Rice Genome Resequencing Reveals a Major Quantitative Trait Locus for Resistance to Bakanae Disease Caused by Fusarium fujikuroi

Abstract
:1. Introduction
2. Results
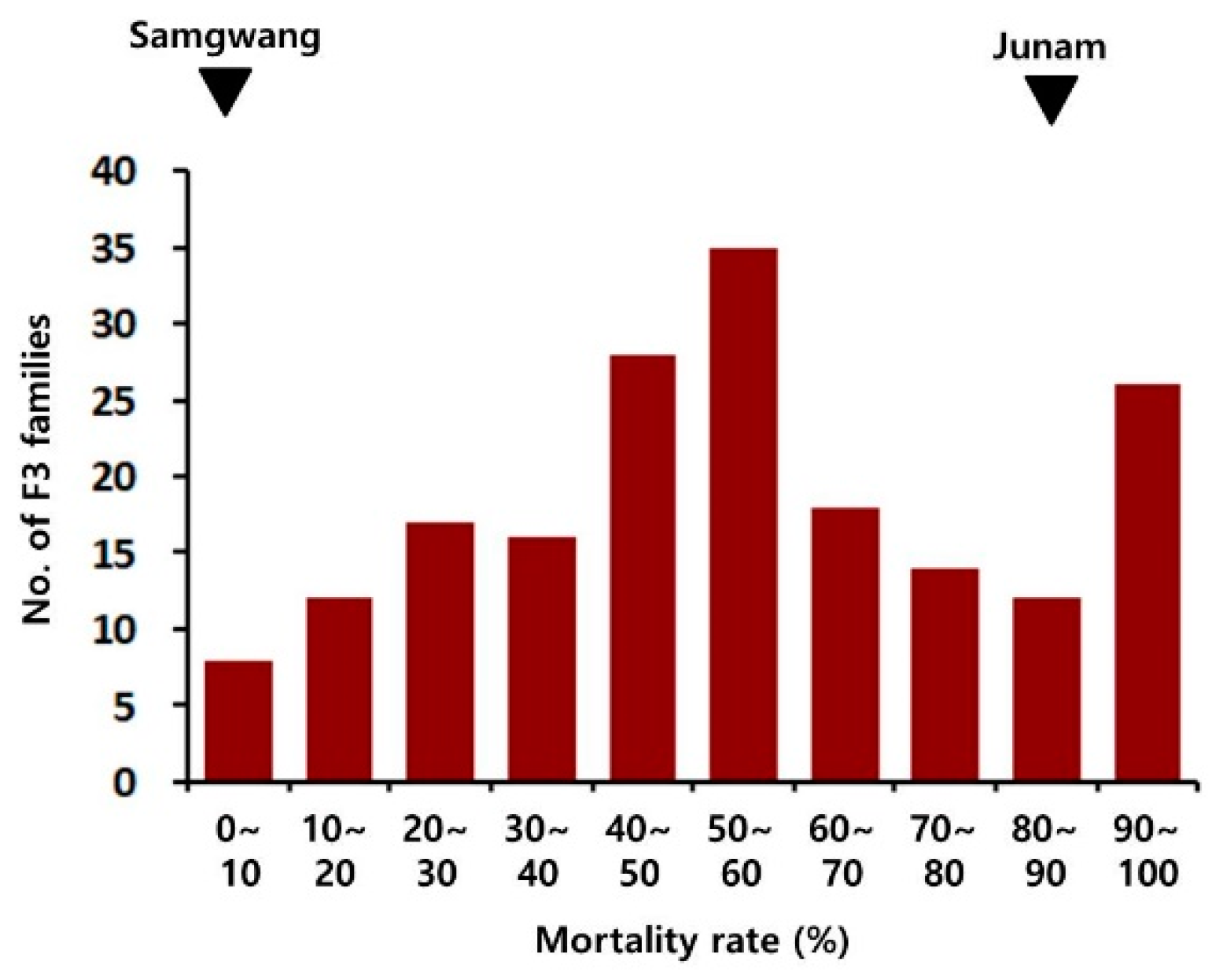
2.1. Phenotype of Parental Varieties and Progenies
2.2. Parental Variety Resequencing
2.3. Genetic Map Construction and Quantitative Trait Locus (QTL) Mapping
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Bakanae Disease (BD) Bioassay
4.2. Parental Variety Resequencing and Marker Development
4.3. Genetic Map Construction and QTL Mapping
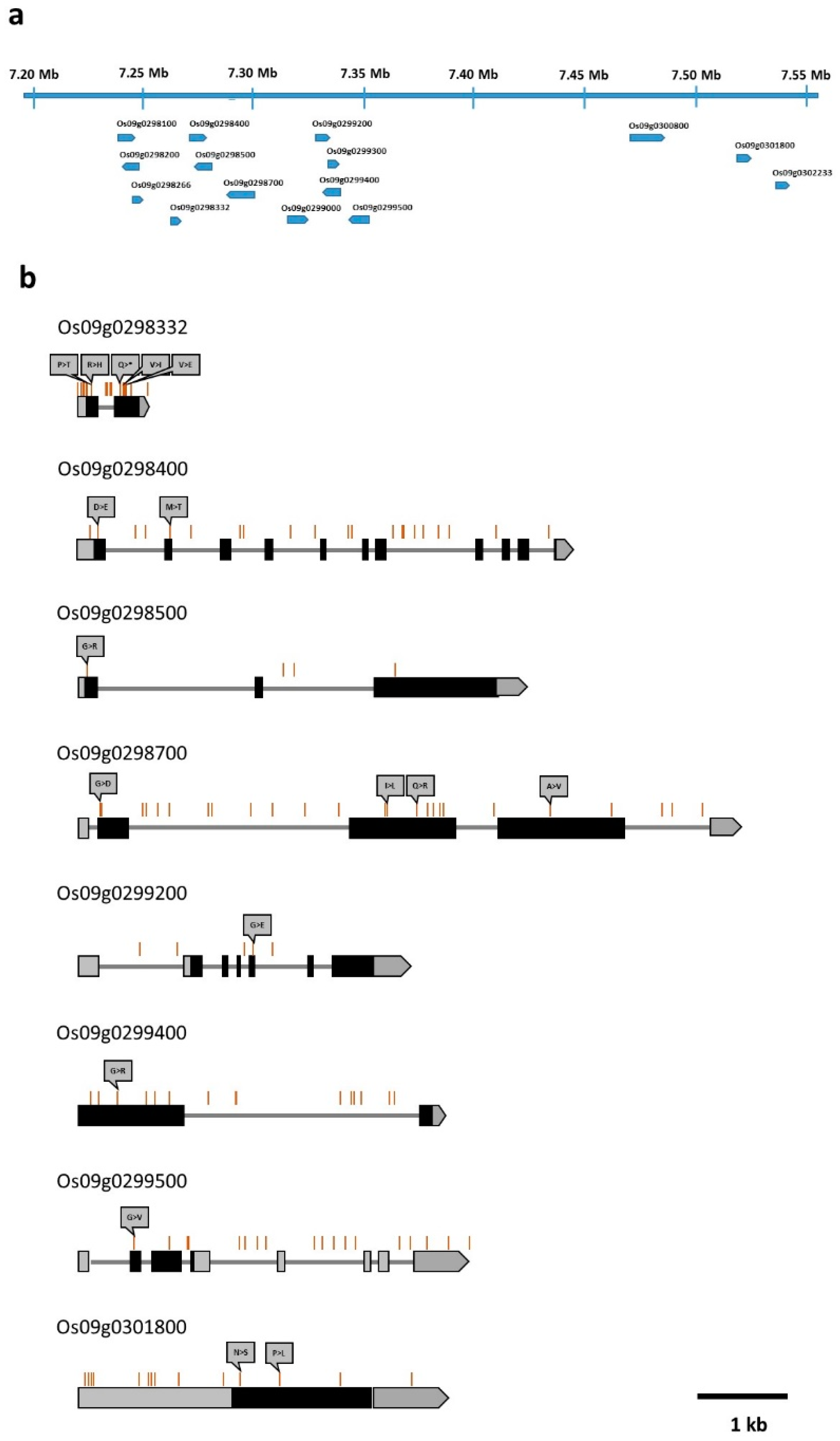
4.4. Candidate Gene Analysis in the Identified QTL Region
5. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Carter, L.L.; Leslie, J.F.; Webster, R.K. Population structure of Fusarium fujikuroi from California rice and water grass. Phytopathology 2008, 98, 992–998. [Google Scholar] [CrossRef] [PubMed]
- Desjardins, A.E.; Manandhar, H.K.; Plattner, R.D.; Manandhar, G.G.; Poling, S.M.; Maragos, C.M. Fusarium species from nepalese rice and production of mycotoxins and gibberellic acid by selected species. Appl. Environ. Microbiol. 2000, 66, 1020–1025. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.K.; Solanki, I.S.; Bashyal, B.M.; Singh, Y.; Srivastava, K. Bakanae of rice—An emerging disease in Asia. J. Anim. Plant Sci. 2015, 25, 1499–1514. [Google Scholar]
- Matic, S.; Gullino, M.L.; Spadaro, D. The puzzle of bakanae disease through interactions between Fusarium fujikuroi and rice. Front. Biosci. 2017, 9, 333–344. [Google Scholar]
- Amatulli, M.T.; Spadaro, D.; Gullino, M.L.; Garibaldi, A. Molecular identification of Fusarium spp. associated with bakanae disease of rice in Italy and assessment of their pathogenicity. Plant Pathol. 2010, 59, 839–844. [Google Scholar] [CrossRef]
- Fiyaz, R.A.; Yadav, A.K.; Krishnan, S.G.; Ellur, R.K.; Bashyal, B.M.; Grover, N.; Bhowmick, P.K.; Nagarajan, M.; Vinod, K.K.; Singh, N.K.; et al. Mapping quantitative trait loci responsible for resistance to Bakanae disease in rice. Rice (N Y) 2016, 9, 45. [Google Scholar] [CrossRef]
- Chen, Z.; Gao, T.; Liang, S.; Liu, K.; Zhou, M.; Chen, C. Molecular mechanism of resistance of Fusarium fujikuroi to benzimidazole fungicides. FEMS Microbiol. Lett. 2014, 357, 77–84. [Google Scholar] [CrossRef]
- Kim, S.H.; Park, M.R.; Kim, Y.C.; Lee, S.W.; Choi, B.R.; Lee, S.W.; Kim, I.S. Degradation of prochloraz by rice Bakanae disease pathogen Fusarium fujikuroi with differing sensitivity: A possible explanation for resistance mechanism. J. Korean Soc. Appl. Biol. Chem. 2010, 53, 433–439. [Google Scholar] [CrossRef]
- Yang, Y.-R.; Lee, S.-W.; Lee, S.-W.; Kim, I.-S. Morphological changes of fungal cell wall and ABC transporter as resistance responses of rice Bakanae disease pathogen Fusarium fujikuroi CF337 to prochloraz. Korean J. Environ. Agric. 2012, 31, 20–36. [Google Scholar] [CrossRef]
- Fiyaz, R.A.; Krishnan, S.G.; Rajashekara, H.; Yadav, A.K.; Bashyal, B.M.; Bhowmick, P.K.; Singh, N.K.; Prabhu, K.V.; Singh, A.K. Development of high throughput screening protocol and identification of novel sources of resistance against bakanae disease in rice (Oryza sativa L.). Indian J. Genet. Plant Breed. 2014, 74, 414–422. [Google Scholar] [CrossRef]
- Kim, M.H.; Hur, Y.J.; Lee, S.B.; Kwon, T.; Hwang, U.H.; Park, S.K.; Yoon, Y.N.; Lee, J.H.; Cho, J.H.; Shin, D.; et al. Large-scale screening of rice accessions to evaluate resistance to bakanae disease. J. Gen. Plant Pathol. 2014, 80, 408–414. [Google Scholar] [CrossRef]
- Hur, Y.J.; Lee, S.B.; Shin, D.J.; Kim, T.H.; Cho, J.H.; Han, S.I.; Oh, S.H.; Lee, J.Y.; Son, Y.B.; Lee, J.H.; et al. Screening of rice germplasm for Bakanae disease resistance in rice. Korean J. Breed. Sci. 2016, 48, 22–28. [Google Scholar] [CrossRef]
- Lee, Y.H.; Lee, M.J.; Choi, H.W.; Kim, S.T.; Park, J.W.; Myung, I.S.; Park, K.; Lee, S.W. Development of in vitro seedling screening method for selection of resistant rice against Bakanae disease. Res. Plant Dis. 2011, 17, 288–294. [Google Scholar] [CrossRef]
- Yang, C.-D.; Guo, L.-B.; Li, X.-M.; Ji, Z.-J.; Ma, L.-Y.; Qian, Q. Analysis of QTLs for resistance to rice bakanae disease. Chin. J. Rice Sci. 2006, 20, 657–659. [Google Scholar]
- Hur, Y.J.; Lee, S.B.; Kim, T.H.; Kwon, T.; Lee, J.H.; Shin, D.J.; Park, S.K.; Hwang, U.H.; Cho, J.H.; Yoon, Y.N.; et al. Mapping of qBK1, a major QTL for bakanae disease resistance in rice. Mol. Breed. 2015, 35, 78. [Google Scholar] [CrossRef]
- Ji, H.; Kim, T.H.; Lee, G.S.; Kang, H.J.; Lee, S.B.; Suh, S.C.; Kim, S.L.; Choi, I.; Baek, J.; Kim, K.H. Mapping of a major quantitative trait locus for bakanae disease resistance in rice by genome resequencing. Mol. Genet. Genom. 2018, 293, 579–586. [Google Scholar] [CrossRef]
- Lee, S.-B.; Hur, Y.-J.; Cho, J.-H.; Lee, J.-H.; Kim, T.-H.; Cho, S.-M.; Song, Y.-C.; Seo, Y.-S.; Lee, J.; Kim, T.-S.; et al. Molecular mapping of qBK1WD, a major QTL for bakanae disease resistance in rice. Rice (N Y) 2018, 11, 3. [Google Scholar] [CrossRef]
- Volante, A.; Tondelli, A.; Aragona, M.; Valente, M.T.; Biselli, C.; Desiderio, F.; Bagnaresi, P.; Matic, S.; Gullino, M.L.; Infantino, A.; et al. Identification of bakanae disease resistance loci in japonica rice through genome wide association study. Rice (N Y) 2017, 10, 29. [Google Scholar] [CrossRef]
- Matic, S.; Bagnaresi, P.; Biselli, C.; Orru, L.; Amaral Carneiro, G.; Siciliano, I.; Vale, G.; Gullino, M.L.; Spadaro, D. Comparative transcriptome profiling of resistant and susceptible rice genotypes in response to the seedborne pathogen Fusarium fujikuroi. BMC Genom. 2016, 17, 608. [Google Scholar] [CrossRef]
- Siciliano, I.; Carneiro, G.A.; Spadaro, D.; Garibaldi, A.; Gullino, M.L. Jasmonic acid, abscisic acid, and salicylic acid are involved in the phytoalexin responses of rice to Fusarium fujikuroi, a high gibberellin producer pathogen. J. Agric. Food Chem. 2015, 63, 8134–8142. [Google Scholar] [CrossRef]
- Wiemann, P.; Sieber, C.M.; von Bargen, K.W.; Studt, L.; Niehaus, E.M.; Espino, J.J.; Huss, K.; Michielse, C.B.; Albermann, S.; Wagner, D.; et al. Deciphering the cryptic genome: Genome-wide analyses of the rice pathogen Fusarium fujikuroi reveal complex regulation of secondary metabolism and novel metabolites. PLoS Pathog. 2013, 9, e1003475. [Google Scholar] [CrossRef]
- Navarro, L.; Bari, R.; Achard, P.; Lison, P.; Nemri, A.; Harberd, N.P.; Jones, J.D. DELLAs control plant immune responses by modulating the balance of jasmonic acid and salicylic acid signaling. Curr. Biol. 2008, 18, 650–655. [Google Scholar] [CrossRef]
- Sun, T.P. The molecular mechanism and evolution of the GA-GID1-DELLA signaling module in plants. Curr. Biol. 2011, 21, R338–R345. [Google Scholar] [CrossRef]
- Cheon, K.-S.; Baek, J.; Cho, Y.-i.; Jeong, Y.-M.; Lee, Y.-Y.; Oh, J.; Won, Y.J.; Kang, D.-Y.; Oh, H.; Kim, S.L.; et al. Single Nucleotide Polymorphism (SNP) Discovery and Kompetitive Allele-Specific PCR (KASP) Marker Development with Korean Japonica Rice Varieties. Plant Breed. Biotechnol. 2018, 6, 391–403. [Google Scholar] [CrossRef]
- Lai, Y.; Cuzick, A.; Lu, X.M.; Wang, J.; Katiyar, N.; Tsuchiya, T.; Roch, K.L.; McDowell, J.M.; Holub, E.; Eulgem, T. The Arabidopsis RRM domain protein EDM3 mediates racespecific disease resistance by controlling H3K9me2-dependent alternative polyadenylation of RPP7 immune receptor transcripts. Plant J. 2019, 97, 646–660. [Google Scholar] [CrossRef]
- Lorieux, M. MapDisto: Fast and efficient computation of genetic linkage maps. Mol. Breed. 2012, 30, 1231–1235. [Google Scholar] [CrossRef]
- Basten, C.J.; Weir, B.S.; Zeng, Z.-B. QTL Cartographer: A Reference Manual and Tutorial for QTL Mapping; Department of Statistics, North Carolina State University: Raleigh, NC, USA, 1996. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Source | Df * | Sum of Squares | Mean Square | F Value | Probability |
|---|---|---|---|---|---|
| line | 185 ** | 365,459.5 | 1975.5 | 16.76 | 4.1 × 10−110 |
| replication | 2 | 863.8 | 431.9 | 3.66 | 0.026545 |
| error | 370 | 43,603.3 | 117.8 |
| Variety | Raw Sequencing Data | After Quality Trimming (Q20 *) | After Read Mapping | |||||
|---|---|---|---|---|---|---|---|---|
| No. of Reads (×106) | Nucleotides (Gbp **) | No. of Reads (×106) | Nucleotides (Gbp) | Sequencing Depth (×) | No. of Reads (×106) | Nucleotides (Gbp) | Average Mapping Depth (×) | |
| ‘Junam’ | 424.65 | 55.20 | 385.75 | 49.67 | 133.09 | 322.89 | 41.74 | 111.83 |
| ‘Samgwang’ | 31.18 | 30.98 | 241.59 | 23.39 | 62.68 | 199.99 | 19.38 | 51.94 |
| QTL Name | Chromosome | Location (cm) | Closest Marker | QTL Interval * (cM) | LOD ** | Additive Effect | Dominance Effect | R2 |
|---|---|---|---|---|---|---|---|---|
| qFfR9 | 9 | 30.1 | KJ09_024, 9FC30 | 29.9–31.2 | 60.3 | 35.15 | −1.27 | 0.59 |
| Variety | Ecotype | Origin | Response to Bakanae Disease | Parental Cross Combination in Genealogy |
|---|---|---|---|---|
| ‘Junam’ | Temperate japonica | Korea | Susceptible | ‘Hwayeong’/‘Sangju’/‘Ilpum’ |
| ‘Samgwang’ | Temperate japonica | Korea | Resistant | ‘Suwon361’/‘Hwayeong’ |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, D.-Y.; Cheon, K.-S.; Oh, J.; Oh, H.; Kim, S.L.; Kim, N.; Lee, E.; Choi, I.; Baek, J.; Kim, K.-H.; et al. Rice Genome Resequencing Reveals a Major Quantitative Trait Locus for Resistance to Bakanae Disease Caused by Fusarium fujikuroi. Int. J. Mol. Sci. 2019, 20, 2598. https://doi.org/10.3390/ijms20102598
Kang D-Y, Cheon K-S, Oh J, Oh H, Kim SL, Kim N, Lee E, Choi I, Baek J, Kim K-H, et al. Rice Genome Resequencing Reveals a Major Quantitative Trait Locus for Resistance to Bakanae Disease Caused by Fusarium fujikuroi. International Journal of Molecular Sciences. 2019; 20(10):2598. https://doi.org/10.3390/ijms20102598
Chicago/Turabian StyleKang, Do-Yu, Kyeong-Seong Cheon, Jun Oh, Hyoja Oh, Song Lim Kim, Nyunhee Kim, Eungyeong Lee, Inchan Choi, Jeongho Baek, Kyung-Hwan Kim, and et al. 2019. "Rice Genome Resequencing Reveals a Major Quantitative Trait Locus for Resistance to Bakanae Disease Caused by Fusarium fujikuroi" International Journal of Molecular Sciences 20, no. 10: 2598. https://doi.org/10.3390/ijms20102598