Insights into the Structural Requirements of 2(S)-Amino-6-Boronohexanoic Acid Derivatives as Arginase I Inhibitors: 3D-QSAR, Docking, and Interaction Fingerprint Studies


Abstract
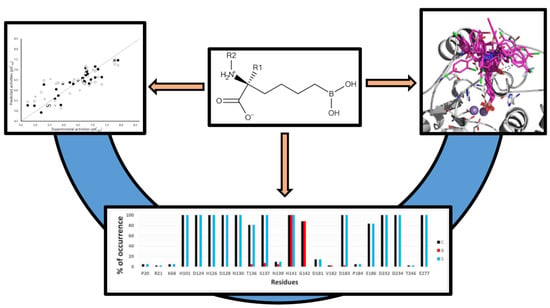
:
1. Introduction
2. Results
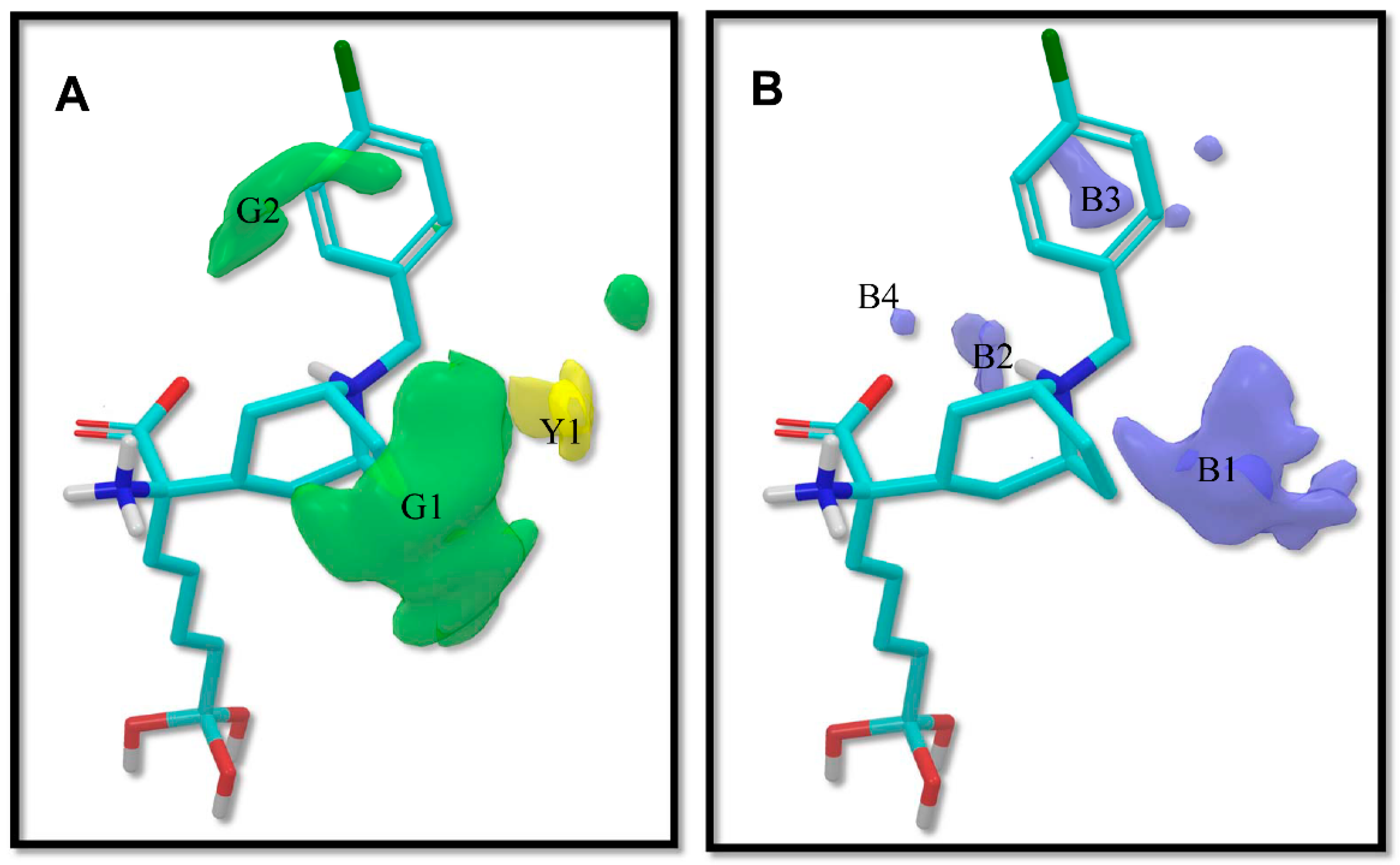
2.1. Analysis of 3D-QSAR Models
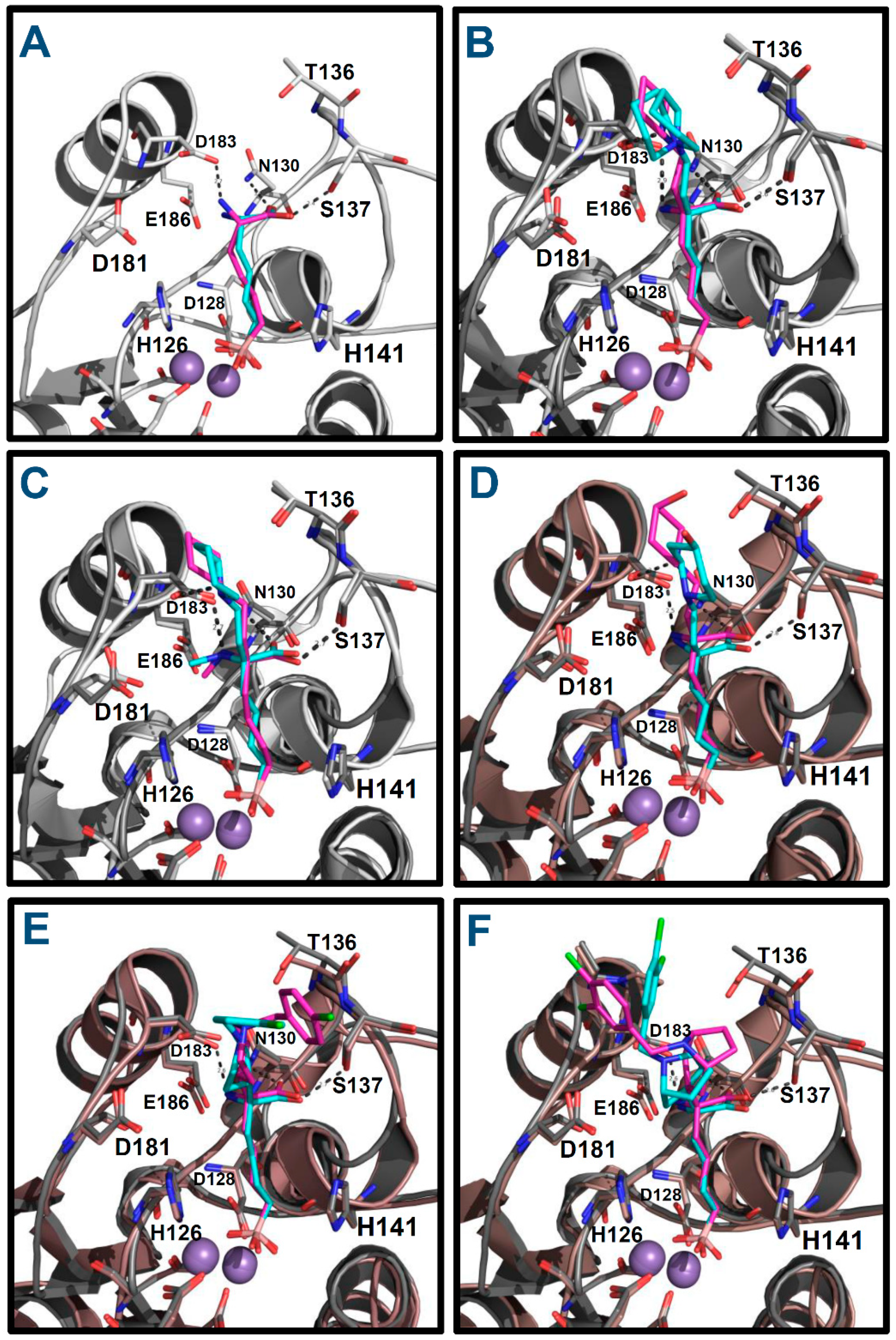
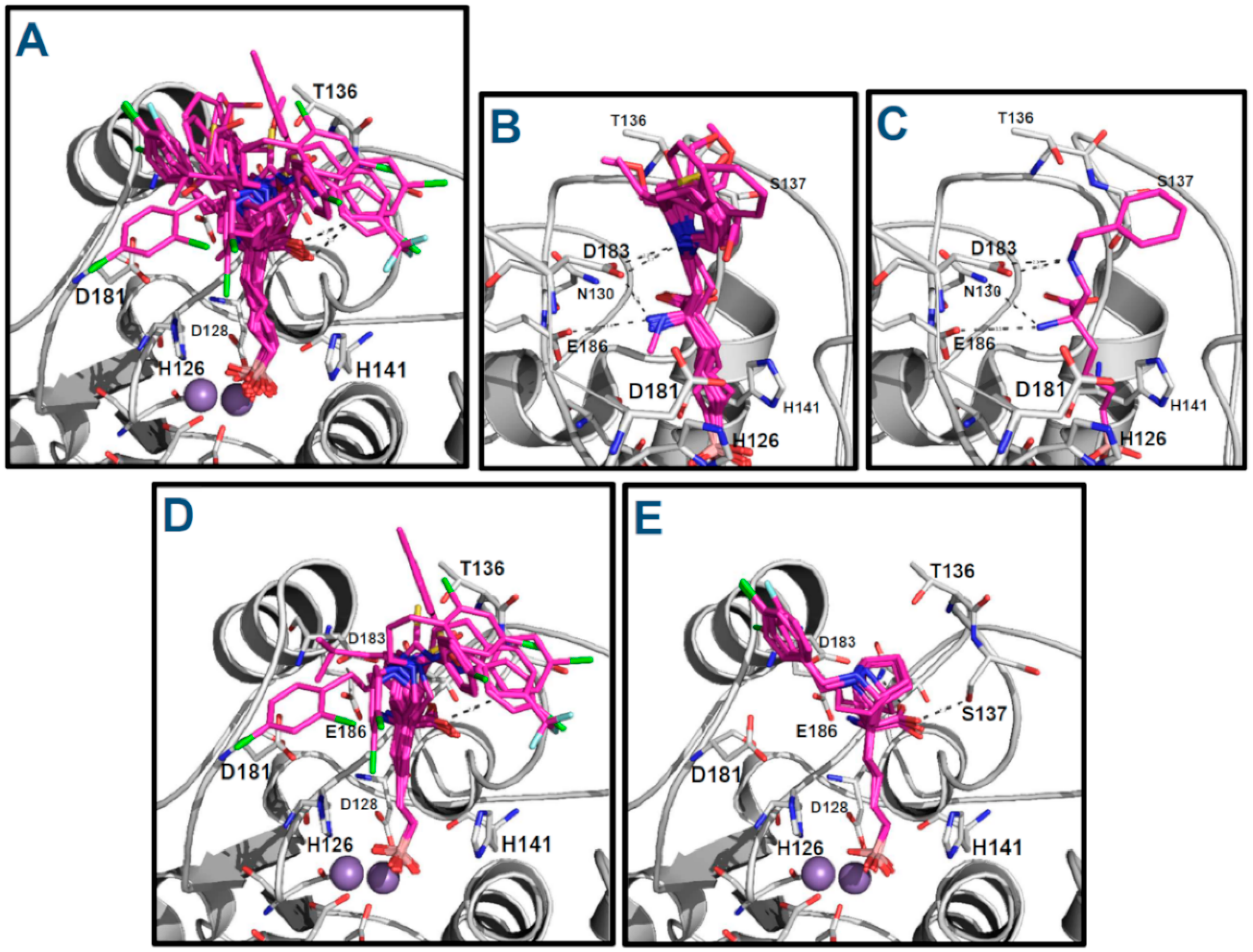
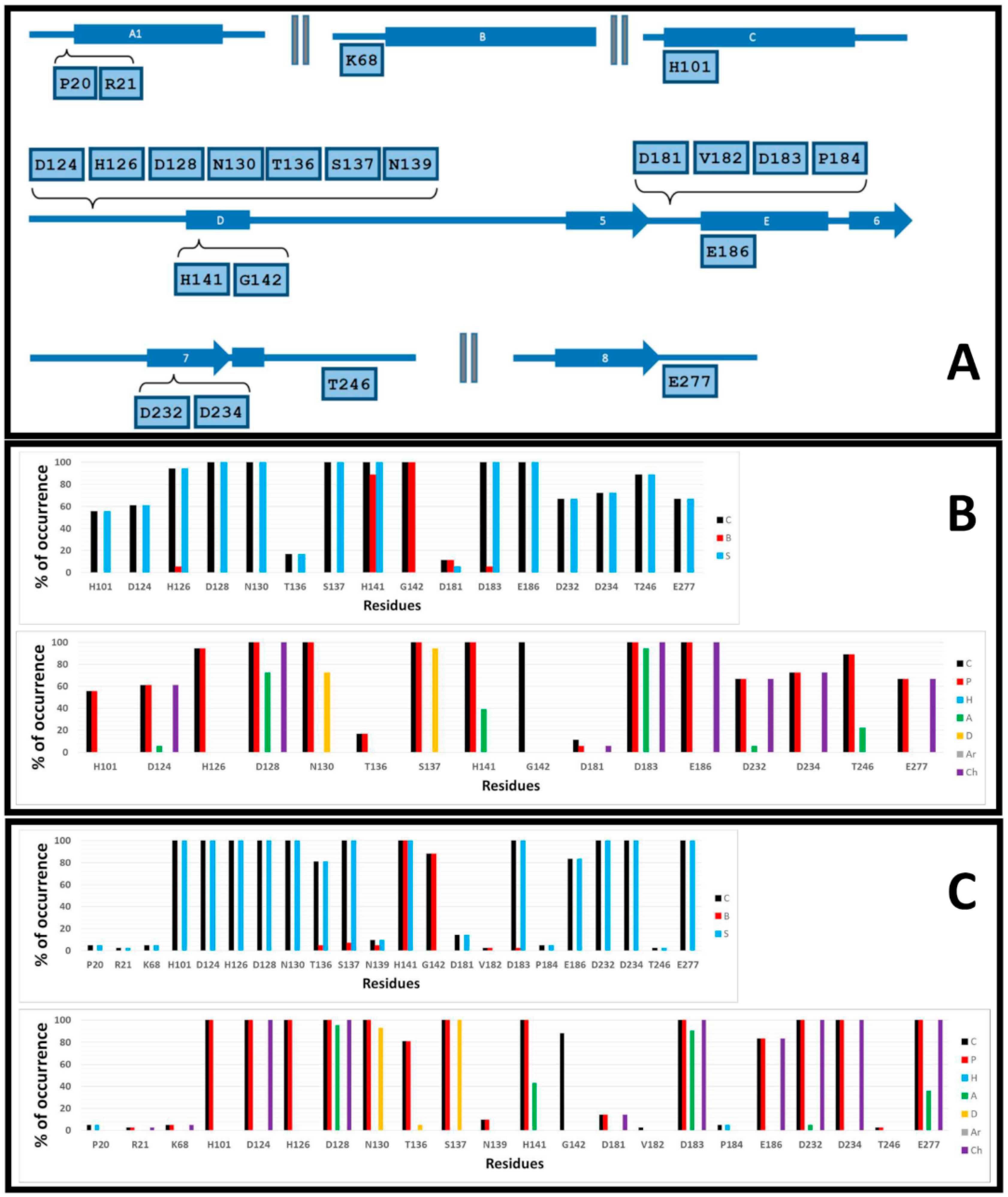
2.2. Prediction of the Binding Modes
3. Materials and Methods
3.1. Dataset Collection
3.2. QSAR Modeling
3.3. Molecular Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 3D | three-dimensional |
| ABH | 2(S)-amino-6-boronohexanoic acid |
| hARGI | human arginase I |
| IFP | interaction fingerprints |
| LOO | leave-one-out |
| PDB | Protein Data Bank |
| RMSD | root mean square deviation |
| SAR | structure–activity relationship |
References
- Morris, S.M. Recent advances in arginine metabolism: Roles and regulation of the arginases. Br. J. Pharmacol. 2009, 157, 922–930. [Google Scholar] [CrossRef] [PubMed]
- Pernow, J.; Jung, C. Arginase as a potential target in the treatment of cardiovascular disease: Reversal of arginine steal? Cardiovasc. Res. 2013, 98, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Xu, H.; Du, X.; Lit, L.; Walker, W.; Lu, A.; Ran, R.; Gregg, J.P.; Reilly, M.; Pancioli, A.; et al. Gene expression in blood changes rapidly in neutrophils and monocytes after ischemic stroke in humans: A microarray study. J. Cereb. Blood Flow Metab. 2006, 26, 1089–1102. [Google Scholar] [CrossRef] [PubMed]
- Pudlo, M.; Demougeot, C.; Girard-Thernier, C. Arginase Inhibitors: A Rational Approach Over One Century. Med. Res. Rev. 2017, 37, 475–513. [Google Scholar] [CrossRef] [PubMed]
- Shemyakin, A.; Kövamees, O.; Rafnsson, A.; Böhm, F.; Svenarud, P.; Settergren, M.; Jung, C.; Pernow, J. Arginase Inhibition Improves Endothelial Function in Patients With Coronary Artery Disease and Type 2 Diabetes MellitusClinical Perspective. Circulation 2012, 126, 2943–2950. [Google Scholar] [CrossRef] [PubMed]
- Golebiowski, A.; Paul Beckett, R.; Van Zandt, M.; Ji, M.K.; Whitehouse, D.; Ryder, T.R.; Jagdmann, E.; Andreoli, M.; Mazur, A.; Padmanilayam, M.; et al. 2-Substituted-2-amino-6-boronohexanoic acids as arginase inhibitors. Bioorg. Med. Chem. Lett. 2013, 23, 2027–2030. [Google Scholar] [CrossRef] [PubMed]
- Golebiowski, A.; Whitehouse, D.; Beckett, R.P.; Van Zandt, M.; Ji, M.K.; Ryder, T.R.; Jagdmann, E.; Andreoli, M.; Lee, Y.; Sheeler, R.; et al. Synthesis of quaternary α-amino acid-based arginase inhibitors via the Ugi reaction. Bioorg. Med. Chem. Lett. 2013, 23, 4837–4841. [Google Scholar] [CrossRef] [PubMed]
- Van Zandt, M.C.; Whitehouse, D.L.; Golebiowski, A.; Ji, M.K.; Zhang, M.; Beckett, R.P.; Jagdmann, G.E.; Ryder, T.R.; Sheeler, R.; Andreoli, M.; et al. Discovery of (R)-2-amino-6-borono-2-(2-(piperidin-1-yl)ethyl)hexanoic acid and congeners as highly potent inhibitors of human arginases I and II for treatment of myocardial reperfusion injury. J. Med. Chem. 2013, 56, 2568–2580. [Google Scholar] [CrossRef] [PubMed]
- Ramírez, D.; Caballero, J. Is It Reliable to Take the Molecular Docking Top Scoring Position as the Best Solution without Considering Available Structural Data? Molecules 2018, 23, 1038. [Google Scholar] [CrossRef] [PubMed]
- Gohlke, H.; Hendlich, M.; Klebe, G. Knowledge-based scoring function to predict protein-ligand interactions. J. Mol. Biol. 2000, 295, 337–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navarro-Retamal, C.; Caballero, J. Molecular Modeling of Tau Proline-Directed Protein Kinase (PDPK) Inhibitors. In Computational Modeling of Drugs Against Alzheimer’s Disease; Humana Press: Neuromethods, NY, USA, 2018; pp. 305–345. [Google Scholar]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A.; et al. PubChem Substance and Compound databases. Nucleic Acids Res. 2016, 44, D1202–D1213. [Google Scholar] [CrossRef] [PubMed]
- Tropsha, A. Best Practices for QSAR Model Development, Validation, and Exploitation. Mol. Inform. 2010, 29, 476–488. [Google Scholar] [CrossRef] [PubMed]
- Tosco, P.; Balle, T. Open3DQSAR: A new open-source software aimed at high-throughput chemometric analysis of molecular interaction fields. J. Mol. Model. 2011, 17, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A. Merck molecular force field. II. MMFF94 van der Waals and electrostatic parameters for intermolecular interactions. J. Comput. Chem. 1996, 17, 520–552. [Google Scholar] [CrossRef]
- Bandyopadhyaya, A.K.; Tiwari, R.; Tjarks, W. Comparative molecular field analysis and comparative molecular similarity indices analysis of boron-containing human thymidine kinase 1 substrates. Bioorg. Med. Chem. 2006, 14, 6924–6932. [Google Scholar] [CrossRef] [PubMed]
- Kastenholz, M.A.; Pastor, M.; Cruciani, G.; Haaksma, E.E.; Fox, T. GRID/CPCA: A new computational tool to design selective ligands. J. Med. Chem. 2000, 43, 3033–3044. [Google Scholar] [CrossRef] [PubMed]
- Boháč, M.; Loeprecht, B.; Damborský, J.; Schüürmann, G. Impact of Orthogonal Signal Correction (OSC) on the Predictive Ability of CoMFA Models for the Ciliate Toxicity of Nitrobenzenes. Quant. Struct.-Act. Relationsh. 2002, 21, 3–11. [Google Scholar] [CrossRef]
- Pastor, M.; Cruciani, G.; Clementi, S. Smart region definition: A new way to improve the predictive ability and interpretability of three-dimensional quantitative structure-activity relationships. J. Med. Chem. 1997, 40, 1455–1464. [Google Scholar] [CrossRef] [PubMed]
- Caballero, J.; Zilocchi, S.; Tiznado, W.; Collina, S.; Rossi, D. Binding studies and quantitative structure-activity relationship of 3-amino-1H-indazoles as inhibitors of GSK3β. Chem. Biol. Drug Des. 2011, 78, 631–641. [Google Scholar] [CrossRef] [PubMed]
- Wendt, B.; Cramer, R.D. Challenging the gold standard for 3D-QSAR: Template CoMFA versus X-ray alignment. J. Comput. Aided Mol. Des. 2014, 28, 803–824. [Google Scholar] [CrossRef] [PubMed]
- Caballero, J.; Fernández, M.; González-Nilo, F.D. Structural requirements of pyrido[2,3-d]pyrimidin-7-one as CDK4/D inhibitors: 2D autocorrelation, CoMFA and CoMSIA analyses. Bioorg. Med. Chem 2008, 16, 6103–6115. [Google Scholar] [CrossRef] [PubMed]
- Munoz, C.; Adasme, F.; Alzate-Morales, J.H.; Vergara-Jaque, A.; Kniess, T.; Caballero, J. Study of differences in the VEGFR2 inhibitory activities between semaxanib and SU5205 using 3D-QSAR, docking, and molecular dynamics simulations. J. Mol. Graph. Model. 2012, 32, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Caballero, J.; Fernández, M.; Saavedra, M.; González-Nilo, F.D. 2D Autocorrelation, CoMFA, and CoMSIA modeling of protein tyrosine kinases’ inhibition by substituted pyrido[2,3-d]pyrimidine derivatives. Bioorg. Med. Chem 2008, 16, 810–821. [Google Scholar] [CrossRef] [PubMed]
- Di Costanzo, L.; Sabio, G.; Mora, A.; Rodriguez, P.C.; Ochoa, A.C.; Centeno, F.; Christianson, D.W. Crystal structure of human arginase I at 1.29-A resolution and exploration of inhibition in the immune response. Proc. Natl. Acad. Sci. USA 2005, 102, 13058–13063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Glisic, S.; Sencanski, M.; Perovic, V.; Stevanovic, S.; García-Sosa, A.T. Arginase Flavonoid Anti-Leishmanial in Silico Inhibitors Flagged against Anti-Targets. Molecules 2016, 21, 589. [Google Scholar] [CrossRef] [PubMed]
- Banks, J.L.; Beard, H.S.; Cao, Y.; Cho, A.E.; Damm, W.; Farid, R.; Felts, A.K.; Halgren, T.A.; Mainz, D.T.; Maple, J.R.; et al. Integrated Modeling Program, Applied Chemical Theory (IMPACT). J. Comput. Chem. 2005, 26, 1752–1780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef] [PubMed]
- Quesada-Romero, L.; Mena-Ulecia, K.; Tiznado, W.; Caballero, J. Insights into the interactions between maleimide derivates and GSK3β combining molecular docking and QSAR. PLoS ONE 2014, 9, e102212. [Google Scholar] [CrossRef] [PubMed]
- Quesada-Romero, L.; Caballero, J. Docking and quantitative structure–activity relationship of oxadiazole derivates as inhibitors of GSK3beta. Mol. Divers. 2014, 18, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Mena-Ulecia, K.; Tiznado, W.; Caballero, J. Study of the Differential Activity of Thrombin Inhibitors Using Docking, QSAR, Molecular Dynamics, and MM-GBSA. PLoS ONE 2015, 10, e0142774. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Gutierrez, C.; Adasme-Carreño, F.; Fuentes, E.; Palomo, I.; Caballero, J. Computational study of the binding orientation and affinity of PPARγ agonists: Inclusion of ligand-induced fit by cross-docking. RSC Adv. 2016, 6, 64756–64768. [Google Scholar] [CrossRef]
- Deng, Z.; Chuaqui, C.; Singh, J. Structural interaction fingerprint (SIFt): A novel method for analyzing three-dimensional protein-ligand binding interactions. J. Med. Chem. 2004, 47, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.; Deng, Z.; Narale, G.; Chuaqui, C. Structural interaction fingerprints: A new approach to organizing, mining, analyzing, and designing protein-small molecule complexes. Chem. Biol. Drug. Des. 2006, 67, 5–12. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
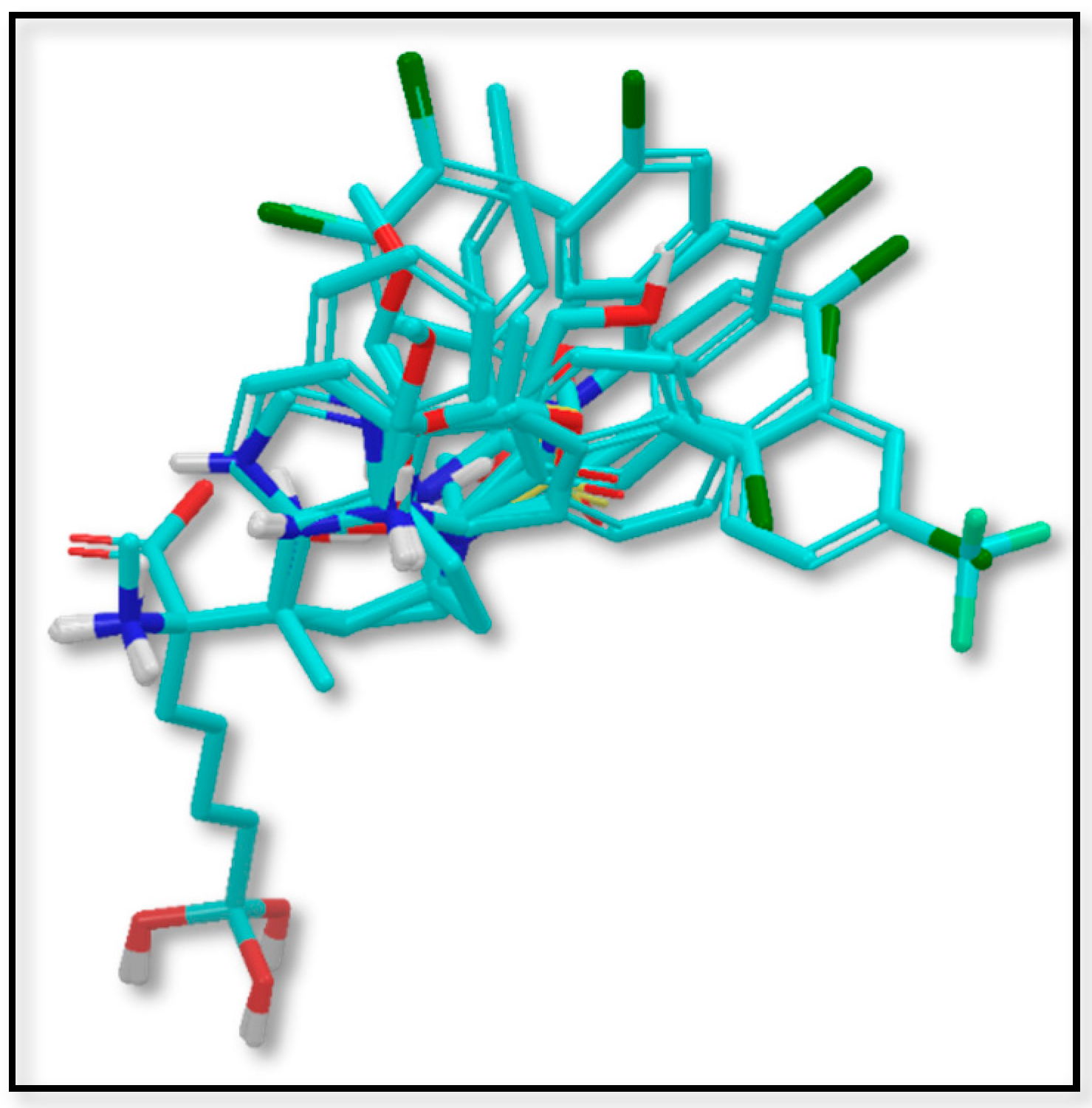{kind=link}
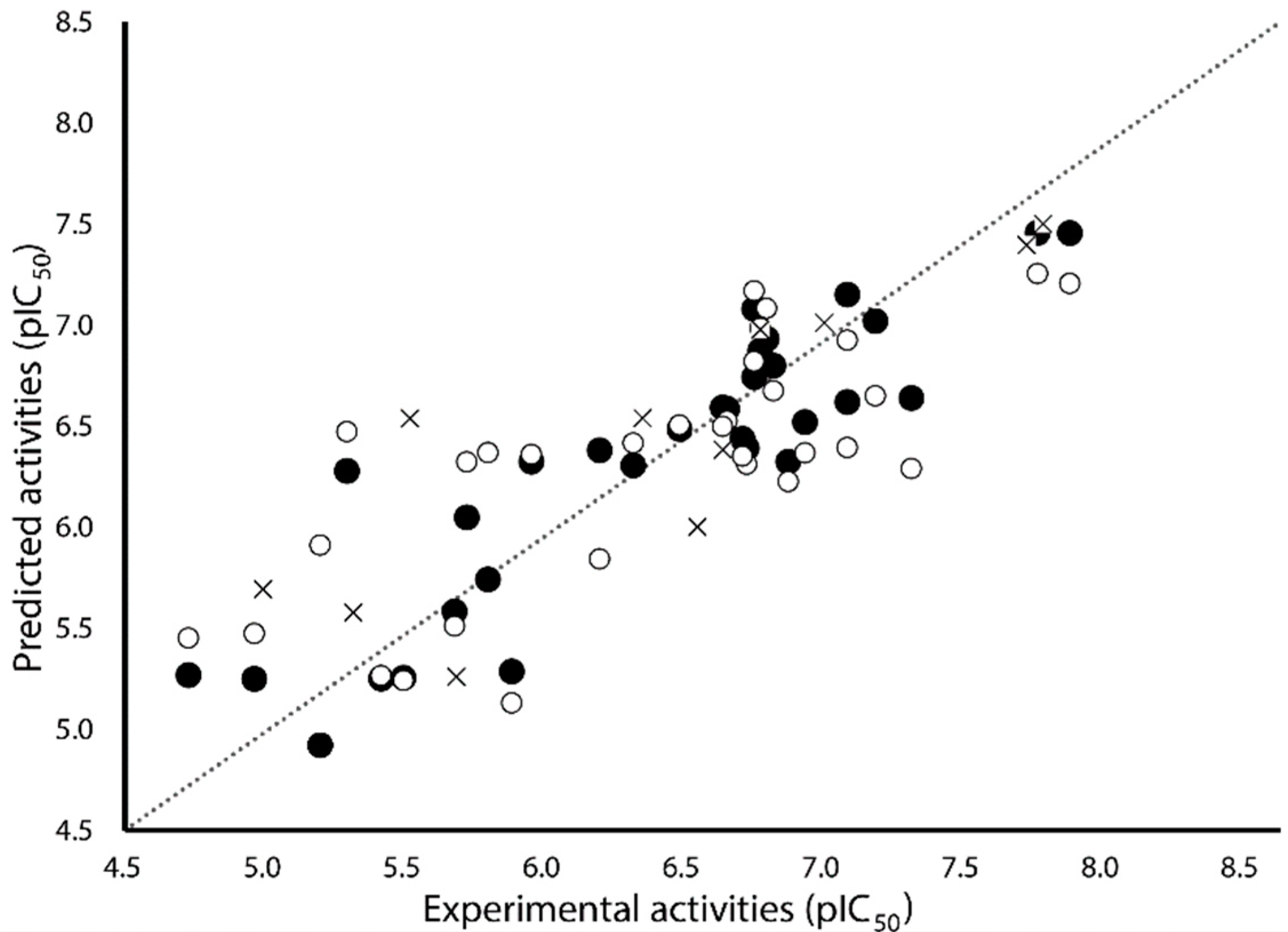{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||
|---|---|---|---|---|---|
| Compound | R1 | R2 | Experimental pIC50 (hARGI) | Predicted pIC50 (hARGI) | Scoring Energies (kcal/mol) |
| ABH |  | | 5.839 | 5.286 | −7.519 |
| p1_9 |  | | 6.652 | 6.390 | −5.375 |
| p1_14 |  |  | 7.222 | 6.639 | −7.654 |
| p1_16 |  | | 5.907 | 6.324 | −5.617 |
| p1_17 |  | | 6.638 | 6.436 | −4.563 |
| p1_181 |  | | 6.292 | 6.538 | −5.527 |
| p1_19 |  | | 6.796 | 6.323 | −6.365 |
| p1_20 |  | | 6.585 | 6.585 | −5.238 |
| p1_21 |  | | 6.678 | 6.742 | −4.378 |
| p1_22 |  | | 5.757 | 5.741 | −5.582 |
| p1_23 |  | | 6.569 | 6.591 | −5.649 |
| p1_24 |  | | 6.260 | 6.306 | −4.098 |
| p1_251 |  | | 6.569 | 6.382 | −5.631 |
| p1_26 |  | | 6.854 | 6.520 | −5.731 |
| p1_27 |  | | 7.000 | 6.619 | −5.697 |
| p2_1b |  | | 5.465 | 5.254 | −2.985 |
| p2_1c |  | | 5.387 | 5.251 | −3.064 |
| p2_1d |  | | 4.949 | 5.249 | −3.258 |
| p2_1e1 |  | | 4.979 | 5.693 | −3.605 |
| p2_1f1 |  | | 5.648 | 5.260 | −2.868 |
| p2_1g |  | | 4.721 | 5.268 | −2.682 |
| p2_1i1 |  | | 5.291 | 5.578 | −2.791 |
| p2_1j |  | | 5.642 | 5.583 | −3.166 |
| p2_1k |  | | 5.684 | 6.048 | −2.993 |
| p2_1l1 |  | | 5.487 | 6.538 | −5.414 |
| p2_1m |  | | 5.269 | 6.277 | −4.513 |
| p3_2a |  | | 6.420 | 6.485 | −8.167 |
| p3_2b |  | | 6.745 | 6.799 | −6.672 |
| p3_2c |  | | 6.699 | 6.873 | −7.551 |
| p3_2d |  | | 6.721 | 6.932 | −7.622 |
| p3_2e1 |  | | 6.699 | 6.977 | −6.540 |
| p3_2f |  | | 6.678 | 7.079 | −6.116 |
| p3_2g |  | | 7.000 | 7.150 | −6.206 |
| p3_2h1 |  | | 6.921 | 7.011 | −6.763 |
| p3_2i |  | | 5.177 | 4.921 | −4.324 |
| p3_2j |  | | 6.143 | 6.380 | −5.062 |
| p3_2k1 |  | | 6.481 | 6.001 | −4.897 |
| p3_11a |  | | 7.097 | 7.020 | −8.395 |
| p3_11b1 |  | | 7.620 | 7.396 | −8.075 |
| p3_11c |  | | 7.770 | 7.455 | −8.060 |
| p3_11d1 |  | | 7.678 | 7.500 | −7.980 |
| p3_11e |  | | 7.658 | 7.456 | −6.961 |
| Model | NC | R2 | S | Q2 | SCV | Fraction | |
|---|---|---|---|---|---|---|---|
| Steric | Electrostatic | ||||||
| S | 5 | 0.860 | 0.285 | 0.570 | 0.497 | 1 | 0 |
| E | 3 | 0.784 | 0.354 | 0.464 | 0.557 | 0 | 1 |
| SE | 3 | 0.802 | 0.339 | 0.572 | 0.497 | 0.460 | 0.540 |
| PDB Code of the Complex | Co-Crystallized Inhibitor | Co-Crystallized Enzyme | RMSD Value (Å) |
|---|---|---|---|
| 2AEB | ABH | hARGI | 1.07 |
| 4HWW | p1_9 | hARGI | 1.09 |
| 4HXQ | p1_14 | hARGI | 1.04 |
| 4IE3 | p1_17 | hARGII | 1.59 |
| 4IXV | p3_2d | hARGII | 1.55 |
| 4IXU | p3_11d | hARGII | 1.91 |
| ABH as Reference in 2AEB | p1_9 as Reference in 4HWW | p1_14 as Reference in 4HXQ | p1_17 as Reference in 4IE3 | p3_2d as Reference in 4IXV | p3_11d as Reference in 4IXU | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Compound | RMSD2AEB 1 (Å) | %RefMatch 2 | %MolMatch 3 | RMSD4HWW 1 (Å) | %RefMatch 2 | %MolMatch 3 | RMSD4HXQ 1 (Å) | %RefMatch 2 | %MolMatch 3 | RMSD4IE3 1 (Å) | %RefMatch 2 | %MolMatch 3 | RMSD4IXV 1 (Å) | %RefMatch 2 | %MolMatch 3 | RMSD4IXU 1 (Å) | %RefMatch 2 | %MolMatch 3 |
| ABH | 1.07 4 | 100 | 100 | 1.06 | 62 | 100 | 1.05 | 59 | 100 | 1.13 | 59 | 100 | 1.08 | 48 | 100 | 1.08 | 43 | 100 |
| p1_9 | 0.89 | 100 | 62 | 1.09 4 | 100 | 100 | 1.02 | 95 | 100 | 1.53 | 95 | 100 | 0.91 | 52 | 67 | 0.87 | 47 | 67 |
| p1_14 | 0.92 | 100 | 59 | 1.11 | 100 | 95 | 1.04 4 | 100 | 100 | 1.55 | 95 | 95 | 0.88 | 52 | 64 | 0.94 | 47 | 64 |
| p1_16 | 0.89 | 100 | 62 | 1.13 5 | 100 | 100 | 1.04 5 | 95 | 100 | 1.56 5 | 95 | 100 | 0.90 | 52 | 67 | 0.87 | 47 | 67 |
| p1_17 | 0.91 | 100 | 59 | 1.07 | 100 | 95 | 0.99 | 95 | 95 | 1.59 4 | 100 | 100 | 0.94 | 52 | 64 | 0.90 | 47 | 64 |
| p1_18 | 0.89 | 100 | 52 | 1.01 | 100 | 84 | 0.96 | 95 | 84 | 1.46 | 95 | 84 | 0.91 | 52 | 56 | 0.87 | 47 | 56 |
| p1_19 | 0.95 | 100 | 65 | 0.93 | 76 | 80 | 0.96 | 73 | 80 | 1.13 | 73 | 80 | 0.95 | 52 | 70 | 0.93 | 47 | 70 |
| p1_20 | 0.89 | 100 | 59 | 0.85 | 76 | 73 | 0.85 | 73 | 73 | 1.10 | 73 | 73 | 0.91 | 52 | 64 | 0.87 | 47 | 64 |
| p1_21 | 0.30 | 100 | 57 | 0.41 | 76 | 70 | 0.44 | 73 | 70 | 0.81 | 73 | 70 | 0.47 | 52 | 61 | 0.35 | 47 | 61 |
| p1_22 | 0.95 | 100 | 57 | 0.89 | 76 | 70 | 0.92 | 73 | 70 | 1.07 | 73 | 70 | 0.94 | 52 | 61 | 0.93 | 47 | 61 |
| p1_23 | 0.89 | 100 | 54 | 0.88 | 76 | 67 | 0.90 | 73 | 67 | 1.11 | 73 | 67 | 0.92 | 52 | 58 | 0.87 | 47 | 58 |
| p1_24 | 0.37 | 100 | 62 | 0.44 | 76 | 76 | 0.44 | 73 | 76 | 0.87 | 73 | 76 | 0.51 | 52 | 67 | 0.41 | 47 | 67 |
| p1_25 | 0.25 | 100 | 65 | 0.37 | 76 | 80 | 0.38 | 73 | 80 | 0.80 | 73 | 80 | 0.40 | 52 | 70 | 0.28 | 47 | 70 |
| p1_26 | 0.32 | 100 | 65 | 0.46 | 76 | 80 | 0.48 | 73 | 80 | 0.86 | 73 | 80 | 0.44 | 52 | 70 | 0.34 | 47 | 70 |
| p1_27 | 0.23 | 100 | 68 | 0.35 | 76 | 84 | 0.37 | 73 | 84 | 0.78 | 73 | 84 | 0.37 | 52 | 74 | 0.27 | 47 | 74 |
| p2_1b | 0.98 | 100 | 93 | 1.00 | 67 | 100 | 0.98 | 64 | 100 | 1.05 | 64 | 100 | 0.98 | 52 | 100 | 1.06 | 47 | 100 |
| p2_1c | 0.98 | 100 | 87 | 0.99 | 71 | 100 | 0.96 | 68 | 100 | 1.06 | 68 | 100 | 0.97 | 52 | 93 | 1.04 | 47 | 93 |
| p2_1d | 0.92 | 100 | 81 | 0.87 | 71 | 94 | 0.88 | 68 | 94 | 1.13 | 68 | 94 | 0.94 | 52 | 88 | 0.93 | 47 | 88 |
| p2_1e | 0.88 | 100 | 65 | 0.92 | 71 | 75 | 0.94 | 68 | 75 | 1.06 | 68 | 75 | 0.91 | 52 | 70 | 0.88 | 47 | 70 |
| p2_1f | 0.96 | 100 | 87 | 0.98 | 67 | 93 | 0.97 | 64 | 93 | 1.03 | 64 | 93 | 0.96 | 52 | 93 | 1.03 | 47 | 93 |
| p2_1g | 0.89 | 100 | 81 | 0.85 | 71 | 94 | 0.85 | 68 | 94 | 1.07 | 68 | 94 | 0.91 | 52 | 88 | 0.88 | 47 | 88 |
| p2_1i | 0.24 | 100 | 72 | 0.27 | 71 | 83 | 0.31 | 68 | 83 | 0.69 | 68 | 83 | 0.25 | 52 | 78 | 0.34 | 47 | 78 |
| p2_1j | 0.92 | 100 | 68 | 0.91 | 71 | 79 | 0.89 | 68 | 79 | 1.12 | 68 | 79 | 0.90 | 52 | 74 | 1.00 | 47 | 74 |
| p2_1k | 0.32 | 100 | 87 | 0.36 | 67 | 93 | 0.38 | 64 | 93 | 0.70 | 64 | 93 | 0.44 | 52 | 93 | 0.34 | 47 | 93 |
| p2_1l | 0.35 | 100 | 59 | 0.39 | 67 | 64 | 0.48 | 64 | 64 | 0.64 | 64 | 64 | 0.40 | 52 | 64 | 0.37 | 47 | 64 |
| p2_1m | 0.55 | 100 | 62 | 1.03 5 | 100 | 100 | 0.95 5 | 95 | 100 | 1.53 5 | 95 | 100 | 0.63 | 52 | 67 | 0.56 | 47 | 67 |
| p3_2a | 0.87 | 100 | 68 | 0.86 | 67 | 74 | 0.87 | 64 | 74 | 1.06 | 64 | 74 | 0.87 | 70 | 100 | 1.20 | 63 | 100 |
| p3_2b | 0.98 | 100 | 54 | 1.01 | 67 | 58 | 0.97 | 64 | 58 | 1.23 | 64 | 58 | 1.11 | 78 | 88 | 1.60 | 70 | 88 |
| p3_2c | 0.19 | 100 | 50 | 0.22 | 67 | 54 | 0.28 | 64 | 54 | 0.67 | 64 | 54 | 1.41 | 96 | 100 | 3.36 | 87 | 100 |
| p3_2d | 0.88 | 100 | 48 | 0.86 | 67 | 52 | 0.86 | 64 | 52 | 1.06 | 64 | 52 | 1.55 4 | 100 | 100 | 3.86 | 90 | 100 |
| p3_2e | 0.89 | 100 | 46 | 0.89 | 67 | 50 | 0.88 | 64 | 50 | 1.10 | 64 | 50 | 1.44 | 100 | 96 | 4.22 | 93 | 100 |
| p3_2f | 1.02 | 100 | 45 | 1.03 | 67 | 48 | 1.01 | 64 | 48 | 1.22 | 64 | 48 | 1.38 | 78 | 72 | 1.36 | 70 | 72 |
| p3_2g | 0.49 | 100 | 43 | 0.57 | 67 | 47 | 0.65 | 64 | 47 | 0.36 | 64 | 47 | 1.29 | 78 | 70 | 1.91 | 70 | 70 |
| p3_2h | 0.88 | 100 | 41 | 0.87 | 67 | 44 | 0.89 | 64 | 44 | 1.05 | 64 | 44 | 1.24 | 78 | 66 | 1.81 | 70 | 66 |
| p3_2i | 0.32 | 100 | 45 | 0.31 | 67 | 48 | 0.32 | 64 | 48 | 0.73 | 64 | 48 | 0.63 | 70 | 66 | 1.11 | 63 | 66 |
| p3_2j | 0.91 | 100 | 45 | 0.88 | 67 | 48 | 0.87 | 64 | 48 | 1.10 | 64 | 48 | 1.15 | 74 | 69 | 1.70 | 67 | 69 |
| p3_2k | 0.26 | 100 | 43 | 0.30 | 67 | 47 | 0.31 | 64 | 47 | 0.71 | 64 | 47 | 0.68 | 74 | 67 | 1.31 | 67 | 67 |
| p3_11a | 0.24 | 100 | 62 | 0.23 | 67 | 67 | 0.31 | 64 | 67 | 0.69 | 64 | 67 | 0.87 | 70 | 90 | 0.89 | 70 | 100 |
| p3_11b | 0.27 | 100 | 46 | 0.28 | 67 | 50 | 0.36 | 64 | 50 | 0.62 | 64 | 50 | 2.94 | 96 | 93 | 1.51 | 93 | 100 |
| p3_11c | 0.91 | 100 | 45 | 0.87 | 67 | 48 | 0.85 | 64 | 48 | 1.12 | 64 | 48 | 3.79 | 100 | 93 | 1.88 | 97 | 100 |
| p3_11d | 0.27 | 100 | 43 | 0.25 | 67 | 47 | 0.23 | 64 | 47 | 0.71 | 64 | 47 | 3.66 | 100 | 90 | 1.91 4 | 100 | 100 |
| p3_11e | 0.29 | 100 | 43 | 0.31 | 67 | 47 | 0.41 | 64 | 47 | 0.61 | 64 | 47 | 2.80 | 96 | 87 | 1.32 | 93 | 93 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Velázquez-Libera, J.L.; Navarro-Retamal, C.; Caballero, J. Insights into the Structural Requirements of 2(S)-Amino-6-Boronohexanoic Acid Derivatives as Arginase I Inhibitors: 3D-QSAR, Docking, and Interaction Fingerprint Studies. Int. J. Mol. Sci. 2018, 19, 2956. https://doi.org/10.3390/ijms19102956
Velázquez-Libera JL, Navarro-Retamal C, Caballero J. Insights into the Structural Requirements of 2(S)-Amino-6-Boronohexanoic Acid Derivatives as Arginase I Inhibitors: 3D-QSAR, Docking, and Interaction Fingerprint Studies. International Journal of Molecular Sciences. 2018; 19(10):2956. https://doi.org/10.3390/ijms19102956
Chicago/Turabian StyleVelázquez-Libera, José Luis, Carlos Navarro-Retamal, and Julio Caballero. 2018. "Insights into the Structural Requirements of 2(S)-Amino-6-Boronohexanoic Acid Derivatives as Arginase I Inhibitors: 3D-QSAR, Docking, and Interaction Fingerprint Studies" International Journal of Molecular Sciences 19, no. 10: 2956. https://doi.org/10.3390/ijms19102956