract 819: High response rates with entospletinib in patients with t(v;11q23.3)KMT2A rearranged acute myeloid leukemia and acute lymphoblastic leukemia. Cancer Res 2018, 78, 819. [Google Scholar]
- Jie, Y.; Jessica, H.; Matthew, T.; Helen, H.; Stephen, T.; Mengkun, Z.; Karuppiah, K. Anti-tumor activity of TAK-659, a dual inhibitor of SYK and FLT-3 kinases, in AML models. J. Clin. Oncol. 2016, 34, e14091. [Google Scholar]
- Kaplan, J.B.; Bixby, D.L.; Morris, J.C.; Frankfurt, O.; Altman, J.; Wise-Draper, T.; Burke, P.W.; Collins, S.; Kannan, K.; Wang, L.; et al. A phase 1b/2 study of TAK-659, an investigational dual SYK and FLT-3 inhibitor, in patients (Pts) with relapsed or refractory acute myelogenous leukemia (R/R AML). Blood 2016, 128, 2834. [Google Scholar]
- Ozawa, Y.; Williams, A.H.; Estes, M.L.; Matsushita, N.; Boschelli, F.; Jove, R.; List, A.F. Src family kinases promote AML cell survival through activation of signal transducers and activators of transcription (STAT). Leuk. Res. 2008, 32, 893–903. [Google Scholar] [CrossRef] [PubMed]
- Marhall, A.; Kazi, J.U.; Ronnstrand, L. The Src family kinase LCK cooperates with oncogenic FLT3/ITD in cellular transformation. Sci. Rep. 2017, 7, 13734. [Google Scholar] [CrossRef] [PubMed]
- Roginskaya, V.; Zuo, S.; Caudell, E.; Nambudiri, G.; Kraker, A.J.; Corey, S.J. Therapeutic targeting of Src-kinase Lyn in myeloid leukemic cell growth. Leukemia 1999, 13, 855–861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hussein, K.; von Neuhoff, N.; Büsche, G.; Buhr, T.; Kreipe, H.; Bock, O. Opposite expression pattern of Src kinase Lyn in acute and chronic haematological malignancies. Ann. Hematol. 2009, 88, 1059–1067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, L.J.; Xue, J.; Corey, S.J. Src family tyrosine kinases are activated by Flt3 and are involved in the proliferative effects of leukemia-associated Flt3 mutations. Exp. Hematol. 2005, 33, 469–479. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, C.; Demur, C.; Bardet, V.; Prade-Houdellier, N.; Payrastre, B.; Recher, C. A critical role for Lyn in acute myeloid leukemia. Blood 2008, 111, 2269–2279. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, M.; Hayakawa, F.; Miyata, Y.; Watamoto, K.; Emi, N.; Abe, A.; Kiyoi, H.; Towatari, M.; Naoe, T. Lyn is an important component of the signal transduction pathway specific to FLT3/ITD and can be a therapeutic target in the treatment of AML with FLT3/ITD. Leukemia 2007, 21, 403–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leischner, H.; Albers, C.; Grundler, R.; Razumovskaya, E.; Spiekermann, K.; Bohlander, S.; Ronnstrand, L.; Gotze, K.; Peschel, C.; Duyster, J. SRC is a signaling mediator in FLT3-ITD- but not in FLT3-TKD-positive AML. Blood 2012, 119, 4026–4033. [Google Scholar] [CrossRef]
- Ingley, E. Functions of the Lyn tyrosine kinase in health and disease. Cell Commun. Sign. CCS 2012, 10, 21. [Google Scholar] [CrossRef]
- Leischner, H.; Grundler, R.; Albers, C.; Illert, A.L.; Gotze, K.; Peschel, C.; Duyster, J. Combination of c-SRC and FLT3 inhibitors has an additive inhibitory effect on FLT3 ITD but not on FLT3 TKD positive cells. Blood 2010, 116, 2892. [Google Scholar]
- Gozgit, J.M.; Wong, M.J.; Wardwell, S.; Tyner, J.W.; Loriaux, M.M.; Mohemmad, Q.K.; Narasimhan, N.I.; Shakespeare, W.C.; Wang, F.; Druker, B.J.; et al. Potent activity of Ponatinib (AP24534) in models of FLT3-driven acute myeloid leukemia and other hematologic malignancies. Mol. Cancer Ther. 2011, 10, 1028–1035. [Google Scholar] [CrossRef] [PubMed]
- Bourrié, B.; Brassard, D.L.; Cosnier-Pucheu, S.; Zilberstein, A.; Yu, K.; Levit, M.; Morrison, J.G.; Perreaut, P.; Jegham, S.; Hilairet, S.; et al. SAR103168: A tyrosine kinase inhibitor with therapeutic potential in myeloid leukemias. Leuk. Lymphoma 2013, 54, 1488–1499. [Google Scholar] [CrossRef] [PubMed]
- Weir, M.C.; Hellwig, S.; Tan, L.; Liu, Y.; Gray, N.S.; Smithgall, T.E. Dual inhibition of Fes and Flt3 tyrosine kinases potently inhibits Flt3-ITD+ AML cell growth. PLoS ONE 2017, 12, e0181178. [Google Scholar] [CrossRef] [PubMed]
- Weir, M.C.; Shu, S.T.; Patel, R.K.; Hellwig, S.; Chen, L.; Tan, L.; Gray, N.S.; Smithgall, T.E. Selective inhibition of the myeloid Src-family kinase Fgr potently suppresses AML cell growth in vitro and in vivo. ACS Chem. Biol. 2018, 13, 1551–1559. [Google Scholar] [CrossRef] [PubMed]
- Kentsis, A.; Reed, C.; Rice, K.L.; Sanda, T.; Rodig, S.J.; Tholouli, E.; Christie, A.; Valk, P.J.M.; Delwel, R.; Ngo, V.; et al. Autocrine activation of the MET receptor tyrosine kinase in acute myeloid leukemia. Nat. Med. 2012, 18, 1118–1122. [Google Scholar] [CrossRef] [PubMed]
- Fialin, C.; Larrue, C.; Vergez, F.; Sarry, J.E.; Bertoli, S.; Mansat-De Mas, V.; Demur, C.; Delabesse, E.; Payrastre, B.; Manenti, S.; et al. The short form of RON is expressed in acute myeloid leukemia and sensitizes leukemic cells to cMET inhibitors. Leukemia 2013, 27, 325–335. [Google Scholar] [CrossRef] [PubMed]
- McGee, S.F.; Kornblau, S.M.; Qiu, Y.; Look, A.T.; Zhang, N.; Yoo, S.Y.; Coombes, K.R.; Kentsis, A. Biological properties of ligand-dependent activation of the MET receptor kinase in acute myeloid leukemia. Leukemia 2015, 29, 1218. [Google Scholar] [CrossRef]
- Mulgrew, N.M.; Kettyle, L.M.J.; Ramsey, J.M.; Cull, S.; Smyth, L.J.; Mervyn, D.M.; Bijl, J.J.; Thompson, A. c-Met inhibition in a HOXA9/Meis1 model of CN-AML. Dev. Dyn. 2014, 243, 172–181. [Google Scholar] [CrossRef]
- Smith, C.I.E.; Islam, T.C.; Mattsson, P.T.; Mohamed, A.J.; Nore, B.F.; Vihinen, M. The Tec family of cytoplasmic tyrosine kinases: Mammalian Btk, Bmx, Itk, Tec, Txk and homologs in other species. Bioessays 2001, 23, 436–446. [Google Scholar] [CrossRef]
- Tang, B.; Mano, H.; Yi, T.; Ihle, J.N. Tec kinase associates with c-kit and is tyrosine phosphorylated and activated following stem cell factor binding. Mol. Cell. Biol. 1994, 14, 8432–8437. [Google Scholar] [CrossRef]
- Van Dijk, T.B.; van den Akker, E.; Parren-van Amelsvoort, M.; Mano, H.; Löwenberg, B.; von Lindern, M. Stem cell factor induces phosphatidylinositol 3-kinase-dependent Lyn/Tec/Dok-1 complex formation in hematopoietic cells. Blood 2000, 96, 3406–3413. [Google Scholar] [PubMed]
- Rushworth, S.A.; Pillinger, G.; Abdul-Aziz, A.; Piddock, R.; Shafat, M.S.; Murray, M.Y.; Zaitseva, L.; Lawes, M.J.; MacEwan, D.J.; Bowles, K.M. Activity of Bruton’s tyrosine-kinase inhibitor ibrutinib in patients with CD117-positive acute myeloid leukaemia: A mechanistic study using patient-derived blast cells. Lancet Haematol. 2015, 2, e204–e211. [Google Scholar] [CrossRef]
- Pillinger, G.; Abdul-Aziz, A.; Zaitseva, L.; Lawes, M.; MacEwan, D.J.; Bowles, K.M.; Rushworth, S.A. Targeting BTK for the treatment of FLT3-ITD mutated acute myeloid leukemia. Sci. Rep. 2015, 5, 12949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaitseva, L.; Murray, M.Y.; Shafat, M.S.; Lawes, M.J.; MacEwan, D.J.; Bowles, K.M.; Rushworth, S.A. Ibrutinib inhibits SDF1/CXCR4 mediated migration in AML. Oncotarget 2014, 5, 9930–9938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rushworth, S.A.; Murray, M.Y.; Zaitseva, L.; Bowles, K.M.; MacEwan, D.J. Identification of Bruton’s tyrosine kinase as a therapeutic target in acute myeloid leukemia. Blood 2014, 123, 1229–1238. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Hu, C.; Wang, A.; Weisberg, E.L.; Wang, W.; Chen, C.; Zhao, Z.; Yu, K.; Liu, J.; Wu, J.; et al. Ibrutinib selectively targets FLT3-ITD in mutant FLT3-positive AML. Leukemia 2016, 30, 754–757. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Drug, TK Targeted and Development Status in AML | NCT Number | ||
|---|---|---|---|
| Midostaurin | FLT3 | FDA approved in newly diagnosed AML | - |
| Gilteritinib | FLT3 | FDA approved in R/R AML | - |
| Quizartinib | FLT3 | MHLW of Japan approved in R/R AML | - |
| Crenolanib | FLT3 | 7 phases 1 to 3 studies recruiting or active not recruiting in Clinical trials | - |
| Sorafenib | FLT3 | 15 phases 1 to 3 studies recruiting or active not recruiting in Clinical trials | - |
| Sunitinib | FLT3 | Phase 1 and 1/2 recruiting and active | NCT01620216 |
| Lestaurtinib | FLT3 | Phase 1/2 completed study in R/R AML | NCT00469859 |
| FF-10101 | FLT3 | Phase 1/2 recruiting study in R/R AML | NCT03194685 |
| SEL24-B489 | FLT3 | Phase 1/2 recruiting study in newly diagnosed or R/R AML | NCT03008187 |
| TAK-659 | FLT3 | Phase 1/2 completed study in newly diagnosed or R/R AML | NCT02323113 |
| Dasatinib | KIT | 13 phases 1 to 3 studies recruiting or active not recruiting in Clinical trials | - |
| SU5416 | KIT | Phase 1/2 completed study in newly diagnosed or R/R AML | NCT00005942 |
| Bemcentinib | AXL | Phase 2 recruiting study in newly diagnosed AML | NCT03824080 |
| Cabozantinib | AXL | Phase 1 completed study in newly diagnosed or R/R AML | NCT01961765 |
| Entospletinib | SYK | Phase 1/2 completed study in newly diagnosed or R/R AML | NCT02343939 |
| SAR103168 | SFK | Phase 1 completed study in R/R AML | NCT00981240 |
| Ibrutinib | Btk | Phase 1 completed study in R/R AML | NCT02635074 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernandez, S.; Desplat, V.; Villacreces, A.; Guitart, A.V.; Milpied, N.; Pigneux, A.; Vigon, I.; Pasquet, J.-M.; Dumas, P.-Y. Targeting Tyrosine Kinases in Acute Myeloid Leukemia: Why, Who and How? Int. J. Mol. Sci. 2019, 20, 3429. https://doi.org/10.3390/ijms20143429
Fernandez S, Desplat V, Villacreces A, Guitart AV, Milpied N, Pigneux A, Vigon I, Pasquet J-M, Dumas P-Y. Targeting Tyrosine Kinases in Acute Myeloid Leukemia: Why, Who and How? International Journal of Molecular Sciences. 2019; 20(14):3429. https://doi.org/10.3390/ijms20143429
Chicago/Turabian StyleFernandez, Solène, Vanessa Desplat, Arnaud Villacreces, Amélie V. Guitart, Noël Milpied, Arnaud Pigneux, Isabelle Vigon, Jean-Max Pasquet, and Pierre-Yves Dumas. 2019. "Targeting Tyrosine Kinases in Acute Myeloid Leukemia: Why, Who and How?" International Journal of Molecular Sciences 20, no. 14: 3429. https://doi.org/10.3390/ijms20143429