
Development of a QuEChERS–HPLC–FLD Procedure for the Simultaneous Detection of Residues of Florfenicol, Its Metabolite Florfenicol Amine, and Three Fluoroquinolones in Eggs
 ,
,  ,
,
Abstract
:1. Introduction
2. Results and Discussion
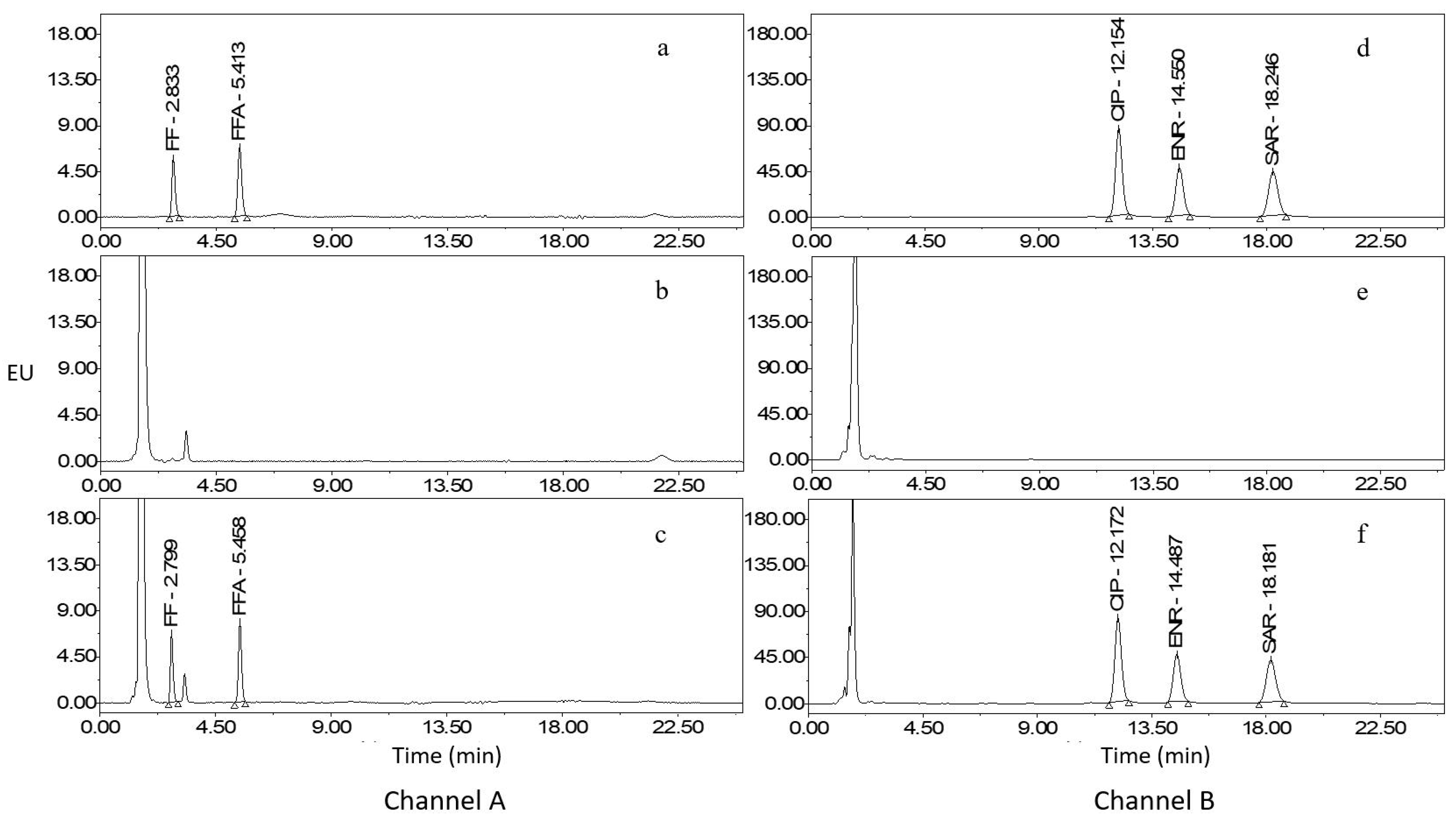
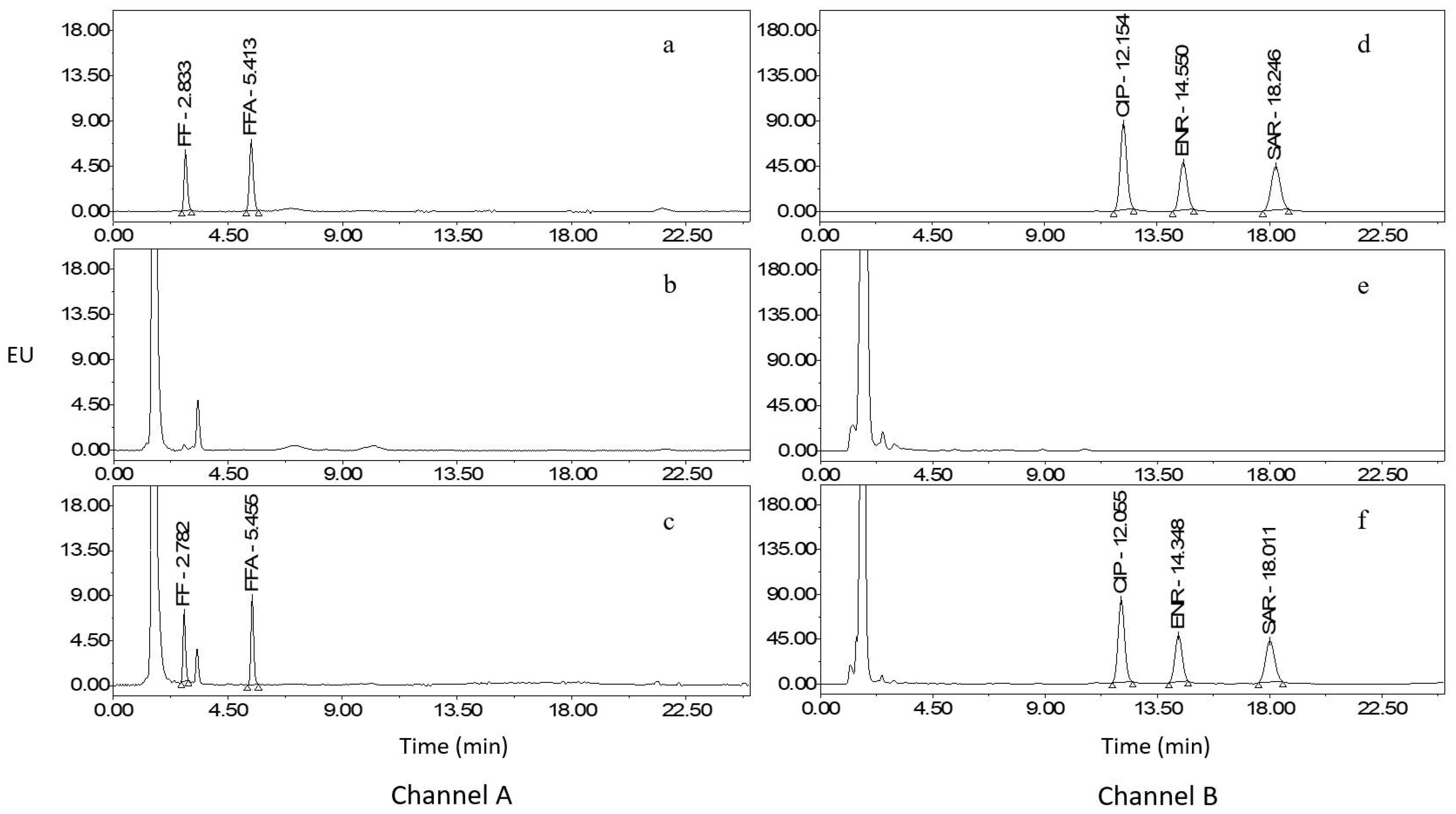
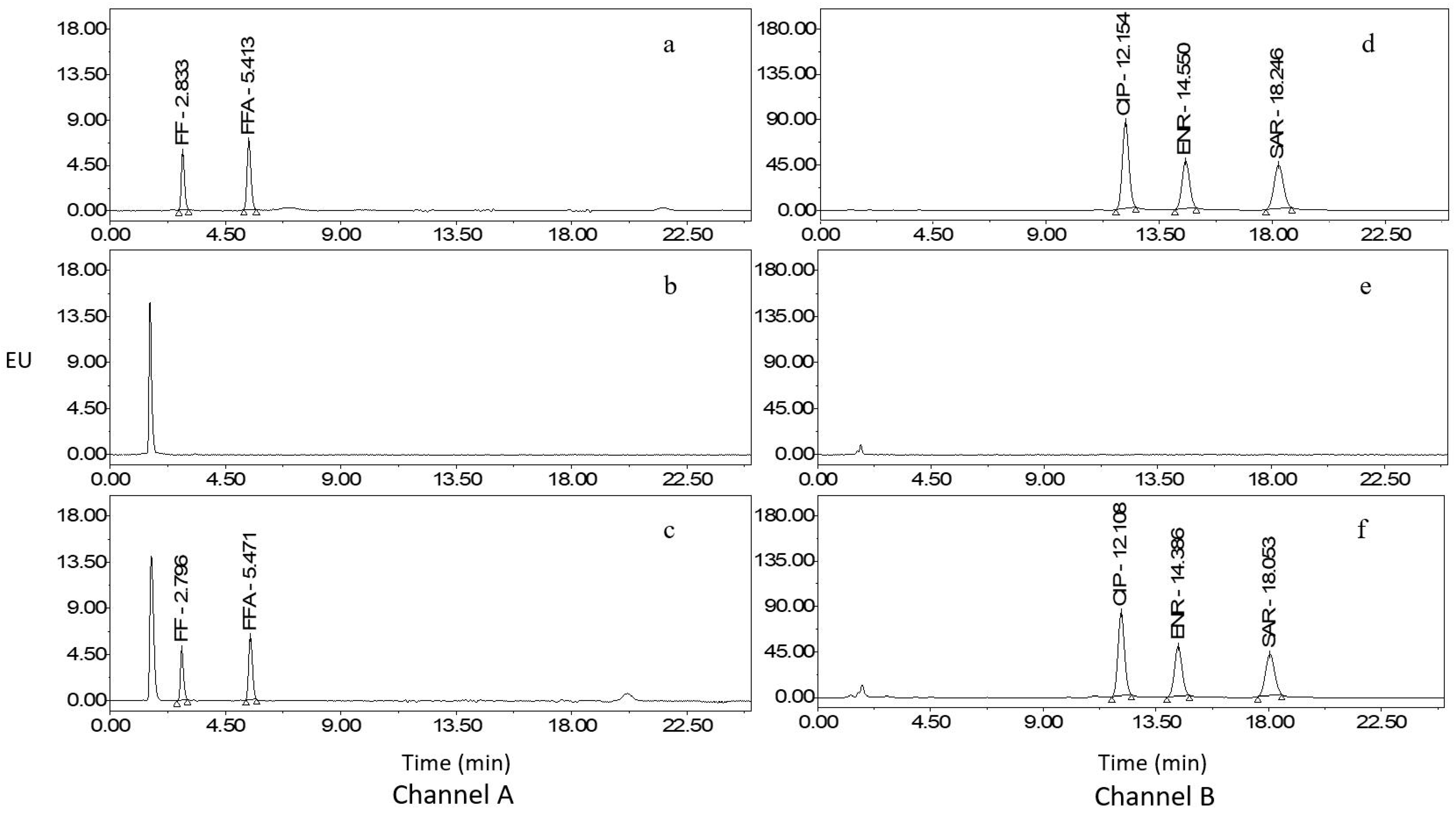
2.1. Optimization of Extraction and Cleanup Procedures
2.2. Optimization of HPLC–FLD
2.3. Method Validation
2.4. Comparison with Reported Methods
2.5. Analysis of Real Samples
3. Materials and Methods
3.1. Reagents, Materials and Solutions
3.2. Instrumental Conditions
3.3. Extraction and Cleanup Procedures
3.3.1. QuEChERS Procedure
3.3.2. Liquid–Liquid Extraction
3.4. Method Validation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Correction Statement
References
- Ben, Y.; Hu, M.; Zhong, F.; Du, E.; Li, Y.; Zhang, H.; Andrews, C.; Zheng, C. Human daily dietary intakes of antibiotic residues: Dominant sources and health risks. Environ. Res. 2022, 212, 113387. [Google Scholar] [CrossRef] [PubMed]
- Piatkowska, M.; Jedziniak, P.; Zmudzki, J. Multiresidue method for the simultaneous determination of veterinary medicinal products, feed additives and illegal dyes in eggs using liquid chromatography-tandem mass spectrometry. Food Chem. 2015, 197, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, S.; Chatterjee, S. Fluoroquinolone antibiotics: Occurrence, mode of action, resistance, environmental detection, and remediation—A comprehensive review. Environ. Pollut. 2022, 315, 120440. [Google Scholar] [CrossRef] [PubMed]
- GB 31650.1-2022; National Food Safety Standard: Maximum Residue Limits of 41 Veterinary Drugs in Food. China Standard Press: Beijing, China, 2022.
- Fedeniuk, R.W.; Mizuno, M.; Neiser, C.; O’Byrne, C. Development of LC-MS/MS methodology for the detection/determination and confirmation of chloramphenicol, chloramphenicol 3-O-d-glucuronide, florfenicol, florfenicol amine and thiamphenicol residues in bovine, equine and porcine liver. J. Chromatogr. B 2015, 991, 68–78. [Google Scholar] [CrossRef]
- Jiméneza, V.; Rubiesb, A.; Centrichb, F.; Companyóa, R.; Guiterasa, J. Development and validation of a multiclass method for the analysis of antibiotic residues in eggs by liquid chromatography-tandem mass spectrometry. J. Chromatogr. A 2015, 1218, 1443–1451. [Google Scholar] [CrossRef]
- Ortiz-Bolsico, C.; Ruiz-Angel, M.J.; Garcia-Alvarez-Coque, M.C. Adsorption of the anionic surfactant sodium dodecyl sulfate on a C18 column under micellar and high submicellar conditions in reversed-phase liquid chromatography. J. Sep. Sci. 2015, 38, 550–555. [Google Scholar] [CrossRef]
- Groeneveld, I.; Pirok, B.W.J.; Molenaar, S.R.A. The development of a generic analysis method for natural and synthetic dyes by ultra-high-pressure liquid chromatography with photo-diode-array detection and triethylamine as an ion-pairing agent. J. Chromatogr. A 2022, 1673, 463038. [Google Scholar] [CrossRef]
- Guo, Y.; He, Z.; Chen, J. Simultaneous determination of tetracyclines and fluoroquinolones in poultry eggs by UPLC integrated with dual-channel-fluorescence detection method. Molecules 2021, 26, 5684. [Google Scholar] [CrossRef]
- SANTE/11813/2017; Guidance Document on Analytical Quality Control and Method Validation Procedures for Pesticide Residues and Analysis in Food and Feed. European Commission Directorate General for Health and Food Safety: Brussels, Belgium, 2017.
- U.S. Department of Health and Human Services; Food and Drug Administration; Center for Drug Evaluation and Research; Center for Veterinary Medicine. Guidance for Industry: Bioanalytical Method Validation; U.S. Department of Health and Human Services: Washington, DC, USA, 2018.
- Tsai, M.; Lin, C.; Yang, W.; Lin, C.; Hung, K.; Chang, G. Health Risk Assessment of Banned Veterinary Drugs and Quinolone Residues in Shrimp through Liquid Chromatography-Tandem Mass Spectrometry. Appl. Sci. 2019, 9, 2463. [Google Scholar] [CrossRef]
- Chen, Y.; Xia, S.; Han, X.; Fu, Z. Simultaneous determination of malachite green, chloramphenicols, sulfonamides, and fluoroquinolones residues in fish by liquid chromatography-mass spectrometry. J. Anal. Methods Chem. 2020, 2020, 3725618. [Google Scholar] [CrossRef]
- Gibbs, R.; Murray, S.; Watson, L.; Nielsen, B.; Potter, R.; Murphy, C. Development and validation of a hybrid screening and quantitative method for the analysis of eight classes of therapeutants in aquaculture products by liquid chromatography-tandem mass spectrometry. J. Agric. Food Chem. 2018, 66, 4997–5008. [Google Scholar] [CrossRef] [PubMed]
- Moretti, S.; Dusi, G.; Giusepponi, D.; Pellicciotti, S.; Rossi, R.; Saluti, G.; Cruciani, G.; Galarini, R. Screening and confirmatory method for multiclass determination of 62 antibiotics in meat. J. Chromatogr. A 2015, 1429, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhao, W.; Guo, W.; Li, Y.; Jiang, R.; Li, H.; Wang, S.; Li, Z. Simultaneous screening and analysis of 155 veterinary drugs in livestock foods using ultra-high performance liquid chromatography tandem quadrupole linear-ion-trap mass spectrometry. Food Chem. 2022, 393, 133260. [Google Scholar] [CrossRef]
- Smith, S.; Gieseker, C.; Reimschuessel, R.; Decker, C.; Carson, M. Simultaneous screening and confirmation of multiple classes of drug residues in fish by liquid chromatography-ion trap mass spectrometry. J. Chromatogr. A 2009, 1216, 8224–8232. [Google Scholar] [CrossRef] [PubMed]
- Turnipseed, S.; Clark, S.; Storey, J.; Carr, J. Analysis of veterinary drug residues in frog legs and other aquacultured species using liquid chromatography quadrupole time-of-flight mass spectrometry. J. Agric. Food Chem. 2012, 60, 4430–4439. [Google Scholar] [CrossRef] [PubMed]
- Xie, K.; Jia, L.; Yao, Y.; Dong, X.; Chen, S. Simultaneous determination of thiamphenicol, florfenicol and florfenicol amine in eggs by reversed-phase high-performance liquid chromatography with fluorescence detection. J. Chromatogr. B 2011, 879, 2351–2354. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Wang, B.; Zhao, X. Determination of thiamphenicol, florfenicol and florfenicol amine residues in poultry meat and pork via ASE-UPLC-FLD. J. Food Compos. Anal. 2019, 81, 19–27. [Google Scholar] [CrossRef]
- Wang, B.; Xie, X.; Zhao, X. Development of an accelerated solvent extraction-ultra-performance liquid chromatography-fluorescence detection method for quantitative analysis of thiamphenicol, florfenicol and florfenicol amine in poultry eggs. Molecules 2019, 24, 1830. [Google Scholar] [CrossRef]
- Yang, J.J.; Sun, G.Z.; Qian, M.R. Development of a high-performance liquid chromatography method for the determination of florfenicol in animal feedstuffs. J. Chromatogr. B 2017, 1068–1069, 9–14. [Google Scholar] [CrossRef]
- Šandor, K.; Andrišić, M.; Žarković, I.; Perak Junaković, E.; Vujnović, A.; Terzić, S. Development of an SPE-HPLC-DAD method for the experimental study of florfenicol and florfenicol amine in pig cerebrospinal fluid. Vet. Stanica 2020, 51, 129–138. [Google Scholar]
- Hassouan, M.; Ballesteros, O.; Vílchez, J. Simple multiresidue determination of fluoroquinolones in bovine milk by liquid chromatography with fluorescence detection. Anal. Lett. 2007, 40, 779–791. [Google Scholar] [CrossRef]
- Herrera-Herrera, A.V.; Hernández-Borges, J.; Rodríguez-Delgado, M. Fluoroquinolone antibiotic determination in bovine, ovine and caprine milk using solid-phase extraction and high-performance liquid chromatography-fluorescence detection with ionic liquids as mobile phase additives. J. Chromatogr. A 2009, 1216, 7281–7287. [Google Scholar] [CrossRef] [PubMed]
- Samanidou, V.F.; Christodoulou, E.A.; Papadoyannis, I.N. Determination of fluoroquinolones in edible animal tissue samples by high performance liquid chromatography after solid phase extraction. J. Sep. Sci. 2015, 28, 555–565. [Google Scholar] [CrossRef] [PubMed]
- Marazuela, M.D.; Moreno-Bondi, M.C. Multiresidue determination of fluoroquinolones in milk by column liquid chromatography with fluorescence and ultraviolet absorbance detection. J. Chromatogr. A 2004, 1034, 25–32. [Google Scholar] [CrossRef]
- Barani, A.; Fallah, A.A. HPLC analysis of some allowable-antibiotic multiresidues in farmed rainbow trout in Iran. Toxin Rev. 2015, 34, 206–209. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Analyte | LOD (µg/kg) | LOQ (µg/kg) | Linearity Range (µg/L) | Linearity | R2 |
|---|---|---|---|---|---|
| FF | 1.5 | 5.0 | 10.0~320.0 | y = 6591.3x − 44,059 | 0.9998 |
| FFA | 0.5 | 2.0 | 4.0~160.0 | y = 16,409x − 53,809 | 0.9999 |
| CIP | 0.05 | 0.1 | 0.2~80.0 | y = 333,491x − 42,526 | 0.9999 |
| ENR | 0.03 | 0.1 | 0.2~20.0 | y = 475,677x + 21,687 | 0.9999 |
| SAR | 0.1 | 0.2 | 0.4~80.0 | y = 151,149x + 2720.6 | 0.9998 |
| Matrix | Analyte | Fortified Level (µg/kg) | Recovery | Between-Day RSD | Within-Day RSD |
|---|---|---|---|---|---|
| Whole egg | FF | 5.0~20.0 | 83.9~94.6 | 3.4~4.1 | 3.4~4.5 |
| FFA | 2.0~20.0 | 71.9~82.4 | 3.1~4.6 | 3.6~5.1 | |
| CIP | 0.1~20.0 | 76.0~84.1 | 3.5~5.1 | 3.6~6.1 | |
| ENR | 0.1~20.0 | 87.1~94.8 | 4.0~5.1 | 3.9~6.2 | |
| SAR | 0.2~10.0 | 82.0~87.5 | 3.3~4.9 | 4.1~5.2 | |
| Egg yolk | FF | 5.0~20.0 | 80.4~90.5 | 3.6~4.6 | 3.7~5.3 |
| FFA | 2.0~20.0 | 73.2~81.9 | 3.9~4.7 | 3.9~4.9 | |
| CIP | 0.1~20.0 | 72.6~83.8 | 3.9~4.6 | 3.7~5.8 | |
| ENR | 0.1~20.0 | 86.2~94.4 | 3.9~6.6 | 4.4~7.3 | |
| SAR | 0.2~10.0 | 73.5~80.7 | 3.5~5.1 | 3.9~5.1 | |
| Egg albumen | FF | 5.0~20.0 | 87.4~92.2 | 3.9~4.5 | 4.1~5.2 |
| FFA | 2.0~20.0 | 75.1~81.5 | 3.1~4.9 | 3.4~5.8 | |
| CIP | 0.1~20.0 | 75.3~83.7 | 3.4~4.5 | 4.6~5.2 | |
| ENR | 0.1~20.0 | 86.7~94.6 | 3.9~6.3 | 4.6~6.4 | |
| SAR | 0.2~10.0 | 82.4~89.3 | 3.6~6.1 | 4.2~7.0 |
| Matrix | Analyte | Detection Method | Sample Pretreatment | LOQ (µg/kg) | Recovery (%) |
|---|---|---|---|---|---|
| Egg [2] | FF, FFA, CIP, ENR, SAR, and other analytes | HPLC–MS/MS | Extraction with 0.1% formic acid in acetonitrile: water (8:2, V/V) and 0.1 M EDTA | - | 75.0~108.0 |
| Shrimp [12] | FF, ENR, SAR, and other analytes | HPLC–MS/MS | Extraction with N, N, N′, N′-tetramethyl-1,4-phenylenediamine dihydrochloride and acetonitrile | FF: 0.25 µg/L ENR and SAR: 1.0 µg/L | FF: 88.67~92.35 ENR and SAR: 75.21~103.31 |
| Fish [13] | FF, CIP, ENR, SAR, and other analytes | HPLC–MS/MS | Extraction with 0.4% hydrochloric acid in acetonitrile and ethyl acetate, SPE clean-up with Cleanert Alumina N column (500 mg) and HLB cartridge | 0.3~1.0 | 67.7~112.8 |
| Aquaculture products [14] | FF, FFA, CIP, ENR, SAR, and other analytes | UHPLC–MS/MS | Extraction with 0.1% formic acid in 80% acetonitrile, followed by 80% acetonitrile | 0.1~30.0 | FF and FFA: 92~106 CIP, ENR and SAR: 85~107 |
| Meat [15] | FF, FFA, CIP, ENR, SAR, and other analytes | HPLC–high resolution MS/MS | Extraction with 0.1 M EDTA and acetonitrile: water (8:2, V/V), followed by acetonitrile | FF and FFA: 10 CIP, ENR and SAR: 3.3 | 71~95 |
| Pork, beef, and mutton [16] | FF, CIP, ENR, SAR, and other analytes | UHPLC–linear-ion-trap MS | Extraction with acetonitrile: water: 0.1% formic acid (80:19: 0.2, V/V/V), SPE clean-up with HLB cartridge | FF: 20 CIP, ENR and SAR: 2 | FF: 84.5~94.1 CIP, ENR and SAR: 67.4~118.8 |
| Fish [17] | FFA, ENR, SAR, and other analytes | HPLC–ion-trap MS | Extraction with acetonitrile | FFA: 0.1 ppm ENR and SAR: 0.01 ppm | >50 |
| Frog legs and other aquacultured species [18] | FF, CIP, and ENR | HPLC–quadrupole-time of flight MS | Extraction with 1% acetic acid, acetonitrile, sodium chloride, and ceramic homogenizers | FF: 1.4 * CIP: 0.5 ENR: 0.8 | 80~130 |
| Rainbow trout muscle [28] | FF, FFA, CIP, and ENR | HPLC–UVD | Extraction with ethyl acetate, SPE clean-up with C18 cartridge | 17.7~39.1 | 71.1~94.7 |
| Egg (This study) | FF, FFA, CIP, ENR, and SAR | HPLC–FLD | Extraction with 0.1 M EDTA disodium solution, water and acetonitrile, followed by a QuEChERS procedure | FF and FFA: 2.0~5.0 CIP, ENR and SAR: 0.1~0.2 | 71.9~94.8 |
| Sample | Brand | Source | Test Results (µg/kg) |
|---|---|---|---|
| 1~6 | free-range egg brand 1 | wholesale market | Not detected |
| 7~12 | free-range egg brand 2 | e-commerce platform | Not detected |
| 13~19 | feedlot industrial egg brand 1 | wholesale market | Not detected |
| 20~24 | feedlot industrial egg brand 2 | supermarket | Not detected |
| 25~30 | feedlot industrial egg brand 3 | supermarket | Not detected |
| 31~36 | feedlot industrial egg brand 4 | retail store | Not detected |
| 37~42 | feedlot industrial egg brand 5 | e-commerce platform | Not detected |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, Y.; Hong, L.; Gao, P.; Liu, S.; Zhu, Y.; Xie, X.; Zhang, G.; Xie, K. Development of a QuEChERS–HPLC–FLD Procedure for the Simultaneous Detection of Residues of Florfenicol, Its Metabolite Florfenicol Amine, and Three Fluoroquinolones in Eggs. Molecules 2024, 29, 252. https://doi.org/10.3390/molecules29010252
Guo Y, Hong L, Gao P, Liu S, Zhu Y, Xie X, Zhang G, Xie K. Development of a QuEChERS–HPLC–FLD Procedure for the Simultaneous Detection of Residues of Florfenicol, Its Metabolite Florfenicol Amine, and Three Fluoroquinolones in Eggs. Molecules. 2024; 29(1):252. https://doi.org/10.3390/molecules29010252
Chicago/Turabian StyleGuo, Yawen, Lu Hong, Pengfei Gao, Shuyu Liu, Yali Zhu, Xing Xie, Genxi Zhang, and Kaizhou Xie. 2024. "Development of a QuEChERS–HPLC–FLD Procedure for the Simultaneous Detection of Residues of Florfenicol, Its Metabolite Florfenicol Amine, and Three Fluoroquinolones in Eggs" Molecules 29, no. 1: 252. https://doi.org/10.3390/molecules29010252