3.2. Synthesis of the New Compounds
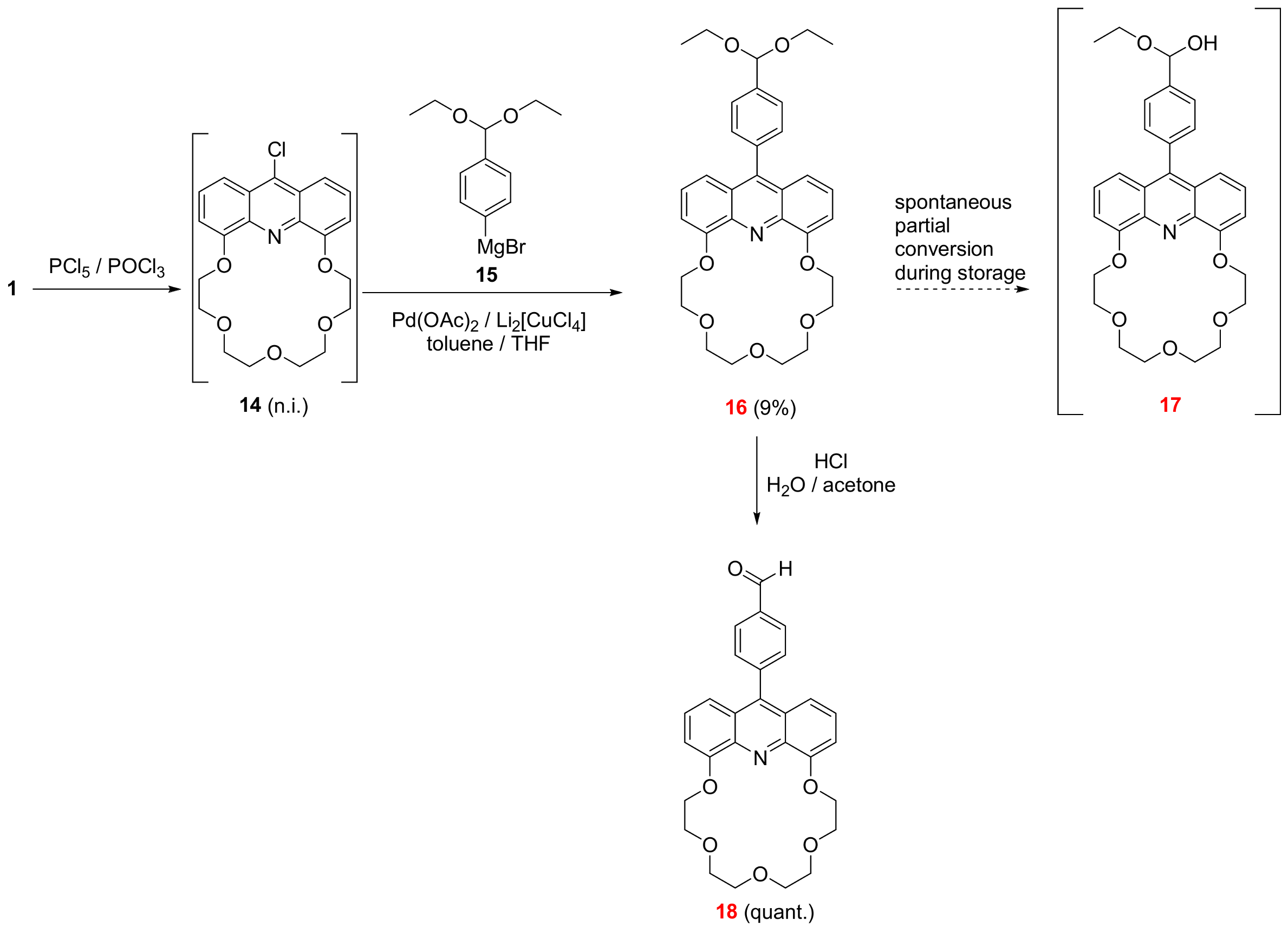
3.2.1. 27-[4-(Diethoxymethyl)phenyl]-6,9,12,15,18-pentaoxa-25-azatetracyclo[21.3.1.05,26.019,24]heptacosa-1,3,5(26),19,21,23(27),24-heptaene (See 16 in Scheme 1)
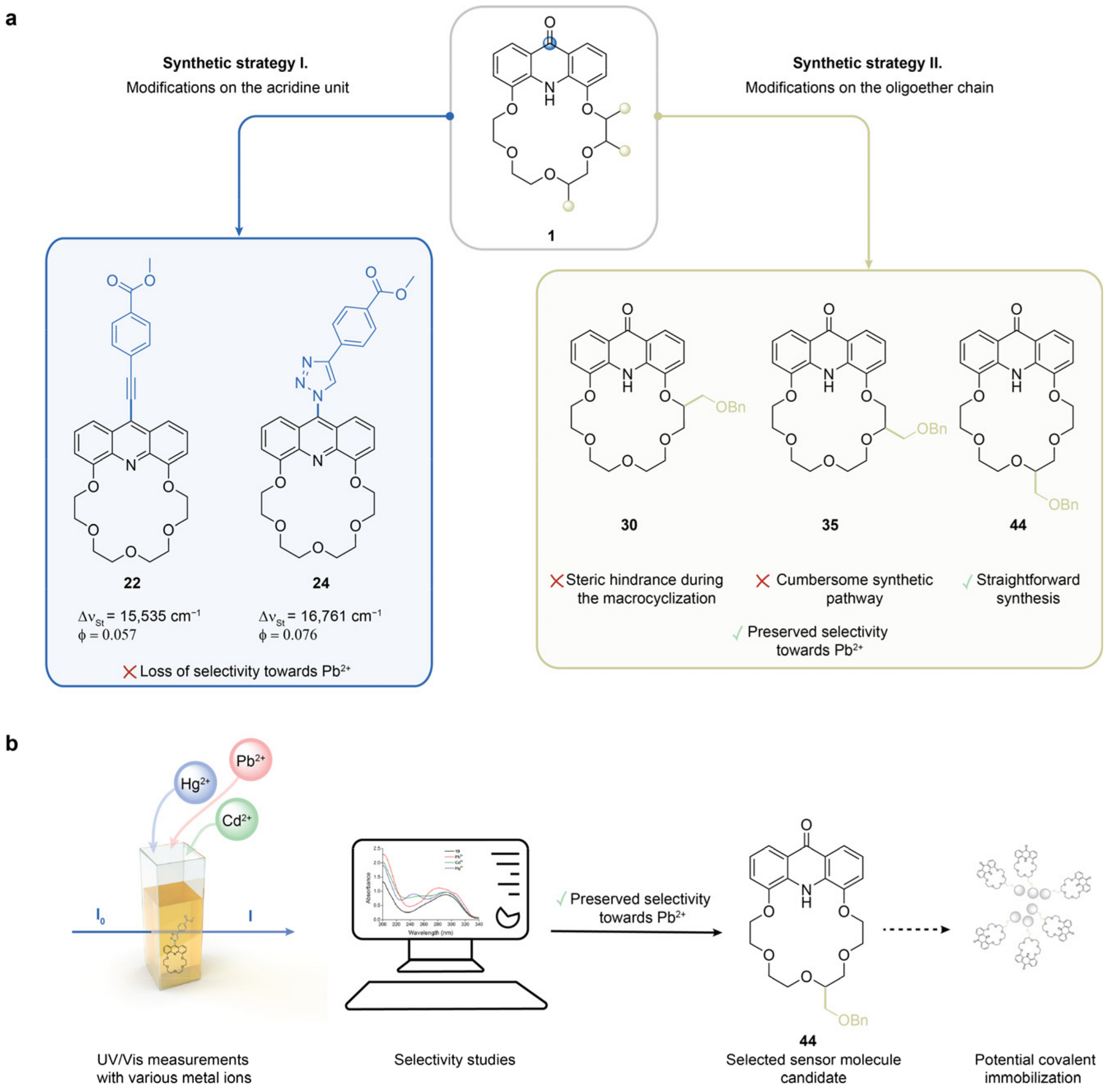
Acridono-18-crown-6 ether
1 (100 mg, 0.26 mmol) was converted into its chloro-acridino-derivative
14 according to a reported procedure [
25].
Grignard-reagent
15 was prepared from 1-bromo-4-(diethoxymethyl)benzene (672 mg, 2.60 mmol) [
26].
A solution of crude chloroacridine 14 (105 mg, 0.26 mmol) in a mixture of dry and pure toluene (15 mL) was added dropwise to a stirred solution of Grignard-reagent 15 (2.60 mmol in 10 mL THF), palladium acetate (4 mg, 0.018 mmol), dilithium tetrachlorocuprate (1.0 M in THF, 6 µL, 0.026 mmol) under an argon atmosphere at room temperature. The temperature of the resulting reaction mixture was raised to 60 °C and kept at this temperature for 20 h then cooled down to 20 °C. The solvent was removed, and the residue was taken up in ethyl acetate (25 mL) and ice-cold water (25 mL). The pH of the aqueous phase was adjusted to 7 using aqueous hydrochloric acid (5 m/m%). The phases were shaken well and separated. The aqueous phase was extracted with ethyl acetate (3 × 15 mL). The combined organic phase was dried over magnesium sulfate, filtered and the solvent was removed. The crude product was purified using PTLC on neutral aluminum-oxide adsorbent using an eluent mixture of ethanol:toluene 1:50 to provide the title compound 16 as a yellow solid (13 mg, 9%).
M.p. = 113 °C. Rf = 0.49 (Al2O3, ethanol:toluene 1:20). 1H-NMR (CDCl3): δ [ppm]: 7.83 (d, J = 8.2 Hz, 2H, ArCH), 7.78 (d, J = 8.2 Hz, 2H, ArCH), 7.66 (d, J = 8.2 Hz, 2H, ArCH), 7.62 (d, J = 8.2 Hz, 2H, ArCH), 7.38 (t, J = 8.2 Hz, 2H, acridine ArCH), 5.59 (s, 1H, protected formyl CH), 3.76–3.52 (m, 20H, ethereal OCH2), 1.29 (t, J = 7.1 Hz, 6H, acetal CH3). 13C-NMR (CDCl3): δ [ppm]: 158.47, 139.27, 135.99, 135.24, 130.36, 130.28, 128.03, 127.68, 127.36, 127.21, 126.63, 101.24, 64.90, 61.19, 61.03, 58.43, 15.24. IR: νmax [cm−1]: 3350 (broad, in the case of the hemiacetal 17), 3064, 1563, 1512, 1449, 1390, 1313, 1281, 1210, 1169, 1106, 1090, 1007, 975, 861, 813, 736, 659, 604, 551, 500, 408. HRMS: m/z = [MH+]: 548.2572, (Calculated for C32H37NO7, 547.2570).
3.2.2. 4-{6,9,12,15,18-Pentaoxa-25-azatetracyclo[21.3.1.05,26.019,24]heptacosa-1,3,5(26),19,21,23(27),24-heptaen-27-yl}benzaldehyde (See 18 in Scheme 1)
Aqueous hydrochloric acid solution (1 mL, 10 m/m%) was added dropwise to a stirred solution of the diacetal-protected crown ether 16 (13 mg, 0.024 mmol) in acetone (500 µL). The reaction mixture was stirred at 50 °C for 3 h under argon. The solvent was evaporated to provide the title compound 18 as a yellow crystal (12 mg, quantitative yield).
M.p. = 144 °C. Rf = 0.47 (Al2O3, ethanol:toluene 1:20). 1H-NMR (CDCl3): δ [ppm]: 10.07 (s, 1H, formyl H), 7.98 (dd, J = 8.7, 4.6 Hz, 2H, ArCH), 7.94 (dd, J = 8.9, 4.5 Hz, 2H, ArCH), 7.79 (dd, J = 8.9, 4.5 Hz, 2H, ArCH), 7.74 (dd, J = 8.2, 4.7 Hz, 2H, ArCH), 7.34 (t, J = 8.0 Hz, 2H, acridine ArCH), 3.71–3.49 (m, 16H, ethereal OCH2). 13C-NMR (CDCl3): δ [ppm]: 190.71, 157.28, 138.08, 134.80, 134.05, 129.17, 129.08, 126.84, 126.48, 126.16, 126.02, 125.44, 63.71, 60.00, 59.83. IR: νmax [cm−1]: 2964, 2924, 2841, 2742, 1934, 1691, 1602, 1562, 1514, 1449, 1390, 1313, 1282, 1211, 1169, 1106, 1090, 1007, 975, 861, 813, 736, 659, 604, 551, 500. HRMS: m/z = [MH+]: 474.1836, (Calculated for C28H27NO6, 473.1838).
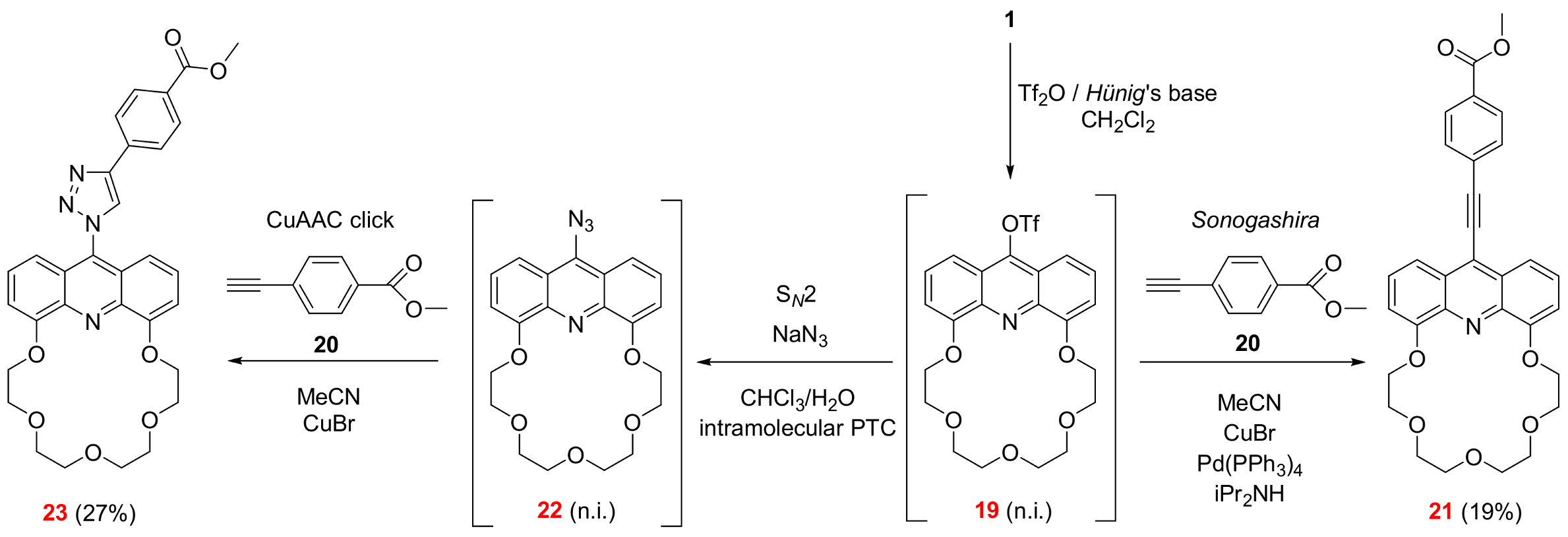
3.2.3. 6,9,12,15,18-Pentaoxa-25-azatetracyclo[21.3.1.05,26.019,24]heptacosa-1,3,5(26),19,21,23(27),24-heptaen-27-yl trifluoromethanesulfonate (See 19 in Scheme 2)
Acridono-crown ether (1, 50 mg, 0.13 mmol) was dissolved in dry dichloromethane (10 mL) in a flame-dried flask equipped with a septum and an argon inlet. The solution was cooled to 0 °C by using an external ice bath. Then, triflic anhydride (66 µL, 110 mg, 0.39 mmol) was added to the stirred solution using a syringe. Hünig’s base (136 µL, 101 mg, 0.78 mmol) was also added using a syringe and the reaction mixture was stirred for 30 min. The volatile components were removed at 20 °C. (If the reaction mixture was poured onto a large excess of 40 m/m% aqueous tetramethylammonium hydroxide at 0 °C and was extracted with dichloromethane, the majority of the product was hydrolyzed). Based on TLC analysis, a quantitative conversion was achieved and the crude product as an orange solid was further reacted without purification to gain functionalized crown ether 21.
3.2.4. Methyl 4-(2-{6,9,12,15,18-pentaoxa-25-azatetracyclo[21.3.1.05,26.019,24]heptacosa-1,3,5(26),19,21,23(27),24-heptaen-27-yl}ethynyl)benzoate (See 21 in Scheme 2)
Freshly prepared triflate (19) (60 mg, 0.12 mmol) was suspended in dry acetonitrile (2.0 mL). Copper(I) bromide (10 mg, 0.07 mmol), tetrakis(triphenylphosphine)palladium(0) (12 mg, 0.01 mmol) and diisopropylamine (842 µL, 6.00 mmol) were added to the suspension, which was stirred at room temperature for 15 min. Acetylene 20 (100 mg, 0.62 mmol) dissolved in acetonitrile (2.0 mL) was added to the stirred reaction mixture at 0 °C under argon. The temperature of the reaction mixture was raised to room temperature and stirred at this temperature for 48 h under argon. The volatile components were removed. The residue was dissolved in a mixture of ethyl acetate (10 mL) and water (10 mL). The phases were shaken well and separated. The aqueous phase was extracted with ethyl acetate (5 × 10 mL). The combined organic phase was dried over magnesium sulfate, filtered and the solvent was removed. The crude product was purified using column chromatography on aluminum oxide adsorbent using a gradient elution of toluene and ethanol (0–10% ethanol). The product needed further purification using PTLC on silica gel using dichloromethane as an eluent to provide 21 (12 mg, 19%) as a yellow solid.
M.p. = 98–99 °C. Rf = 0.30 (Al2O3, ethanol:toluene 1:20). Rf = 0.20 (SiO2, dichloromethane). Rf = 0.87 (SiO2, methanol:dichloromethane 1:10). 1H-NMR (CDCl3): δ [ppm]: 8.09–7.83 (m, 2H, ArCH), 7.63–7.54 (m, 2H, ArCH), 7.50–7.40 (m, 2H, ArCH), 7.35–7.26 (m, 2H, ArCH), 7.19–7.09 (m, 2H, ArCH), 4.47–4.29 (m, 3H, ethereal OCH2), 4.07–3.77 (m, 12H, ethereal OCH2 and ester OCH3), 3.61–3.34 (m, 4H, ethereal OCH2). 13C-NMR (CDCl3): δ [ppm]: 166.23, 146.54, 132.45, 131.44, 130.56, 129.56, 126.09, 120.72, 118.84, 113.13, 81.86, 76.27, 71.37, 69.44, 52.34. IR: νmax [cm−1]: 3420, 3057, 2962, 2927, 2873, 2850, 1719, 1624, 1598, 1577, 1533, 1437, 1262, 1222, 1153, 1100, 1029, 802, 748, 722, 694, 638, 573, 541, 517. HRMS: m/z = [MH+]: 528.1948, (Calculated for C31H29NO7, 527.1944).
3.2.5. 27-Azido-6,9,12,15,18-pentaoxa-25-azatetracyclo[21.3.1.05,26.019,24]heptacosa-1,3,5(26),19,21,23(27),24-heptaene (See 22 in Scheme 2)
Triflate
19 was prepared according to the procedure described in
Section 3.2.3 starting from acridono-crown ether
1 (50 mg, 0.13 mmol).
Freshly prepared triflate (19) (60 mg, 0.12 mmol) was suspended in a mixture of dichloromethane (2.0 mL), water (2.0 mL) and Hünig’s base (63 µL, 0.36 mmol). Sodium azide (78 mg, 1.2 mmol) was added to this suspension at room temperature, then the reaction mixture was stirred at 40 °C for 4 h. The mixture was cooled to room temperature, diluted with water (15 mL) and extracted with dichloromethane (3 × 5.0 mL). The combined organic phase was dried over magnesium sulfate, filtered and the solvent was removed. Based on TLC analysis a quantitative conversion was achieved and the crude product as an orange solid was further reacted without purification to gain functionalized crown ether 23.
3.2.6. Methyl 4-(1-{6,9,12,15,18-pentaoxa-25-azatetracyclo[21.3.1.05,26.019,24]heptacosa-1,3,5(26),19,21,23(27),24-heptaen-27-yl}-1H-1,2,3-triazol-4-yl)benzoate (See 23 in Scheme 2)
Freshly prepared crude azide (22) (50 mg, 0.12 mmol) was suspended in dry acetonitrile (3.0 mL). Copper(I) bromide (10 mg, 0.07 mmol) and acetylene 20 (100 mg, 0.62 mmol) were added to the suspension, which was stirred at room temperature for 15 min under argon. The temperature of the reaction mixture was raised to 50 °C and it was stirred at this temperature for 24 h under argon. The solvent was removed. The residue was dissolved in a mixture of ethyl acetate (10 mL) and water (10 mL). The phases were shaken well and separated. The aqueous phase was extracted with ethyl acetate (5 × 10 mL). The combined organic phase was dried over magnesium sulfate, filtered and the solvent was removed. The crude product was purified using column chromatography on aluminum oxide adsorbent using a gradient elution of toluene and ethanol mixture (0–10% ethanol). The product was further purified using silica gel PTLC using dichloromethane as an eluent to provide 23 (18 mg, 27%) as a yellow solid.
M.p. = 117–118 °C. Rf = 0.27 (Al2O3, ethanol:toluene 1:20). Rf = 0.16 (SiO2, dichloromethane). Rf = 0.78 (SiO2, methanol:dichloromethane 1:10). 1H-NMR (CDCl3): δ [ppm]: 8.37 (s, 1H, triazole ArCH), 8.19 (d, J = 8.0 Hz, 2H, ArCH), 8.11 (d, J = 8.1 Hz, 2H, Ar CH), 7.58–7.41 (m, 2H, ArCH), 7.12 (d, J = 7.1 Hz, 2H, ArCH), 7.05 (d, J = 8.8 Hz, 2H, ArCH), 4.74–4.68 (m, 4H, ethereal OCH2), 4.62–4.56 (m, 4H, ethereal OCH2), 3.96 (s, 3H, ester OCH3), 3.94–3.89 (m, 4H, ethereal OCH2), 3.83–3.76 (m, 4H, ethereal OCH2). 13C-NMR (CDCl3): δ [ppm]: 166.71, 141.10, 140.59, 132.95, 130.39, 129.76, 128.53, 127.05, 65.23, 58.41, 52.35. IR: νmax [cm−1]: 3359 (broad, complexed water), 3112, 3040, 3018, 2962, 2929, 2872, 1718, 1628, 1604, 1483, 1449, 1261, 1156, 1098, 1078, 1016, 864, 800, 750, 745, 681, 638, 551, 459, 442, 405. HRMS: m/z = [MH+]: 571.2110, (Calculated for C31H30N4O7, 570.2114).
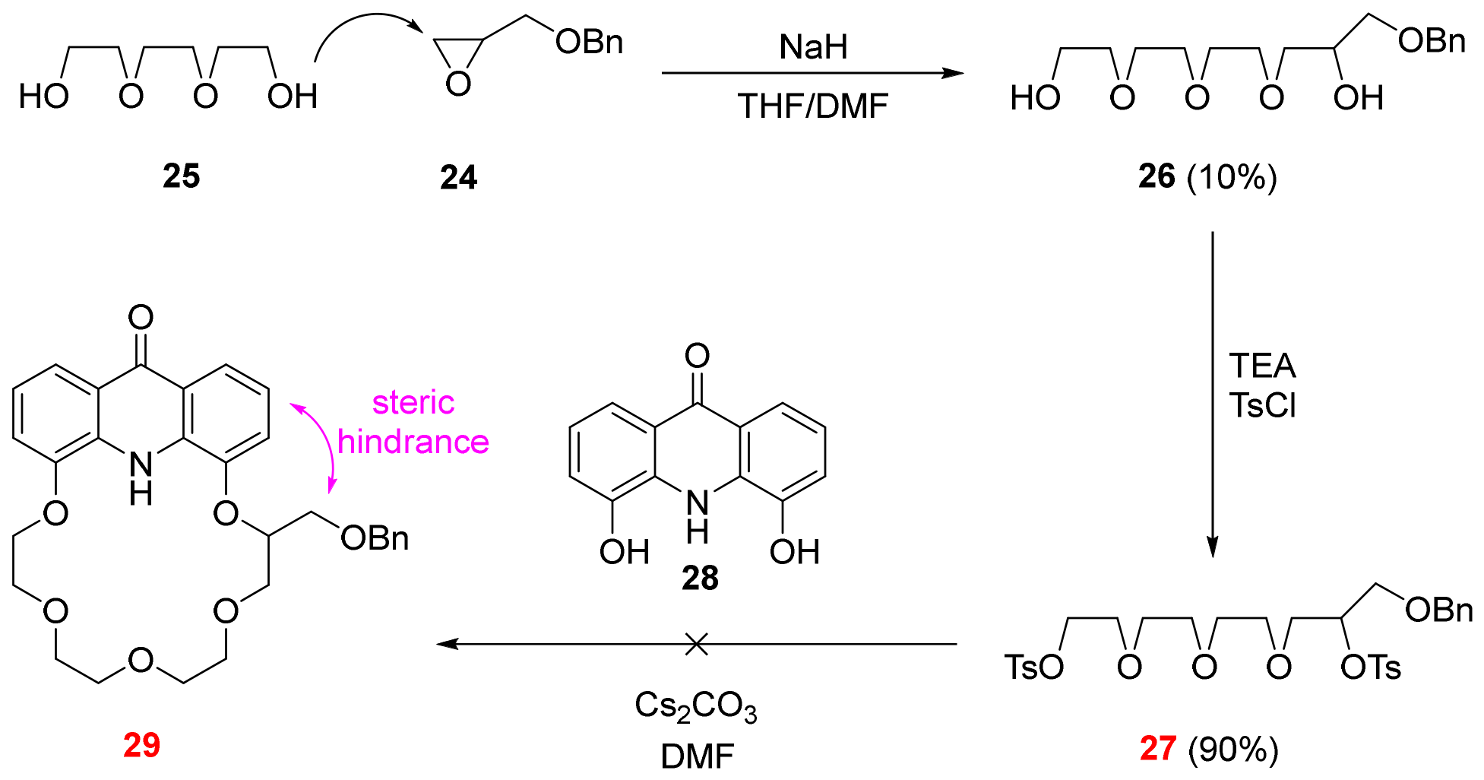
3.2.7. 4,14-Bis[(4-methylbenzenesulfonyl)oxy]-1-phenyl-2,6,9,12-tetraoxatetradecane (See 27 in Scheme 3)
Diol 26 (1.00 g, 3.18 mmol) was dissolved in triethylamine (10 mL) and a solution of tosyl chloride (1.52 g, 7.95 mmol) in triethylamine (10 mL) was added dropwise to its stirred solution. The reaction mixture was stirred at room temperature for 2 days. The volatile components were removed and the residue was dissolved in a mixture of ethyl acetate (100 mL) and water (200 mL). The phases were shaken well and separated. The pH of the aqueous phase was adjusted to 7 with aqueous hydrochloric acid (5 m/m%). The aqueous phase was extracted with ethyl acetate (3 × 100 mL). The combined organic phase was extracted with saturated aqueous sodium chloride solution (100 mL), dried over magnesium sulfate, filtered and the solvent was removed. The crude product was purified using column chromatography on silica gel using ethyl acetate:hexane 1:5 mixture as eluent to provide 27 (1.78 g, 90%) as a colorless oil.
Rf = 0.42 (SiO2, ethyl acetate:hexane 1:4). 1H-NMR (CDCl3): δ [ppm]: 7.71 (d, J = 8.1 Hz, 4H, Ts ArCH), 7.32–7.08 (m, 9H, Ts and Bn ArCH), 4.65 (q, J = 5.1 Hz, 1H, ethereal OCH), 4.34 (d, J = 3.7 Hz, 2H, ethereal OCH2), 4.17–4.09 (m, 4H, ethereal OCH2), 3.63–3.58 (m, 6H, ethereal OCH2), 3.56–3.52 (m, 3H, ethereal OCH2), 3.49–3.45 (m, 3H, ethereal OCH2), 2.32 (s, 6H, Ts p-CH3). 13C-NMR (CDCl3): δ [ppm]: 176.33, 171.13, 144.54, 137.69, 134.00, 129.60, 129.57, 128.35, 128.00, 127.71, 127.61, 125.31, 79.65, 73.32, 70.97, 70.60, 70.57, 70.44, 69.78, 69.14, 68.67, 63.63, 21.65, 20.97. IR: νmax [cm−1]: 2871, 1735, 1598, 1453, 1362, 1236, 1175, 1097, 1048, 921, 815, 776, 665, 553. HRMS: m/z = [MH+]: 623.1980, (Calculated for C30H38O10S2, 622.1906).
3.2.8. General Procedure for Macrocyclizations
A mixture of dihydroxy derivative (1.00 eq.), tetraethylene glycol ditosylate (1.00 eq.) and finely powdered anhydrous caesium carbonate (8.00 eq.) in dry DMF (200 mL/g diol) was stirred vigorously under argon atmosphere at room temperature for 7 days. Water (200 mL/g diol) was added to the reaction mixture, and it was extracted with ethyl acetate (3 × 200 mL/g diol). The combined organic phase was extracted with aqueous lithium bromide solution (6 × 200 mL/g diol, 5 m/m%) and then with saturated aqueous sodium chloride solution (2 × 200 mL/g diol). The organic phase was dried over magnesium sulfate, filtered and the solvent was evaporated.
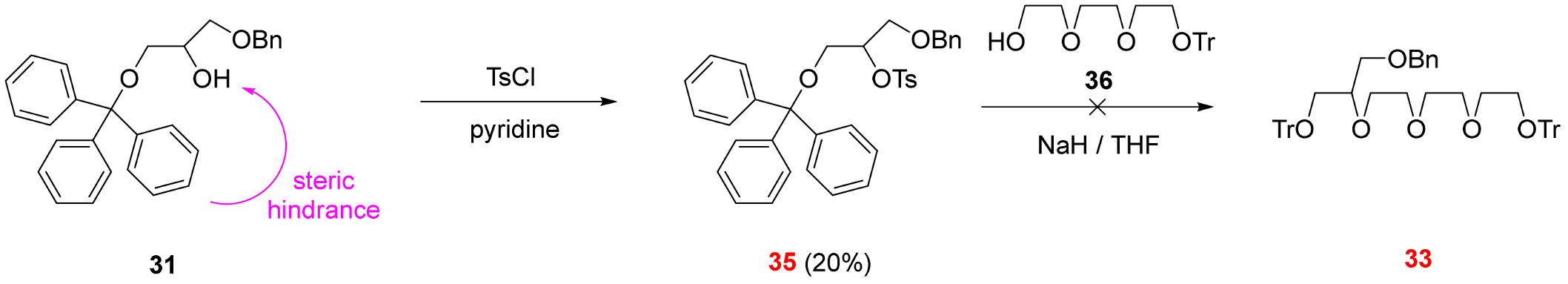
3.2.9. 1-(Benzyloxy)-3-(triphenylmethoxy)propan-2-yl 4-methylbenzene-1-sulfonate (See 35 in Scheme 5)
Alcohol 31 (500 mg, 1.18 mmol) was dissolved in dry pyridine (10 mL) and a solution of tosyl chloride (677 mg, 3.54 mmol) in dry pyridine (10 mL) was added dropwise to its stirred solution. The reaction mixture was stirred at 40 °C for 3 days. The solvent was removed and the residue was dissolved in a mixture of ethyl acetate (100 mL) and water (200 mL). The phases were shaken well and separated. The pH of the cold aqueous phase was adjusted to 7 with cold aqueous hydrochloric acid (5 m/m%). The aqueous phase was extracted with ethyl acetate (3 × 100 mL). The combined organic phase was extracted with saturated aqueous sodium chloride solution (100 mL), dried over magnesium sulfate, filtered and the solvent was removed. The crude product was purified using PTLC on silica gel using ethyl acetate:hexane 1:4 mixture as eluent to provide 35 (136 mg, 20%) as a colorless oil.
Rf = 0.29 (SiO2, ethyl acetate:hexane 1:4). 1H-NMR (CDCl3): δ [ppm]: 7.71 (dd, J = 18.7, 7.6 Hz, 4H, Ts ArCH), 7.29–7.24 (m, 6H, Tr ArCH), 7.23–7.11 (m, 12H, Tr and Bn ArCH), 7.11–7.01 (m, 2H, ArCH), 4.70–4.55 (m, 1H, ethereal OCH), 4.38–4.29 (m, 1H, ethereal OCH2), 4.10–3.99 (m, 3H, ethereal OCH2), 3.62–3.54 (m, 1H, ethereal OCH2), 3.28–3.20 (m, 1H, ethereal OCH2), 2.38 (s, 3H, Ts p-CH3). 13C-NMR (CDCl3): δ [ppm]: 146.88, 143.48, 137.68, 133.36, 129.82, 129.63, 128.63, 128.29, 127.93, 127.87, 127.82, 127.08, 86.86, 80.04, 73.25, 68.85, 66.79, 21.64. IR: νmax [cm−1]: 1598, 1494, 1448, 1355, 1189, 1176, 1096, 1003, 919, 816, 764, 702, 663, 555. HRMS: m/z = [MH+]: 579.2130, (Calculated for C36H34O5S, 578.2127).
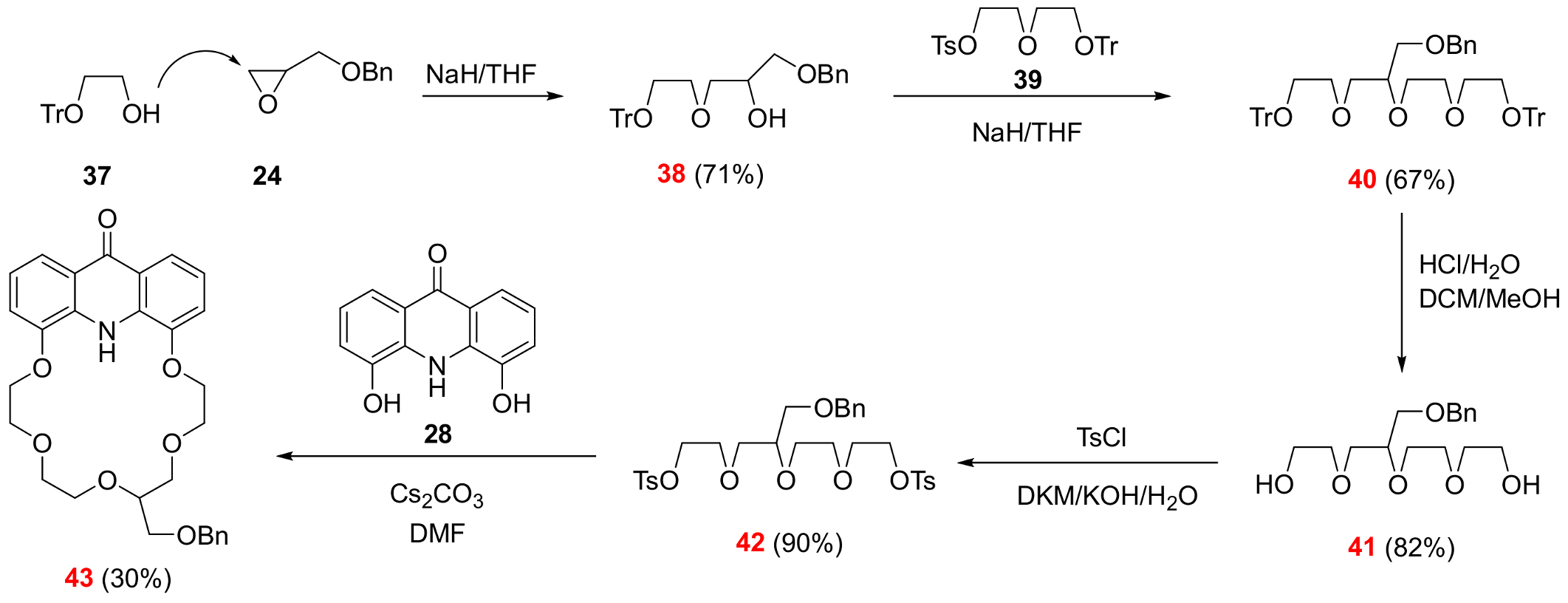
3.2.10. ({2-[3-(Benzyloxy)-2-hydroxypropoxy]ethoxy}diphenylmethyl)benzene (See 38 in Scheme 6)
Sodium hydride (1.58 g, 39.5 mmol, 60 m/m% dispersion in mineral oil) was suspended in dry tetrahydrofuran (30 mL) under argon at room temperature. Solution of alcohol
37 (10 g, 32.9 mmol, [
31]) in tetrahydrofuran (50 mL) was added dropwise to the stirred suspension of sodium hydride. The resulting mixture was refluxed for 30 min. The temperature of the reaction mixture was set to 0 °C with an external ice-water bath, then a solution of benzyl-glycidyl ether
24 (5.4 g, 32.9 mmol) in tetrahydrofuran (50 mL) was added dropwise and it was stirred for 1 h at this temperature. The temperature of the reaction mixture was raised to boiling point and it was refluxed for 24 h. The solvent was evaporated, and the residue was dissolved in a mixture of cold water (100 mL) and diethyl ether (100 mL). The aqueous phase was extracted with diethyl ether (2 × 100 mL). The combined organic phase was dried over magnesium sulfate, filtered and evaporated. The crude product was purified using column chromatography on silica gel using a mixture of ethyl acetate:hexane 1:4 to provide the title compound
38 (10.9 g, 71%) as a colorless oil.
Rf = 0.20 (SiO2, ethyl acetate:hexane 1:4). 1H-NMR (CDCl3): δ [ppm]: 7.47 (d, J = 7.5 Hz, 6H, Tr ArCH), 7.39–7.18 (m, 14H, Tr and Bn ArCH), 4.57 (s, 2H, Bn OCH2C), 4.03 (t, J = 5.4 Hz, 1H, ethereal OCH2), 3.81–3.45 (m, 6H, ethereal OCH2 and OCH), 3.25 (t, J = 4.9 Hz, 2H, ethereal OCH2), 2.61 (s, 1H, OH). 13C-NMR (CDCl3): δ [ppm]: 147.00, 144.11, 128.76, 128.51, 128.02, 127.95, 127.86, 127.26, 73.52, 72.79, 72.50, 71.33, 69.68, 61.68. IR: νmax [cm−1]: 3447 (broad), 3085, 3058, 3030, 2918, 2868, 1596, 1490, 1448, 1217, 1076, 1002, 950, 900, 761, 745, 695, 649, 632, 465. HRMS: m/z = [MH+]: 469.2353, (Calculated for C31H32O4, 468.2301).
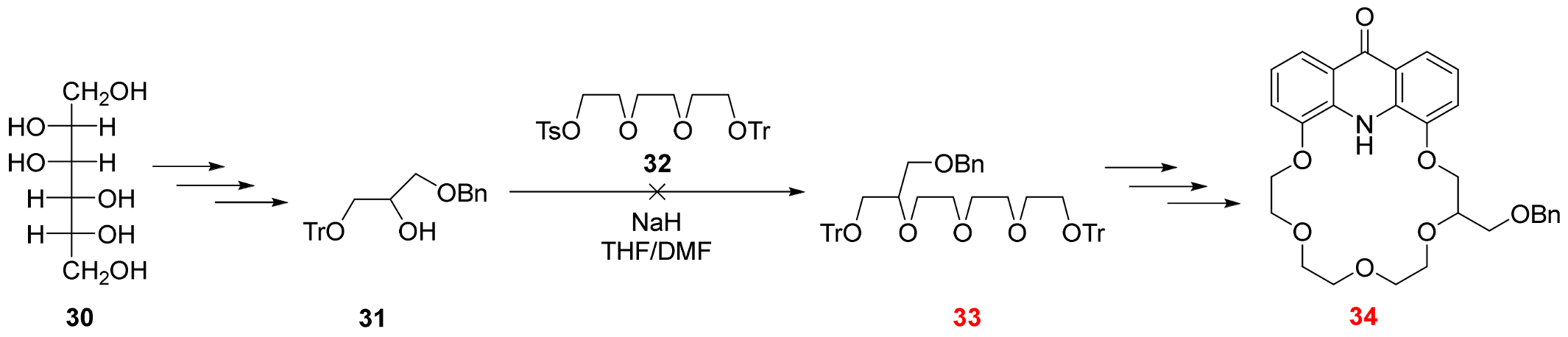
3.2.11. 7-[(Benzyloxy)methyl]-1,1,1,15,15,15-hexaphenyl-2,5,8,11,14-pentaoxapentadecane (See 40 in Scheme 6)
Sodium hydride (390 mg, 16.3 mmol, 60 m/m% dispersion in mineral oil) was suspended in dry tetrahydrofuran (7 mL) under argon at room temperature. Solution of alcohol
38 (5.45 g, 10.8 mmol) in tetrahydrofuran (30 mL) was added dropwise to the stirred suspension of sodium hydride. The resulting mixture was refluxed for 30 min. The temperature of the reaction mixture was set to 0 °C with an external ice-water bath, then a solution of tosylate
39 (5.08 g, 10.8 mmol, [
32]) in tetrahydrofuran (30 mL) was added dropwise and it was stirred for 1 h at this temperature. The temperature of the reaction mixture was raised to 40 °C and it was stirred for 5 days. The solvent was evaporated, and the residue was mixed with ice-cold water (100 mL), then this aqueous mixture was extracted with ethyl acetate (3 × 100 mL). The combined organic phase was dried over magnesium sulfate, filtered and evaporated. The crude product was purified using column chromatography on silica gel adsorbent using a gradient elution of ethyl acetate and hexane (0–20% ethyl acetate) to provide the title compound
40 (5.78 g, 67%) as a colorless oil.
Rf = 0.40 (SiO2, ethyl acetate:hexane 1:4). 1H-NMR (CDCl3): δ [ppm]: 7.43–7.35 (m, 12H, Tr ArCH), 7.23–7.15 (m, 23H, Tr and Bn ArCH), 4.51–4.42 (m, 2H, ethereal OCH2), 3.73–3.50 (m, 13H, ethereal OCH2 and OCH), 3.21–3.11 (m, 4H, ethereal OCH2). 13C-NMR (CDCl3): δ [ppm]: 146.95, 144.18, 138.03, 129.73, 128.78, 128.48, 127.99, 127.96, 127.80, 127.71, 127.28, 126.98, 86.58, 82.05, 78.48, 73.52, 73.05, 72.93, 69.76, 69.71, 63.40, 61.60, 61.50. IR: νmax [cm−1]: 3087, 3060, 3031, 2916, 2870, 2246, 1597, 1490, 1446, 1328, 1078, 1031, 1002, 906, 727, 696, 648, 633, 583, 509, 448. HRMS: m/z = [MH+]: 799.3921, (Calculated for C54H54O6, 798.3920).
3.2.12. 2-[3-(Benzyloxy)-2-[2-(2-hydroxyethoxy)ethoxy]propoxy]ethan-1-ol (See 41 in Scheme 6)
Aqueous hydrochloric acid solution (20 mL, 25 m/m%) was added slowly to a stirred mixture of protected tetraethylene glycol 40 (2.50 g, 3.19 mmol) in dichloromethane (20 mL) and methanol (20 mL). The reaction mixture was stirred at room temperature for 1 day under argon. The volatile components were evaporated, and the residue was taken up in a mixture of ice-cold water (100 mL) and diethyl ether (100 mL). The phases were shaken well and separated. The aqueous phase was extracted with diethyl ether (5 × 100 mL). The combined organic phase was dried over magnesium sulfate, filtered and evaporated to provide title compound 41 (820 mg, 82%) as a colorless oil.
Rf,40 = 0.90 (SiO2, ethyl acetate). Rf,41 = 0.33 (SiO2, ethyl acetate, phosphomolybdic acid stain). 1H-NMR (CDCl3): δ [ppm]: 7.46–7.30 (m, 1H, Bn ArCH), 7.29–7.16 (m, 4H, Bn ArCH), 4.50 (s, 2H, Bn OCH2C), 4.18–4.14 (m, 2H, ethereal OCH2), 3.67–3.53 (m, 15H, ethereal OCH2 and OCH), 2.76 (broad s, 2H, OH). 13C-NMR (CDCl3): δ [ppm]: 138.12, 128.35, 127.64, 127.62, 78.44, 73.39, 72.69, 71.34, 71.12, 70.76, 70.65, 69.77, 69.31, 69.07, 63.58, 61.61. IR: νmax [cm−1]: 3468 (broad), 2868 (broad), 1735, 1453, 1370, 1233, 1101, 1052, 958, 885, 739, 699, 606. HRMS: m/z = [MH+]: 315.1728, (Calculated for C16H26O6, 314.1729).
3.2.13. 1-({2-[3-(Benzyloxy)-2-(2-{2-[(4-methylbenzenesulfonyl)oxy]ethoxy}ethoxy)propoxy]ethoxy}sulfonyl)-4-methylbenzene (See 42 in Scheme 6)
Diol 41 (480 mg, 1.53 mmol) was dissolved in dichloromethane (10 mL) under argon. The temperature of this solution was set to 0 °C with an external ice-water bath, then a cold aqueous solution of potassium hydroxide (20 mL, 40 m/m%) was added to it. The resulting mixture was stirred at 0 °C for 10 min. A solution of tosyl chloride (730 mg, 3.82 mmol) in dichloromethane (10 mL) was added dropwise, then the mixture was stirred at room temperature for 5 days. Water (50 mL) and dichloromethane (50 mL) were added to the reaction mixture, the pH of the aqueous phase was adjusted to 8 with aqueous hydrochloric acid (5 m/m%), then the phases were shaken well and separated. The aqueous phase was further extracted with dichloromethane (3 × 50 mL). The combined organic phase was dried over magnesium sulfate, filtered and evaporated. The crude product was purified using column chromatography on silica gel adsorbent using a gradient elution of ethyl acetate and hexane (0–20% ethyl acetate) to provide the title compound 42 (857 mg, 90%) as a colorless oil.
Rf = 0.41 (SiO2, ethyl acetate:hexane 1:1). 1H-NMR (CDCl3): δ [ppm]: 7.82–7.79 (m, 4H, Ts ArCH), 7.38–7.31 (m, 9H, ArCH), 4.53 (s, 2H, Bn OCH2C), 4.16–4.11 (m, 4H, ethereal OCH2), 3.71–3.66 (m, 6H, ethereal OCH2), 3.63–3.54 (m, 5H, ethereal OCH2 and OCH), 3.53–3.50 (m, 2H, ethereal OCH2), 2.45 (s, 6H, tosyl p-CH3). 13C-NMR (CDCl3): δ [ppm]: 144.78, 138.22, 133.06, 128.37, 127.96, 127.63, 78.42, 73.39, 71.46, 70.93, 69.86, 69.84, 69.29, 69.20, 68.88, 68.65, 21.62. IR: νmax [cm−1]: 2871, 1735, 1598, 1453, 1362, 1236, 1175, 1097, 1048, 921, 815, 776, 665, 606, 553. HRMS: m/z = [MH+]: 623.1908, (Calculated for C30H38O10S2, 622.1906).
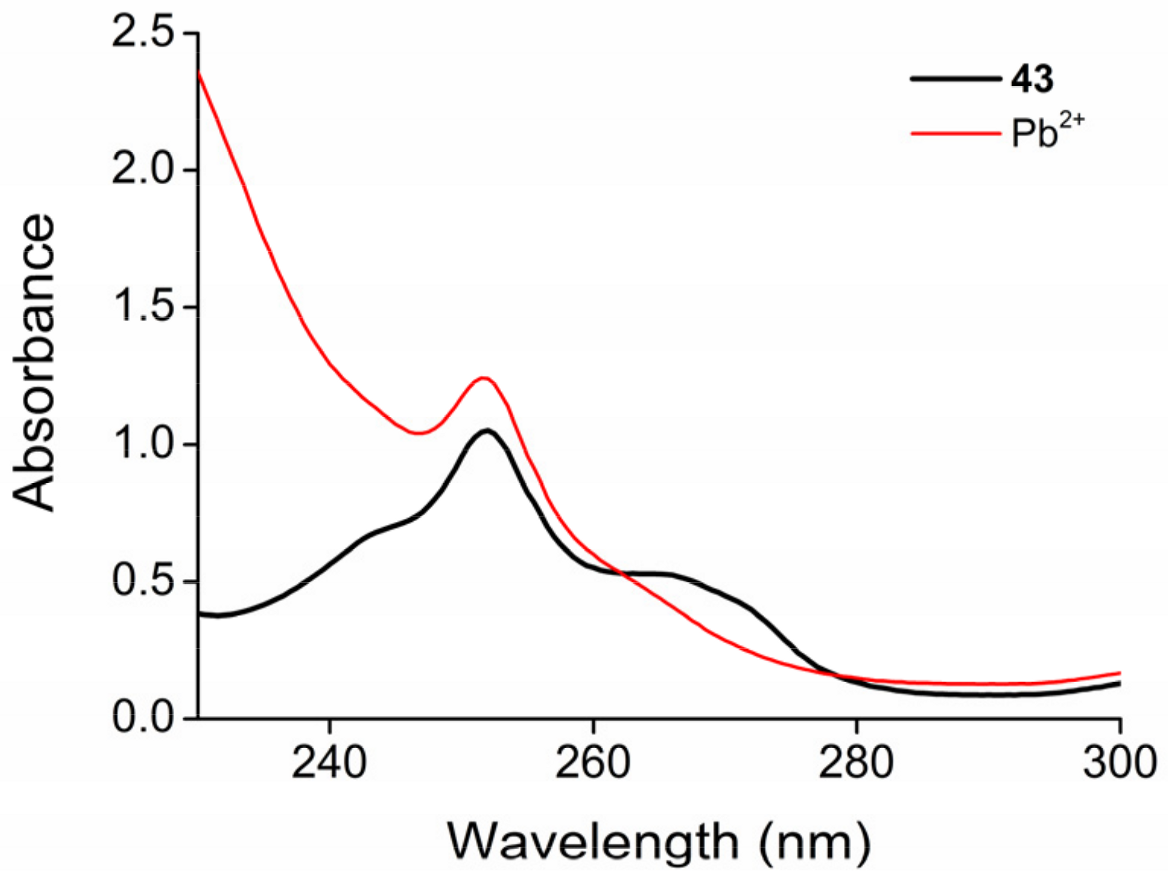
3.2.14. 13-[(Benzyloxy)methyl]-6,9,12,15,18-pentaoxa-25-azatetracyclo[21.3.1.05,26.019,24]heptacosa-1,3,5(26),19,21,23-hexaen-27-one (See 43 in Scheme 6)
Acridono-crown ether
43 was prepared according to the general procedure described in
Section 3.2.8. starting from the previously reported acridone diol
28 (1.00 g, 4.42 mmol, [
18]) and ditosylate
42 (2.75 g, 4.42 mmol). The crude product was purified using column chromatography on silica gel using a gradient elution of dichloromethane and methanol (0–5% methanol). The product needed further purification using PTLC on neutral aluminum oxide using an eluent mixture of ethanol:toluene 1:50. Finally, it was recrystallized from isopropyl alcohol to provide the title compound
43 (670 mg, 30%) as a yellow solid.
M.p. = 63 °C. Rf = 0.41 (SiO2, methanol:dichloromethane 1:30). Rf = 0.53 (Al2O3, ethanol:toluene 1:20). 1H-NMR (CDCl3): δ [ppm]: 9.39 (s, 1H, acridone NH), 8.07 (d, J = 8.2 Hz, 2H, acridone CH), 7.32–7.30 (m, 5H, Bn CH), 7.17 (t, J = 8.0 Hz, 2H, acridone CH), 7.10–7.06 (m, 2H, acridone CH), 4.54 (s, 2H, Bn OCH2C), 4.39–4.29 (m, 5H, ethereal OCH2), 4.12–3.98 (m, 3H, ethereal OCH2), 3.94–3.90 (m, 2H, ethereal OCH2), 3.84–3.77 (m, 4H, ethereal OCH2), 3.71–3.52 (m, 3H, ethereal OCH2 and OCH). 13C-NMR (CDCl3): δ [ppm]: 177.93, 162.51, 146.71, 138.15, 131.39, 128.34, 127.59, 122.11, 120.66, 118.65, 112.30, 112.08, 78.52, 73.42, 72.90, 71.80, 69.97, 69.92, 69.81, 69.38, 68.84. IR: νmax [cm−1]: 2961, 2923, 2653, 2739, 1694, 1602, 1561, 1493, 1465, 1389, 1312, 1259, 1207, 1167, 1087, 1014, 797, 659, 550, 476. HRMS: m/z = [MH+]: 506.2107, (Calculated for C29H31NO7, 505.2101).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}














