Chronic Administration of Hydroxyurea (HU) Benefits Caucasian Patients with Sickle-Beta Thalassemia

 ,
,
Abstract
:1. Introduction
2. Results
2.1. Hydroxyurea Dosing Adjustment Was Common
2.2. Hydroxyurea Led to Similar Beneficial Hematologic Changes in All Sickle Phenotypes
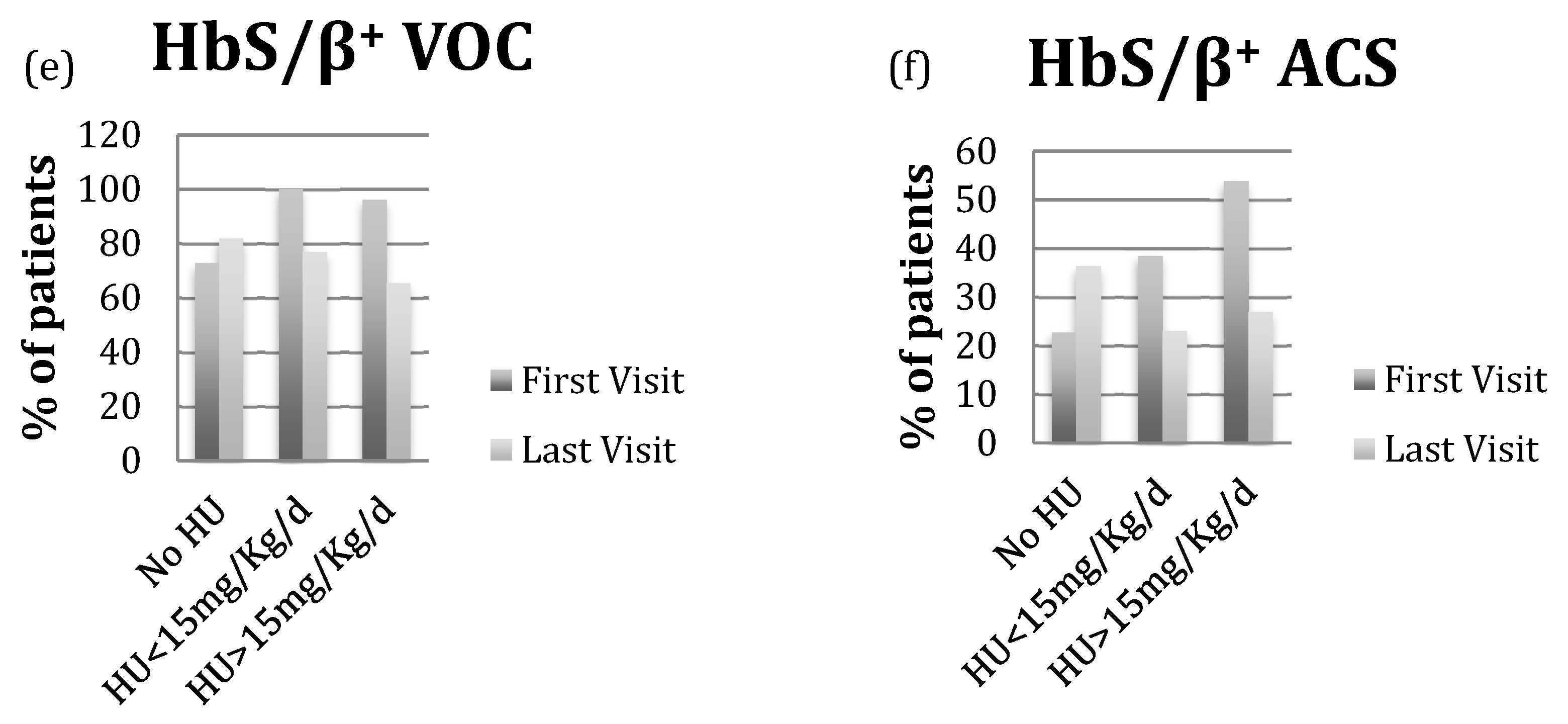
2.3. Hydroxyurea Reduced VOC and ACS Events
2.4. Mortality
3. Discussion
4. Methods
4.1. Patient Population
4.2. Clinical, Laboratory, and Echocardiographic Data
4.3. Hydroxyurea Status and Dose Determination
4.4. Statistical Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- World Health Organization Regional Office for Africa. Sickle-Cell Disease: A Strategy for the WHO African Region; Report of the Regional Director; WHO: Malabo, Equatorial Guinea, 2010; Available online: http://www.afro.who.int/index.php?option=com_docman&task=doc_download&gid=6638 (accessed on 30 October 2011).
- Modell, B.; Darlison, M. Global epidemiology of aemoglobin disorders and derived service indicators. Boll. World Health Organ. 2008, 86, 480–487. [Google Scholar] [CrossRef]
- Piel, F.B.; Patil, A.P.; Howes, R.E.; Nyangiri, O.A.; Gething, P.W.; Dewi, M.; Temperley, W.H.; Williams, T.N.; Weatherall, D.J.; Hay, S.I. Global epidemiology of sickle haemoglobin in neonates: A contemporary geostatical model-based map and population estimates. Lancet 2013, 281, 142–151. [Google Scholar] [CrossRef]
- Weatherall, D.J. The inherited diseases of hemoglobin are an emerging global health burden. Blood 2010, 115, 4331–4336. [Google Scholar] [CrossRef] [PubMed]
- Gaston, M.H.; Verter, J.I.; Woods, G.; Pegelow, C.; Kelleher, J.; Presbury, G.; Zarkowsky, H.; Vichinsky, E.; Iyer, R.; Lobel, J.S.; et al. Prophylaxis with oral penicillin in children with sickle cell anemia. N. Engl. J. Med. 1986, 314, 1593–1599. [Google Scholar] [CrossRef] [PubMed]
- Falletta, J.M.; Woods, G.M.; Verter, J.I.; Buchanan, G.R.; Pegelow, C.H.; Iyer, R.V.; Miller, S.T.; Holbrook, C.T.; Kinney, T.R.; Vichinsky, E. Discontinuing penicillin prophylaxis in children with sickle cell anemia. Prophylactic Penicillin Study II. J. Pediatr. 1995, 127, 685–690. [Google Scholar] [CrossRef]
- Halasa, N.B.; Shankar, S.M.; Talbot, T.R.; Arbogast, P.G.; Mitchel, E.F.; Wang, W.C.; Schaffner, W.; Craig, A.S.; Griffin, M.R. Incidence of invasive pneumococcal disease among individuals with sickle cell disease before and after the introduction of the pneumococcal conjugate vaccine. Clin. Infect. Dis. 2007, 44, 1428–1433. [Google Scholar] [CrossRef] [PubMed]
- McGann, P.T.; Nero, A.C.; Ware, R.E. Current Management of Sickle Cell Anemia. Cold Spring Harb. Perspect. Med. 2013, 3, a011817. [Google Scholar] [CrossRef] [PubMed]
- National Institutes of Health NHLBI. The Management of Sickle Cell Disease; NIH Publication No 02-2117; 2002. Available online: http://www.nhlbi.nih.gov/health/prof/blood/sickle/sc_mngt.pdf (accessed on 28 October 2010).
- Hamideh, D.; Alvarez, O. Sickle cell disease related mortality in the United States (1999–2009). Pediatr. Blood Cancer 2013, 60, 1482–1486. [Google Scholar] [CrossRef] [PubMed]
- Elmariah, H.; Garrett, M.E.; De Castro, L.M.; Jonassaint, J.C.; Ataga, K.I.; Eckman, J.R.; Ashley-Koch, A.E.; Telen, M.J. Factors associated with survival in a contemporary adult sickle cell disease cohort. Am. J. Hematol. 2014, 89, 530–535. [Google Scholar] [CrossRef] [PubMed]
- Charache, S.; Barton, F.B.; Moore, R.D.; Terrin, M.L.; Steinberg, M.H.; Dover, G.J.; Ballas, S.K.; McMahon, R.P.; Castro, O.; Orringer, E.P. Hydroxyurea and sickle cell anemia: Clinical utility of a myelosuppressive “switching” agent. The Multicenter Study of Hydroxyurea in Sickle Cell Anemia. Medicine (Baltim.) 1996, 75, 300–326. [Google Scholar] [CrossRef]
- Kato, G.J.; Gladwin, M.T.; Steinberg, M.H. Deconstructing sickle cell disease: Reappraisal of the role of hemolysis in the development of clinical subphenotypes. Blood Rev. 2007, 21, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Benkerrou, M.; Delarche, C.; Brahimi, L.; Fay, M.; Vilmer, E.; Elion, J.; Gougerot-Pocidalo, M.A.; Elbim, C. Hydroxyurea corrects the dysregulated l-selectin expression and increased h2o2 production of polymorphonuclear neutrophils from patients with sickle cell anemia. Blood 2002, 99, 2297–2303. [Google Scholar] [CrossRef] [PubMed]
- Gladwin, M.T.; Shelhamer, J.H.; Ognibene, F.P.; Pease-Fye, M.E.; Nichols, J.S.; Link, B.; Patel, D.B.; Jankowski, M.A.; Pannell, L.K.; Schechter, A.N.; et al. Nitric oxide donor properties of hydroxyurea in patients with sickle cell disease. Br. J. Haematol. 2002, 116, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Ware, R.E.; Aygun, B. Advances in the use of hydroxyurea. Hematol. Am. Soc. Hematol. Educ. Program 2009, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Dover, G.J.; Charache, S. Hydroxyurea induction of fetal hemoglobin synthesis in sickle-cell disease. Semin. Oncol. 1992, 19 (Suppl. 9), 61–66. [Google Scholar] [PubMed]
- Cokic, V.P.; Smith, R.D.; Beleslin-Cokic, B.B.; Njoroge, J.M.; Miller, J.L.; Gladwin, M.T.; Schechter, A.N. Hydroxyurea induces fetal hemoglobin by the nitric oxide-dependent activation of soluble guanylyl cyclase. J. Clin. Investig. 2003, 111, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Ho, J.A.; Pickens, C.V.; Gamcsik, M.P.; Colvin, O.M.; Ware, R.E. In vitro induction of fetal hemoglobin in human erythroid progenitor cells. Exp. Hematol. 2003, 31, 586–591. [Google Scholar] [CrossRef]
- Charache, S.; Terrin, M.L.; Moore, R.D.; Dover, G.J.; Barton, F.B.; Eckert, S.V.; McMahon, R.P.; Bonds, D.R. Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. Investigators of the Multicenter Study of Hydroxyurea in Sickle Cell Anemia. N. Engl. J. Med. 1995, 332, 1317–1322. [Google Scholar] [CrossRef] [PubMed]
- Ferster, A.; Vermylen, C.; Cornu, G.; Buyse, M.; Corazza, F.; Devalck, C.; Fondu, P.; Toppet, M.; Sariban, E. Hydroxyurea for treatment of severe sickle cell anemia: A pediatric clinical trial. Blood 1996, 88, 1960–1964. [Google Scholar] [PubMed]
- Steinberg, M.H.; McCarthy, W.F.; Castro, O.; Ballas, S.K.; Armstrong, F.D.; Smith, W.; Ataga, K.; Swerdlow, P.; Kutlar, A.; DeCastro, L.; et al. The risks and benefits of long-term use of hydroxyurea in sickle cell anemia: A 17.5 year follow-up. Am. J. Hematol. 2010, 85, 403–408. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.C.; Ware, R.E.; Miller, S.T.; Iyer, R.V.; Casella, J.F.; Minniti, C.P.; Rana, S.; Thornburg, C.D.; Rogers, Z.R.; Kalpatthi, R.V.; et al. Hydroxycarbamide in very young children with sickle-cell anaemia: A multicentre, randomised, controlled trial (BABY HUG). Lancet 2011, 377, 1663–1672. [Google Scholar] [CrossRef]
- Wang, W.; Brugnara, C.; Snyder, C.; Wynn, L.; Rogers, Z.; Kalinyak, K.; Brown, C.; Qureshi, A.; Bigelow, C.; Neumayr, L.; et al. The effects of hydroxycarbamide and magnesium on haemoglobin SC disease: Results of the multi-centre CHAMPS trial. Br. J. Haematol. 2011, 152, 771–776. [Google Scholar] [CrossRef] [PubMed]
- Voskaridou, E.; Christoulas, D.; Bilalis, A.; Plata, E.; Varvagiannis, K.; Stamatopoulos, G.; Sinopoulou, K.; Balassopoulou, A.; Loukopoulos, D.; Terpos, E. The effect of prolonged administration of hydroxyurea on morbidity and mortality in adult patients with sickle cell syndromes: Results of a 17-year, single-center trial (LASHS). Blood 2010, 115, 2354–2363. [Google Scholar] [CrossRef] [PubMed]
- Lobo, C.; Hankins, J.S.; Moura, P.; Plata, E.; Varvagiannis, K.; Stamatopoulos, G.; Sinopoulou, K.; Balassopoulou, A.; Loukopoulos, D.; Terpos, E. Hydroxyurea therapy reduces mortality among children with sickle cell disease. ASH Annu. Meet. Abstr. 2010, 116, 843. [Google Scholar]
- Platt, O.S.; Brambilla, D.J.; Rosse, W.F.; Milner, P.F.; Castro, O.; Steinberg, M.H.; Klug, P.P. Mortality in sickle cell disease: Life expectancy and risk factors for early death. N. Engl. J. Med. 1994, 330, 1639–1644. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, M.H.; Lu, Z.H.; Barton, F.B.; Terrin, M.L.; Charache, S.; Dover, G.J. Fetal hemoglobin in sickle cell anemia: Determinants of response to hydroxyurea. Multicenter Study of Hydroxyurea. Blood 1997, 89, 1078–1088. [Google Scholar] [CrossRef] [PubMed]
- Akinsheye, I. Fetal hemoglobin in sickle cell anemia. Blood 2011, 118, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Rigano, P.; De Franceschi, L.; Sainati, L.; Piga, A.; Piel, F.B.; Cappellini, M.D.; Fidone, C.; Masera, N.; Palazzi, G.; Gianesin, B.; et al. Italian Multicenter Study of Hydroxyurea in Sickle Cell Anemia Investigators Real-life experience with hydroxyurea in sickle cell disease: A multicenter study in a cohort of patients with heterogeneous descent. Blood Cells Mol. Dis. 2018, 69, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, M.H.; Barton, F.; Castro, O.; Ballas, S.K.; Armstrong, F.D.; Smith, W.; Ataga, K.; Swerdlow, P.; Kutlar, A.; DeCastro, L.; et al. Effect of hydroxyurea on mortality and morbidity in adult sickle cell anemia: Risks and benefits up to 9 years of treatment. JAMA 2003, 289, 1645–1651. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, M.H. Predicting clinical severity in sickle cell anemia. Br. J. Hematol. 2005, 129, 464–481. [Google Scholar] [CrossRef] [PubMed]
- Mark, T.; Gladwin, M.D.; Vichinsky, E. Pulmonary Complications of Sickle Cell Disease. N. Engl. J. Med. 2008, 359, 2254–2265. [Google Scholar]
- Castro, O.; Brambilla, D.J.; Thorington, B.; Reindorf, C.A.; Scott, R.B.; Gillette, P.; Vera, J.C.; Levy, P.S. The acute chest syndrome in sickle cell disease: Incidence and risk factors. Blood 1994, 84, 643–649. [Google Scholar] [PubMed]



{kind=link}
{kind=link}
| Variable | All (N = 140) | Alive (N = 125) | Deceased (N = 15) | |
|---|---|---|---|---|
| Age at enrollment (years) | 35 (0.4–61) | 35 (0.4–61) | 42 (20–60) | |
| Gender | Male | 69 (49%) | 59 (47%) | 10 (67%) |
| Female | 71 (51%) | 66 (53%) | 5 (33%) | |
| Hydroxyurea Status | Yes | 90 (72%) | 81 (73%) | 9 (60%) |
| No | 50 (28%) | 44 (27%) | 6 (40%) | |
| Transfusion | Yes | 48 (34%) | 43 (34%) | 5 (33%) |
| No | 92 (66%) | 82 (66%) | 10 (67%) | |
| Chelation | Yes | 40 (29%) | 32 (26%) | 8 (53%) |
| No | 100 (71%) | 93 (74%) | 7 (47%) | |
| Variable | HbSS (N = 25) | HbS/β0 thal (N = 54) | HbS/β+ thal, δβthal, Lepore (N = 61) | ||||
|---|---|---|---|---|---|---|---|
| HU No (N = 10) | HU Yes (N = 15) | HU No (N = 18) | HU Yes (N = 36) | HU No (N = 22) | HU Yes (N = 39) | ||
| Age (years) | 31.5 (22–44) | 32.7 (13–44) | 35.3 (0.41–57) | 31.9 (5–61) | 38 (1–60) | 34.4 (7–56) | |
| Gender | Male | 4 (40%) | 7 (47%) | 7 (39%) | 20 (55%) | 8 (36%) | 23 (59%) |
| Female | 6 (60) | 8 (53%) | 11 (61%) | 16 (45%) | 14 (64%) | 16 (41%) | |
| Survival Status | Alive | 9 (90%) | 15 (100%) | 16 (89%) | 31 (86%) | 19 (86%) | 35 (90%) |
| Deceased | 1 (10%) | 0 (0%) | 1 (11%) | 5 (14%) | 3 (14%) | 4 (10%) | |
| Hydroxyurea Dosage, N (%) | No HU | 10 (100%) | 0 (0%) | 18 (100%) | 0 (0%) | 22 (100%) | 0 (0%) |
| <15 mg/kg/d | 0 (0%) | 4 (16%) | 0 (0%) | 13 (36%) | 0 (0%) | 13 (33%) | |
| ≥15 mg/kg/d | 0 (0%) | 11 (84%) | 0 (0%) | 23 (64%) | 0 (0%) | 26 (67%) | |
| Maximum HbF (%) | No HU | 16.1 ± 12.2 | 0 | 28.7 ± 17.3 | 0 | 11.5 ± 10.1 | 0 |
| <15 mg/kg/d | 0 | 12.8 ± 8.3 | 0 | 25.9 ± 9 | 0 | 9 ± 7 | |
| ≥15 mg/kg/d | 0 | 19.3 ± 7.8 | 0 | 21.6 ± 11.9 | 0 | 19.6 ± 7.6 | |
| Mean HbF (%) | No HU | 11.2 ± 11.8 | 0 | 10.1 ± 9.2 | 0 | 9.4 ± 9.7 | 0 |
| <15 mg/kg/d | 0 | 10.1 ± 4.9 | 0 | 8.8 ± 6.3 | 0 | 4.7 ± 3.1 | |
| ≥15 mg/kg/d | 0 | 10.2 ± 6.9 | 0 | 15 ± 11.1 | 0 | 10.9 ± 6.8 | |
| Maximum MCV | No HU | 101 ± 12 | 0 | 89 ± 14 | 0 | 79 ± 10 | 0 |
| <15 mg/kg/d | 115 ± 14 | 0 | 95 ± 10 | 0 | 86 ± 13 | ||
| ≥15 mg/kg/d | 124 ± 18 | 0 | 94 ± 12 | 0 | 100 ± 9 | ||
| Variable | First Visit | Lst Visit | ||
|---|---|---|---|---|
| Alive | Deceased | Alive | Deceased | |
| White Blood Count (K/µL) | 10.08 ± 1.71 | 8.9 ± 3.13 | 10.87 ± 8.65 | 11.81 ± 9.52 |
| ANC (K/µL) | 5.59 ± 3.13 | 4.73 ± 1.85 | 4.68 ± 2.24 | 4.54 ± 2.95 |
| Hemoglobin (g/dL) | 10.08 ± 1.71 | 9.72 ± 1.38 | 10.87 ± 8.65 | 9.5 ± 1.21 |
| MCV (fL) | 81.12 ± 13.45 | 82.97 ± 11.21 | 85.08 ± 13.65 | 81.82 ± 7.45 |
| Platelet Count (K/µL) | 366.72 ± 207.92 | 299.9 ± 174.84 | 338.54 ± 192.24 | 244.33 ± 112.95 |
| Reticolocyte (%) | 6.87 ± 4.77 | 6.38 ± 2.72 | 6.04 ± 3.60 | 7.65 ± 3.91 |
| Hemoglobin F (%) | 10.7 ± 10.1 | 11.96 ± 9.43 | 10.86 ± 8.98 | 10.68 ± 10.41 |
| Alkaline Phosphatase (U/L) | 134.78 ± 96.08 | 191.93 ± 100.14 | 77.4 ± 53.71 | 197.13 ± 140.42 |
| ALT (U/L) | 29.04 ± 17.32 | 53.66 ± 47.19 | 31.87 ± 20.01 | 46.46 ± 31.04 |
| AST (U/L) | 38.21 ± 21.65 | 69.8 ± 43.43 | 38.38 ± 22.25 | 93.33 ± 63.93 |
| Totl Bilirubin (mg/dL) | 2.44 ± 1.90 | 3.23 ± 2.1 | 2.2 ± 2.06 | 7.24 ± 8.9 |
| Direct Bilirubin (mg/dL) | 0.41 ± 0.35 | 0.98 ± 1.03 | 0.43 ± 0.66 | 3.14 ± 2.98 |
| Creatinine (mg/dL) | 0.65 ± 0.41 | 0.72 ± 0.26 | 0.58 ± | 0.74 ± 0.33 |
| Ejection Fraction (%) | 63.93 ± 5.69 | 64.1 ± 4.98 | 62.51 ± 7.46 | 64.14 ± 6.64 |
| TRV (m/s) | 2.49 ± 0.52 | 2.95 ± 0.36 | 2.67 ± 0.33 | 2.8 ± 0.8 |
| Ferritin (mcg/L) | 536.89 ± 730.40 | 1338.8 ± 1485.95 | 648.65 ± 858.47 | 1927.96 ± 1654.50 |
| Iron (mcg/L) | 109.28 ± 55.67 | 153.86 ± 79.01 | 116.76 ± 53.63 | 160.4 ± 71.23 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Maggio, R.; Hsieh, M.M.; Zhao, X.; Calvaruso, G.; Rigano, P.; Renda, D.; Tisdale, J.F.; Maggio, A. Chronic Administration of Hydroxyurea (HU) Benefits Caucasian Patients with Sickle-Beta Thalassemia. Int. J. Mol. Sci. 2018, 19, 681. https://doi.org/10.3390/ijms19030681
Di Maggio R, Hsieh MM, Zhao X, Calvaruso G, Rigano P, Renda D, Tisdale JF, Maggio A. Chronic Administration of Hydroxyurea (HU) Benefits Caucasian Patients with Sickle-Beta Thalassemia. International Journal of Molecular Sciences. 2018; 19(3):681. https://doi.org/10.3390/ijms19030681
Chicago/Turabian StyleDi Maggio, Rosario, Matthew M. Hsieh, Xiongce Zhao, Giuseppina Calvaruso, Paolo Rigano, Disma Renda, John F. Tisdale, and Aurelio Maggio. 2018. "Chronic Administration of Hydroxyurea (HU) Benefits Caucasian Patients with Sickle-Beta Thalassemia" International Journal of Molecular Sciences 19, no. 3: 681. https://doi.org/10.3390/ijms19030681