Structural Insights into the Molecular Evolution of the Archaeal Exo-β-d-Glucosaminidase

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Structure and Thermostability of GlmA
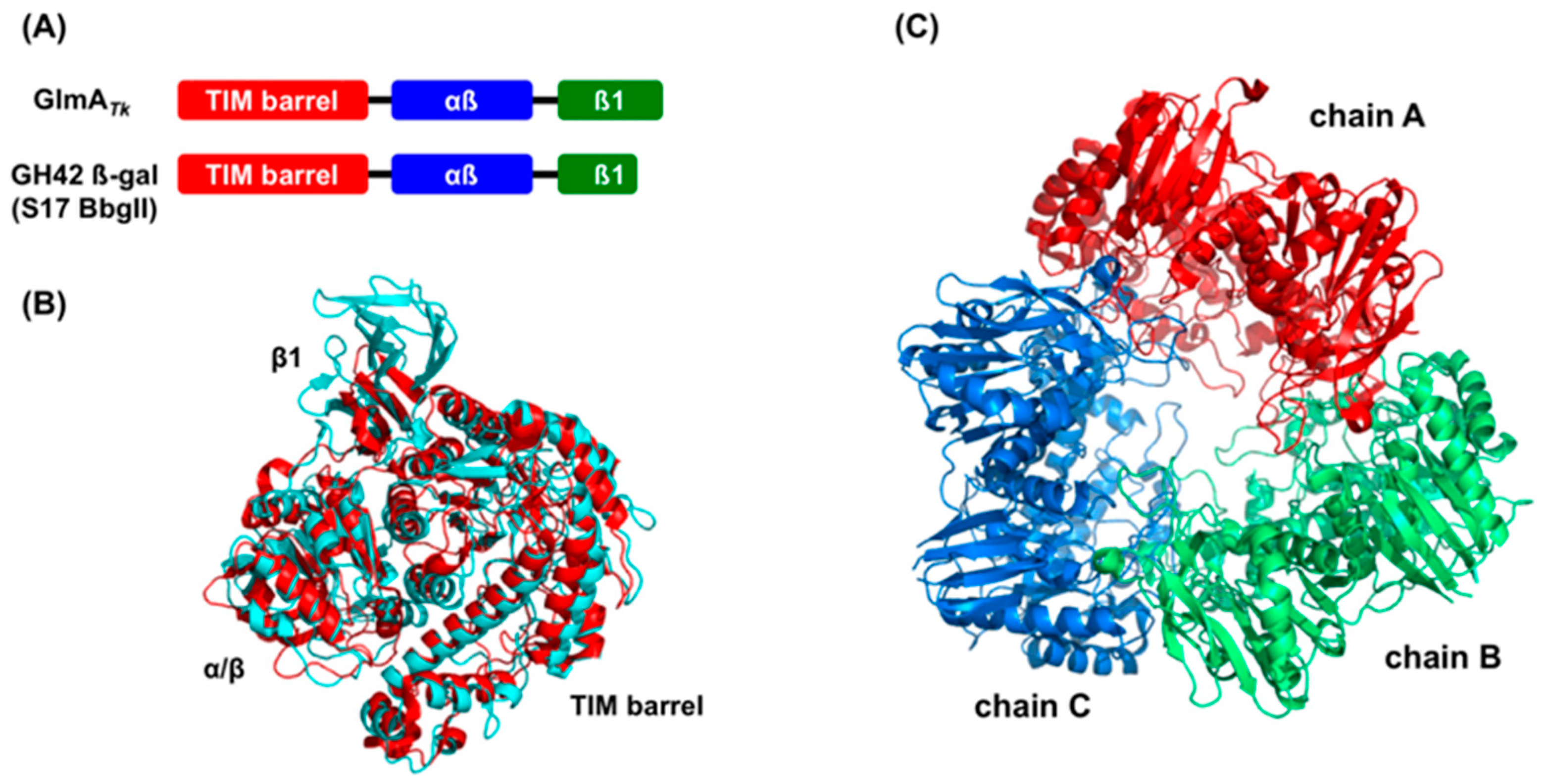
3. Structural Comparison with GlmA Homologous Proteins
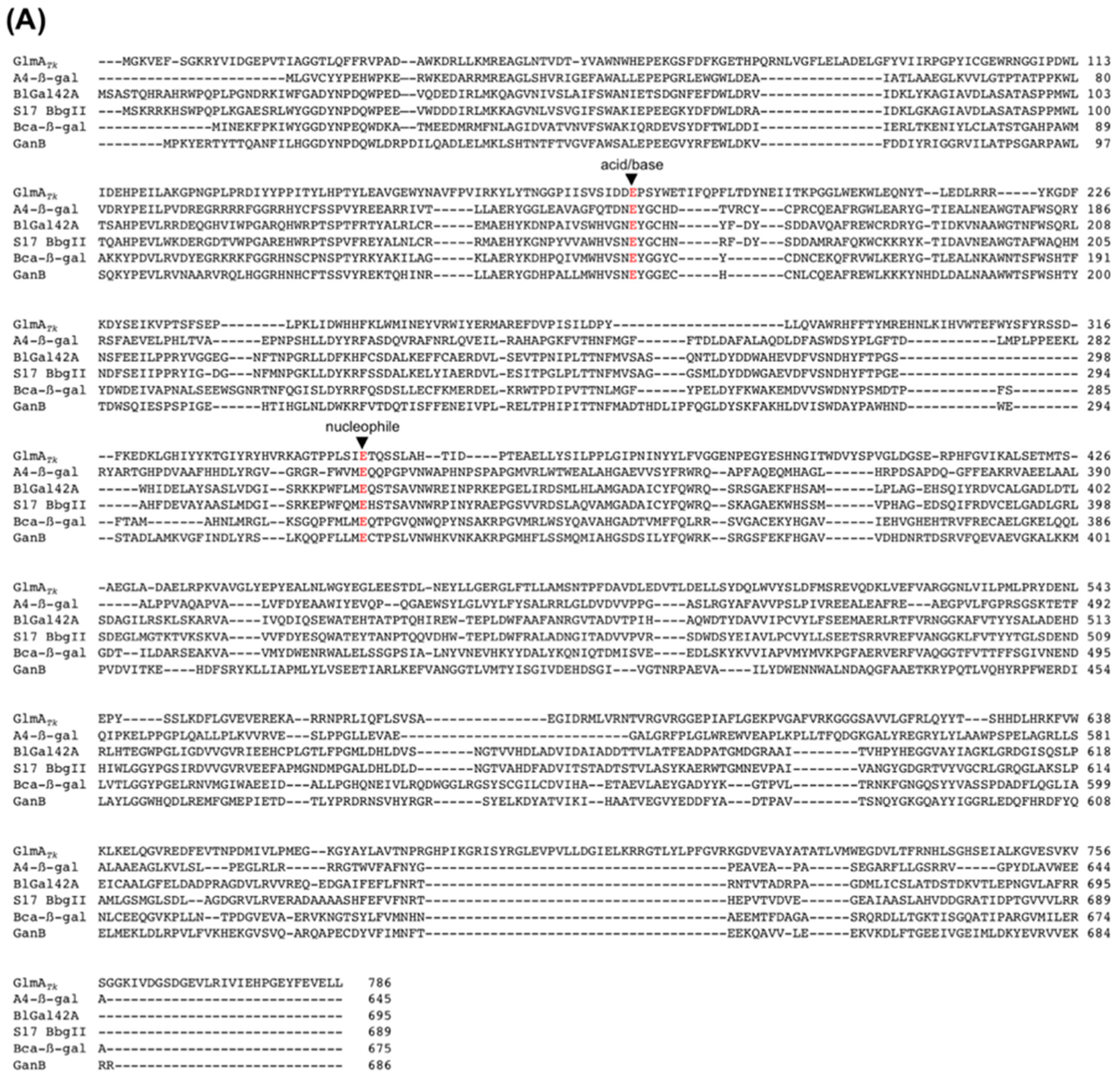
4. GlmA Active Site and Catalytic Mechanism
4.1. The Active-Site Architecture of GlmATk: Comparison with the GH35 β-Galactosidase
4.2. GlmA Catalytic Mechanism Determined through In-Depth Crystallographic Analysis
4.3. The Role of Asp178
4.4. Residue Conservation during Evolution
4.5. GlmA Dimer Structure Influences Substrate Specificity
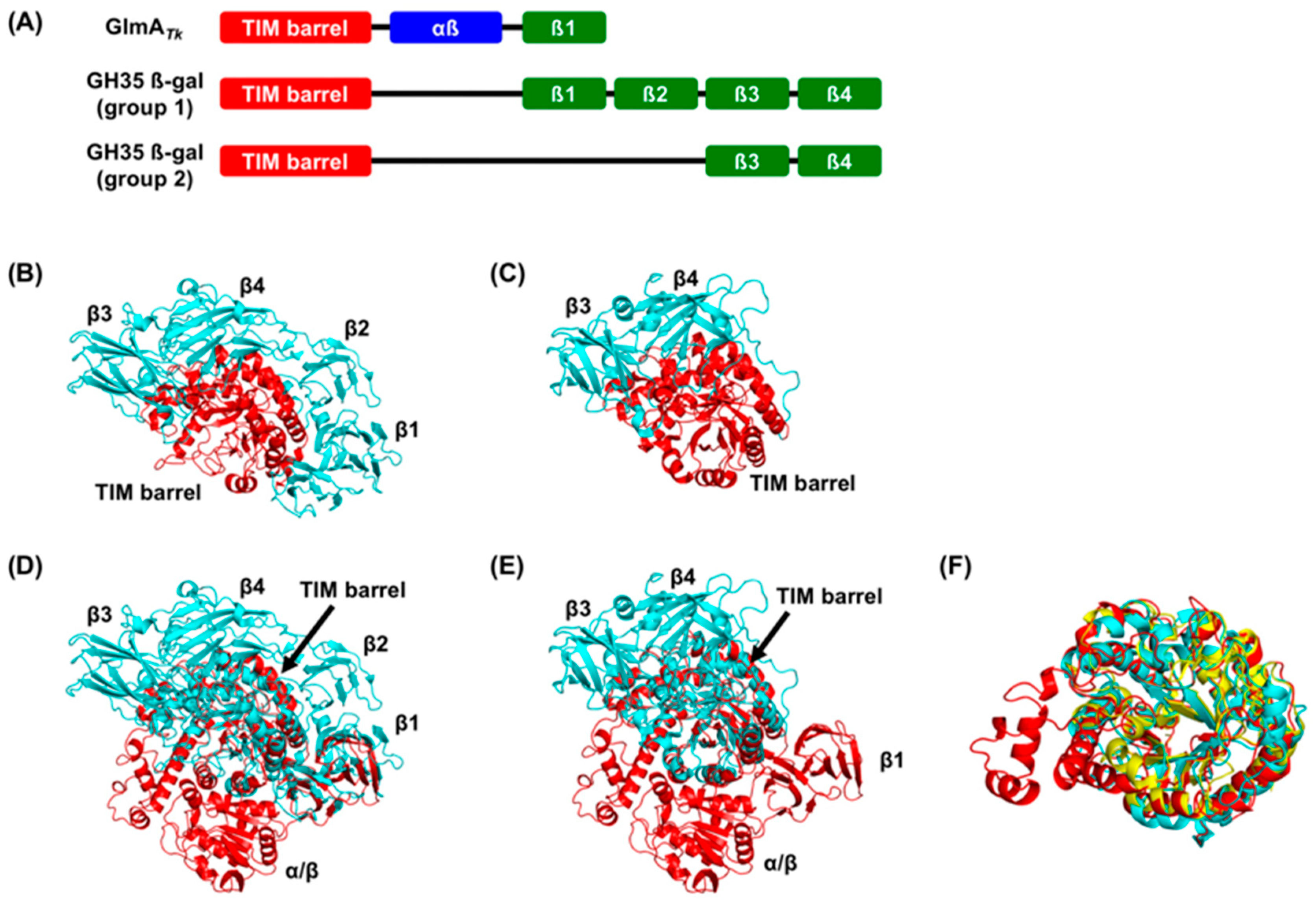
5. Molecular Evolution of GlmAs and β-Galactosidases
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tanaka, T.; Fukui, T.; Fujiwara, S.; Atomi, H.; Imanaka, T. Concerted action of diacetylchitobiose deacetylase and exo-β-D-glucosaminidase in a novel chitinolytic pathway in the hyperthermophilic archaeon Thermococcus kodakaraensis KOD1. J. Biol. Chem. 2004, 279, 30021–30027. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Fukui, T.; Atomi, H.; Imanaka, T. Characterization of an exo-β-D-glucosaminidase involved in a novel chitinolytic pathway from the hyperthermophilic archaeon Thermococcus kodakaraensis KOD1. J. Bac. 2003, 185, 5175–5181. [Google Scholar] [CrossRef]
- Aslam, M.; Horiuchi, A.; Simons, J.R.; Jha, S.; Yamada, M.; Odani, T.; Fujimoto, R.; Yamamoto, Y.; Gunji, R.; Imanaka, T.; et al. Engineering of a hyperthermophilic archaeon, Thermococcus kodakarensis, that displays chitin-dependent hydrogen production. Appl. Environ. Microbiol. 2017, 83, e00280-17. [Google Scholar] [CrossRef] [PubMed]
- Mine, S.; Nakamura, T.; Sato, T.; Ikegami, T.; Uegaki, K. Solution structure of the chitin-binding domain 1 (ChBD1) of a hyperthermophilic chitinase from Pyrococcus furiosus. J. Biochem. 2014, 155, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Mine, S.; Niiyama, M.; Hashimoto, W.; Ikegami, T.; Koma, D.; Ohmoto, T.; Fukuda, Y.; Inoue, T.; Abe, Y.; Ueda, T.; et al. Expression from engineered Escherichia coli chromosome and crystallographic study of archaeal N,N′-diacetylchitobiose deacetylase. FEBS J. 2014, 281, 2584–2596. [Google Scholar] [CrossRef]
- Nakamura, T.; Mine, S.; Hagihara, Y.; Ishikawa, K.; Ikegami, T.; Uegaki, K. Tertiary structure and carbohydrate recognition by the chitin-binding domain of a hyperthermophilic chitinase from Pyrococcus furiosus. J. Mol. Biol. 2008, 381, 670–680. [Google Scholar] [CrossRef]
- Nakamura, T.; Mine, S.; Hagihara, Y.; Ishikawa, K.; Uegaki, K. Structure of the catalytic domain of the hyperthermophilic chitinase from Pyrococcus furiosus. Acta Crystallogr. Sect. F 2007, 63, 7–11. [Google Scholar] [CrossRef]
- Hanazono, Y.; Takeda, K.; Niwa, S.; Hibi, M.; Takahashi, N.; Kanai, T.; Atomi, H.; Miki, K. Crystal structures of chitin binding domains of chitinase from Thermococcus kodakarensis KOD1. FEBS Lett. 2016, 590, 298–304. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Niiyama, M.; Hashimoto, W.; Ida, K.; Abe, M.; Morita, J.; Uegaki, K. Multiple crystal forms of N,N′-diacetylchitobiose deacetylase from Pyrococcus furiosus. Acta Crystallogr. Sect. F 2015, 71, 657–662. [Google Scholar] [CrossRef]
- Nakamura, T.; Yonezawa, Y.; Tsuchiya, Y.; Niiyama, M.; Ida, K.; Oshima, M.; Morita, J.; Uegaki, K. Substrate recognition of N,N′-diacetylchitobiose deacetylase from Pyrococcus horikoshii. J. Struct. Biol. 2016, 195, 286–293. [Google Scholar] [CrossRef] [PubMed]
- Henrissat, B.; Bairoch, A. Updating the sequence-based classification of glycosyl hydrolases. Biochem. J. 1996, 316, 695–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cote, N.; Fleury, A.; Dumont-Blanchette, E.; Fukamizo, T.; Mitsutomi, M.; Brzezinski, R. Two exo-β-D-glucosaminidases/exochitosanases from actinomycetes define a new subfamily within family 2 of glycoside hydrolases. Biochem. J. 2006, 394, 675–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Honda, Y.; Shimaya, N.; Ishisaki, K.; Ebihara, M.; Taniguchi, H. Elucidation of exo-β-D-glucosaminidase activity of a family 9 glycoside hydrolase (PBPRA0520) from Photobacterium profundum SS9. Glycobiology 2011, 21, 503–511. [Google Scholar] [CrossRef]
- Kaper, T.; Verhees, C.H.; Lebbink, J.H.; van Lieshout, J.F.; Kluskens, L.D.; Ward, D.E.; Kengen, S.W.; Beerthuyzen, M.M.; de Vos, W.M.; van der Oost, J. Characterization of β-glycosylhydrolases from Pyrococcus furiosus. Methods Enzymol. 2001, 330, 329–346. [Google Scholar] [PubMed]
- Mine, S.; Watanabe, M.; Kamachi, S.; Abe, Y.; Ueda, T. The structure of an Archaeal β-glucosaminidase provides insight into glycoside hydrolase evolution. J. Biol. Chem. 2017, 292, 4996–5006. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Li, Z.; Hong, Y.; Ni, J.; Sheng, D.; Shen, Y. Cloning, expression and characterization of a thermostable exo-β-D-glucosaminidase from the hyperthermophilic archaeon Pyrococcus horikoshii. Biotechnol. Lett. 2006, 28, 1655–1660. [Google Scholar] [CrossRef]
- Henrissat, B.; Callebaut, I.; Fabrega, S.; Lehn, P.; Mornon, J.P.; Davies, G. Conserved catalytic machinery and the prediction of a common fold for several families of glycosyl hydrolases. Proc. Natl. Acad. Sci. USA 1995, 92, 7090–7094. [Google Scholar] [CrossRef]
- Reardon, D.; Farber, G.K. The structure and evolution of α/β barrel proteins. FASEB J. 1995, 9, 497–503. [Google Scholar] [CrossRef]
- Pucci, F.; Rooman, M. Physical and molecular bases of protein thermal stability and cold adaptation. Curr. Opin. Struct. Biol. 2017, 42, 117–128. [Google Scholar] [CrossRef]
- Akiba, T.; Nishio, M.; Matsui, I.; Harata, K. X-ray structure of a membrane-bound β-glycosidase from the hyperthermophilic archaeon Pyrococcus horikoshii. Proteins 2004, 57, 422–431. [Google Scholar] [CrossRef]
- Walden, H.; Bell, G.S.; Russell, R.J.; Siebers, B.; Hensel, R.; Taylor, G.L. Tiny TIM: A small, tetrameric, hyperthermostable triosephosphate isomerase. J. Mol. Biol. 2001, 306, 745–757. [Google Scholar] [CrossRef]
- Fraser, N.J.; Liu, J.W.; Mabbitt, P.D.; Correy, G.J.; Coppin, C.W.; Lethier, M.; Perugini, M.A.; Murphy, J.M.; Oakeshott, J.G.; Weik, M.; et al. Evolution of protein quaternary structure in response to selective pressure for increased thermostability. J. Mol. Biol. 2016, 428, 2359–2371. [Google Scholar] [CrossRef]
- Rutkiewicz-Krotewicz, M.; Pietrzyk-Brzezinska, A.J.; Sekula, B.; Cieslinski, H.; Wierzbicka-Wos, A.; Kur, J.; Bujacz, A. Structural studies of a cold-adapted dimeric β-D-galactosidase from Paracoccus sp. 32d. Acta Crystallogr. D 2016, 72, 1049–1061. [Google Scholar] [CrossRef] [PubMed]
- Holm, L.; Sander, C. Mapping the protein universe. Science 1996, 273, 595–603. [Google Scholar] [CrossRef]
- Hidaka, M.; Fushinobu, S.; Ohtsu, N.; Motoshima, H.; Matsuzawa, H.; Shoun, H.; Wakagi, T. Trimeric crystal structure of the glycoside hydrolase family 42 β-galactosidase from Thermus thermophilus A4 and the structure of its complex with galactose. J. Mol. Biol. 2002, 322, 79–91. [Google Scholar] [CrossRef]
- Maksimainen, M.; Paavilainen, S.; Hakulinen, N.; Rouvinen, J. Structural analysis, enzymatic characterization, and catalytic mechanisms of β-galactosidase from Bacillus circulans sp. alkalophilus. FEBS J. 2012, 279, 1788–1798. [Google Scholar] [CrossRef]
- Solomon, H.V.; Tabachnikov, O.; Lansky, S.; Salama, R.; Feinberg, H.; Shoham, Y.; Shoham, G. Structure-function relationships in Gan42B, an intracellular GH42 β-galactosidase from Geobacillus stearothermophilus. Acta Crystallogr. D 2015, 71, 2433–2448. [Google Scholar] [CrossRef] [PubMed]
- Viborg, A.H.; Fredslund, F.; Katayama, T.; Nielsen, S.K.; Svensson, B.; Kitaoka, M.; Lo Leggio, L.; Abou Hachem, M. A β1-6/β1-3 galactosidase from Bifidobacterium animalis subsp. lactis Bl-04 gives insight into sub-specificities of β-galactoside catabolism within Bifidobacterium. Mol. Microbiol. 2014, 94, 1024–1040. [Google Scholar] [CrossRef] [PubMed]
- Godoy, A.S.; Camilo, C.M.; Kadowaki, M.A.; Muniz, H.D.; Espirito Santo, M.; Murakami, M.T.; Nascimento, A.S.; Polikarpov, I. Crystal structure of β1→6-galactosidase from Bifidobacterium bifidum S17: Trimeric architecture, molecular determinants of the enzymatic activity and its inhibition by α-galactose. FEBS J. 2016, 283, 4097–4112. [Google Scholar] [CrossRef] [PubMed]
- Maksimainen, M.M.; Lampio, A.; Mertanen, M.; Turunen, O.; Rouvinen, J. The crystal structure of acidic β-galactosidase from Aspergillus oryzae. Int. J. Biol. Macromol. 2013, 60, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Rico-Diaz, A.; Ramirez-Escudero, M.; Vizoso-Vazquez, A.; Cerdan, M.E.; Becerra, M.; Sanz-Aparicio, J. Structural features of Aspergillus niger β-galactosidase define its activity against glycoside linkages. FEBS J. 2017, 284, 1815–1829. [Google Scholar] [CrossRef] [PubMed]
- Maksimainen, M.; Hakulinen, N.; Kallio, J.M.; Timoharju, T.; Turunen, O.; Rouvinen, J. Crystal structures of Trichoderma reesei β-galactosidase reveal conformational changes in the active site. J. Struct. Biol. 2011, 174, 156–163. [Google Scholar] [CrossRef]
- Rojas, A.L.; Nagem, R.A.; Neustroev, K.N.; Arand, M.; Adamska, M.; Eneyskaya, E.V.; Kulminskaya, A.A.; Garratt, R.C.; Golubev, A.M.; Polikarpov, I. Crystal structures of β-galactosidase from Penicillium sp. and its complex with galactose. J. Mol. Biol. 2004, 343, 1281–1292. [Google Scholar] [CrossRef] [PubMed]
- Ohto, U.; Usui, K.; Ochi, T.; Yuki, K.; Satow, Y.; Shimizu, T. Crystal structure of human β-galactosidase: Structural basis of Gm1 gangliosidosis and morquio B diseases. J. Biol. Chem. 2012, 287, 1801–1812. [Google Scholar] [CrossRef] [PubMed]
- Henze, M.; You, D.J.; Kamerke, C.; Hoffmann, N.; Angkawidjaja, C.; Ernst, S.; Pietruszka, J.; Kanaya, S.; Elling, L. Rational design of a glycosynthase by the crystal structure of β-galactosidase from Bacillus circulans (BgaC) and its use for the synthesis of N-acetyllactosamine type 1 glycan structures. J. Biotechnol. 2014, 191, 78–85. [Google Scholar] [CrossRef]
- Cheng, W.; Wang, L.; Jiang, Y.L.; Bai, X.H.; Chu, J.; Li, Q.; Yu, G.; Liang, Q.L.; Zhou, C.Z.; Chen, Y. Structural insights into the substrate specificity of Streptococcus pneumoniae β(1,3)-galactosidase BgaC. J. Biol. Chem. 2012, 287, 22910–22918. [Google Scholar] [CrossRef]
- Suzuki, H.; Ohto, U.; Higaki, K.; Mena-Barragan, T.; Aguilar-Moncayo, M.; Ortiz Mellet, C.; Nanba, E.; Garcia Fernandez, J.M.; Suzuki, Y.; Shimizu, T. Structural basis of pharmacological chaperoning for human β-galactosidase. J. Biol. Chem. 2014, 289, 14560–14568. [Google Scholar] [CrossRef]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef]
- Beecher, C.N.; Larive, C.K. 1H and 15N NMR Characterization of the Amine Groups of Heparan Sulfate Related Glucosamine Monosaccharides in Aqueous Solution. Anal. Chem. 2015, 87, 6842–6848. [Google Scholar] [CrossRef]
- Van Bueren, A.L.; Ghinet, M.G.; Gregg, K.; Fleury, A.; Brzezinski, R.; Boraston, A.B. The structural basis of substrate recognition in an exo-β-D-glucosaminidase involved in chitosan hydrolysis. J. Mol. Biol. 2009, 385, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Pluvinage, B.; Ghinet, M.G.; Brzezinski, R.; Boraston, A.B.; Stubbs, K.A. Inhibition of the exo-β-D-glucosaminidase CsxA by a glucosamine-configured castanospermine and an amino-australine analogue. Org. Biomol. Chem. 2009, 7, 4169–4172. [Google Scholar] [CrossRef]
- Juers, D.H.; Huber, R.E.; Matthews, B.W. Structural comparisons of TIM barrel proteins suggest functional and evolutionary relationships between β-galactosidase and other glycohydrolases. Protein Sci. 1999, 8, 122–136. [Google Scholar] [CrossRef]
- Qian, W.; Zhang, J. Genomic evidence for adaptation by gene duplication. Genome Res. 2014, 24, 1356–1362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, Y.; Gangoiti, J.; Dijkstra, B.W.; Dijkhuizen, L.; Pijning, T. Crystal structure of 4,6-α-glucanotransferase supports diet-driven evolution of GH70 enzymes from α-amylases in oral bacteria. Structure 2017, 25, 231–242. [Google Scholar] [CrossRef]
- Eklof, J.M.; Shojania, S.; Okon, M.; McIntosh, L.P.; Brumer, H. Structure-function analysis of a broad specificity Populus trichocarpa endo-β-glucanase reveals an evolutionary link between bacterial licheninases and plant XTH gene products. J. Biol. Chem. 2013, 288, 15786–15799. [Google Scholar] [CrossRef]
- Gangoiti, J.; Pijning, T.; Dijkhuizen, L. The Exiguobacterium sibiricum 255-15 GtfC enzyme represents a novel glycoside hydrolase 70 subfamily of 4,6-α-glucanotransferase enzymes. Appl. Environ. Microbiol. 2016, 82, 756–766. [Google Scholar] [CrossRef] [PubMed]
- Matthews, B.W. The structure of E. coli β-galactosidase. C. R. Biol. 2005, 328, 549–556. [Google Scholar] [CrossRef]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mine, S.; Watanabe, M. Structural Insights into the Molecular Evolution of the Archaeal Exo-β-d-Glucosaminidase. Int. J. Mol. Sci. 2019, 20, 2460. https://doi.org/10.3390/ijms20102460
Mine S, Watanabe M. Structural Insights into the Molecular Evolution of the Archaeal Exo-β-d-Glucosaminidase. International Journal of Molecular Sciences. 2019; 20(10):2460. https://doi.org/10.3390/ijms20102460
Chicago/Turabian StyleMine, Shouhei, and Masahiro Watanabe. 2019. "Structural Insights into the Molecular Evolution of the Archaeal Exo-β-d-Glucosaminidase" International Journal of Molecular Sciences 20, no. 10: 2460. https://doi.org/10.3390/ijms20102460