
Photochemical Transformations of Diverse Biologically Active Resveratrol Analogs in Batch and Flow Reactors

 ,
,  , , and
, , and
Abstract
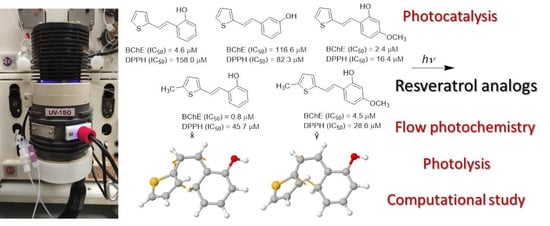
:
1. Introduction
2. Results and Discussion
2.1. Photochemical Transformations of Resveratrol Derivatives 1–5 in Batch Reactor
2.2. Photochemical Transformations of Resveratrol Derivatives 1–5 in Flow Reactor
2.3. Computational Study
3. Materials and Methods
3.1. General Remarks
| Time, min | A, % | B, % | Flow, mL/min |
| 0.00 | 97.00 | 3.00 | 0.900 |
| 1.50 | 0.00 | 100.00 | 0.900 |
| 1.90 | 0.00 | 100.00 | 0.900 |
| 2.00 | 97.00 | 3.00 | 0.900 |
3.2. Synthesis of the Resveratrol Derivatives 1–5
3.3. Photochemistry of the Resveratrol Derivatives 1–5 in a Batch Reactor
3.4. Photochemistry of the Resveratrol Derivatives 1–5 in a Flow Reactor
| Compound 1 | Catalyst | |||
| MnC | MnA | FbC | FbA | |
| τ, min | 10 | 2 | 2 | 10 |
| λ, nm | 365 | 365 | 405 | 420 |
| Compound | c, mol/dm3 | V (acetone), mL | m, mg | n, mol |
| 1 | 0.001 | 20 | 4.05 | 0.0002 |
| 2 | 0.001 | 20 | 4.05 | 0.0002 |
| 3 | 0.001 | 20 | 4.64 | 0.0002 |
| 4 | 0.001 | 20 | 4.32 | 0.0002 |
| 5 | 0.001 | 20 | 4.92 | 0.0002 |
| Catalyst | c, mol/dm3 | V (H2O), mL | m, mg | n, mol |
| MnC | 0.0001 | 20 | 1.82 | 0.00002 |
| MnA | 0.0001 | 20 | 2.06 | 0.00002 |
| FbC | 0.0001 | 20 | 2.73 | 0.00002 |
| FbA | 0.0001 | 20 | 2.08 | 0.00002 |
3.5. Cholinesterase Inhibitory Activity
3.6. Computational Study
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mlakić, M.; Odak, I.; Barić, D.; Talić, S.; Šagud, I.; Štefanić, Z.; Molčanov, K.; Lasić, Z.; Kovačević, B.; Škorić, I. New resveratrol analogs as improved biologically active structures: Design, synthesis and computational modeling. Bioorg. Chem. 2024, 143, 106965. [Google Scholar] [CrossRef] [PubMed]
- Mlakić, M.; Ljubić, A.; Šalić, A.; Zelić, B.; Horváth, O.; Milašinović, V.; Gojun, M.; Molčanov, K.; Škorić, I. Photocatalytic transformations of the resveratrol derivative in microflow reactor. Catalysts 2022, 12, 1510. [Google Scholar] [CrossRef]
- Marchiori, C.F.N.; Damas, G.B.; Araujo, C.M. Tuning the photocatalytic properties of porphyrins for hydrogen evolution reaction: An in-silico design strategy. J. Power Sources Adv. 2022, 15, 100090. [Google Scholar] [CrossRef]
- Rybicka-Jasińska, K.; Wdowik, T.; Łuczak, K.; Wierzba, A.J.; Drapała, O.; Gryko, D. Porphyrins as Promising Photocatalysts for Red-Light-Induced Functionalizations of Biomolecules. ACS Org. Inorg. Au 2022, 2, 422–426. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ren, K.; Wang, L.; Wang, L.; Fan, Z. Porphyrin-based heterogeneous photocatalysts for solar energy conversion. Chinese Chem. Lett. 2022, 33, 33–60. [Google Scholar] [CrossRef]
- Kuramochi, Y.; Satake, A. Porphyrins Acting as Photosensitizers in the Photocatalytic CO2 Reduction Reaction. Catalysts 2023, 13, 282. [Google Scholar] [CrossRef]
- O’Neill, J.S.; Kearney, L.; Brandon, M.P.; Pryce, M.T. Design components of porphyrin-based photocatalytic hydrogen evolution systems: A review. Coord. Chem. Rev. 2022, 467, 214599. [Google Scholar] [CrossRef]
- Wu, Y.; Liu, J.-C.; Li, R.-Z.; Ci, C.-G. Different metal upper porphyrin based self-assembly sensitizers for application in efficient dye-sensitized solar cells. Polyhedron 2022, 211, 115573. [Google Scholar] [CrossRef]
- Zhang, Y.; Higashino, T.; Imahori, H. Molecular designs, synthetic strategies, and properties for porphyrins as sensitizers in dye-sensitized solar cells. J. Mater. Chem. A 2023, 11, 12659–12680. [Google Scholar] [CrossRef]
- Oyim, J.; Amuhaya, E.; Matshitse, R.; Mack, J.; Nyokong, T. Integrated photocatalyst adsorbents based on porphyrin anchored to activated carbon granules for water treatment. Carbon Trends 2022, 8, 100191. [Google Scholar] [CrossRef]
- Luo, H.; Yu, W.; Chen, S.; Wang, Z.; Tian, Z.; He, J.; Liu, Y. Application of metalloporphyrin sensitizers for the treatment or diagnosis of tumors. J. Chem. Res. 2022, 46, 174751982210909. [Google Scholar] [CrossRef]
- Isokuortti, J.; Kuntze, K.; Virkki, M.; Ahmed, Z.; Vuorimaa-Laukkanen, E.; Filatov, M.A.; Turshatov, A.; Laaksonen, T.; Priimagi, A.; Durandin, N.A. Expanding excitation wavelengths for azobenzene photoswitching into the near-infrared range via endothermic triplet energy transfer. Chem. Sci. 2021, 12, 7504–7509. [Google Scholar] [CrossRef]
- Zhang, R.; Horner, J.H.; Newcomb, M. Laser Flash Photolysis Generation and Kinetic Studies of Porphyrin−Manganese−Oxo Intermediates. Rate Constants for Oxidations Effected by Porphyrin−Mn(V)−Oxo Species and Apparent Disproportionation Equilibrium Constants for Porphyrin−Mn(IV)−Oxo Species. J. Am. Chem. Soc. 2005, 127, 6573–6582. [Google Scholar] [CrossRef] [PubMed]
- Newcomb, M.; Zhang, R.; Pan, Z.; Harischandra, D.; Chandrasena, R.; Horner, J.; Martinez II, E. Laser flash photolysis production of metal-oxo derivatives and direct kinetic studies of their oxidation reactions. Catal. Today 2006, 117, 98–104. [Google Scholar] [CrossRef]
- Zhang, R.; Newcomb, M. Laser Flash Photolysis Generation of High-Valent Transition Metal−Oxo Species: Insights from Kinetic Studies in Real Time. Acc. Chem. Res. 2008, 41, 468–477. [Google Scholar] [CrossRef] [PubMed]
- Ahadi, E.; Hosseini-Monfared, H.; Spieß, A.; Janiak, C. Photocatalytic asymmetric epoxidation of trans -stilbene with manganese–porphyrin/graphene-oxide nanocomposite and molecular oxygen: Axial ligand effect. Catal. Sci. Technol. 2020, 10, 3290–3302. [Google Scholar] [CrossRef]
- Horspool, W.M.; Song, P.S. CRC Handbook of Organic Photochemistry and Photobiology, 2nd ed.; CRC Press: Boca Raton, FL, USA, 1995. [Google Scholar]
- Griesbeck, A.; Oelgemöller, M.; Ghetti, F. CRC Handbook of Organic Photochemistry and Photobiology, 3rd ed.; CRC Press: Boca Raton, FL, USA, 2012. [Google Scholar]
- Mlakić, M.; Rajič, L.; Ljubić, A.; Vušak, V.; Zelić, B.; Gojun, M.; Odak, I.; Čule, I.; Šagud, I.; Šalić, A.; et al. Synthesis of new heterocyclic resveratrol analogues in milli- and microreactors: Intensification of the Wittig reaction. J. Flow Chem. 2022, 12, 429–440. [Google Scholar] [CrossRef]
- Galván, I.F.; Delcey, M.G.; Pedersen, T.B.; Aquilante, F.; Lindh, R. Analytical state-average complete-active-space self-consistent field nonadiabatic coupling cevtors: Impletation with density-fitted two-electron integrals and application to conical intersections. J. Chem. Theory Comput. 2016, 12, 3636–3653. [Google Scholar] [CrossRef] [PubMed]
- Ellman, G.L.; Courtnex, K.D.; Andres, V.; Featherstone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Li Manni, G.; Galván, I.F.; Alavi, A.; Aleotti, F.; Aquilante, F.; Autschbach, J.; Avagliano, D.; Baiardi, A.; Bao, J.J.; Battaglia, S.; et al. The OpenMolcas Web: A community-driven approach to advancing computational chemistry. J. Chem. Theory Comput. 2023, 19, 6933–6991. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://github.com/jaimergp/easymecp (accessed on 15 November 2023).
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2019. [Google Scholar]



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AChE | BChE ** | ||
|---|---|---|---|
| IC50/μM | % Inhibition * | IC50/μM | % Inhibition * |
| - | 16.7 ± 0.6 (250) | 9.3 | 83.2 ± 0.9 (250) |
| Reaction Type | Photocatalyst/ Wavelength (nm) | Conversion (%) | Yields of Photoproducts (%) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Irr. Time | cis-1 | 6 | 7 | 8 | 9 | 1 | Undefined | |||
| Direct irradiation | 313 | 80 | 30 min | 52 | 8 | 3 | 1 | 16 | 20 | - |
| 365 | 100 | 30 min | 91 | - | 7 | 2 | - | - | - | |
| Photocatalytic reaction [2] | free base cationic (>380) | 80 | 2 h | 31 | 25 | - | 2 | 1 | 20 | 21 |
| free base anionic (>380) | 97 | 2 h | 15 | 18 | - | - | - | 3 | 64 | |
| cationic Mn(III) (>380) | 93 | 6 h | 74 | - | - | 13 | 1 | 7 | 5 | |
| anionic Mn(III) (>380) | 98 | 6 h | 54 | 10 | - | 3 | 2 | 2 | 29 | |
| Compound | Wavelength (nm) | Conversion (%) | Yields of Photoproducts (%) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Irr. Time | cis- | Dimer | Cycl. * | Cycl.2OH ** | trans- | Undefined | |||
| 1 | 313 | 80 | 30 min | 52 | 8 | 3 | 1 | 20 | 16 |
| 365 | 100 | 30 min | 91 | - | 7 | 2 | - | - | |
| 2 | 313 | 91 | 30 min | 65 | - | 12 | 4 | 19 | - |
| 365 | 93 | 30 min | 62 | - | 9 | 5 | 24 | - | |
| 3 | 313 | 17 | 30 min | 17 | - | - | - | 83 | - |
| 365 | 31 | 30 min | 24 | - | 7 | - | 69 | - | |
| 4 | 313 | 75 | 30 min | 58 | - | 2 | - | 25 | 15 |
| 365 | 83 | 30 min | 83 | - | - | - | 17 | - | |
| 5 | 313 | 90 | 30 min | 45 | - | 15 | - | 10 | 30 |
| 365 | 85 | 30 min | 38 | - | 19 | - | 15 | 28 | |
| Retention Time (min) | Wavelength (nm)/Temp. °C | Flow Rate (mL/min) | Conversion (%) | Selectivity (cis/trans Ratio) | Yields of Photoproducts (%) | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| cis-1 | 6 | 7 | 8 | 1 | Undefined | |||||
| 2 | 450/22 | 0.5 | 5.8 | 0.025 | 2.4 | - | - | - | 94.2 | 3.4 |
| 420/22 | 0.5 | 2.6 | 0.026 | 2.5 | - | - | - | 97.4 | - | |
| 405/22 | 0.5 | 33.8 | 0.421 | 27.9 | - | - | - | 66.2 | 5.9 | |
| 365/22 | 0.5 | 68.7 | 1.885 | 59.0 | - | - | 1.7 | 31.3 | 8.0 | |
| 10 | 450/22 | 0.1 | 4.6 | 0.048 | 4.6 | - | - | - | 95.4 | - |
| 420/22 | 0.1 | 4.0 | 0.042 | 4.0 | - | - | - | 96.0 | - | |
| 405/22 | 0.1 | 60.9 | 1.281 | 50.1 | - | - | - | 39.1 | 10.8 | |
| 365/22 | 0.1 | 81.0 | 3.789 | 72.0 | - | - | 9.0 | 19.0 | - | |
| 10 | 365/40 | 0.1 | 89.2 | 7.806 | 84.3 | - | - | 4.9 | 10.8 | - |
| Retention Time (min) | Wavelength (nm)/Temp. °C | Flow Rate (mL/min) | Conversion (%) | Selectivity (cis/trans Ratio) | Yields of Photoproducts (%) | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| cis-1 | 6 | 7 | 8 | 1 | Undefined | |||||
| 2 | 450/22 | 0.5 | 8.5 | 0.093 | 8.5 | - | - | - | 91.5 | - |
| 420/22 | 0.5 | 11.0 | 0.124 | 11.0 | - | - | - | 89.0 | - | |
| 405/22 | 0.5 | 17.8 | 0.216 | 17.8 | - | - | - | 82.2 | - | |
| 365/22 | 0.5 | 91.9 | 11.346 | 91.9 | - | - | - | 8.1 | - | |
| 10 | 450/22 | 0.1 | 9.0 | 0.099 | 9.0 | - | - | - | 91.0 | - |
| 420/22 | 0.1 | 17.0 | 0.144 | 12.0 | - | - | - | 83.0 | 5.0 | |
| 405/22 | 0.1 | 69.6 | 2.006 | 61.0 | - | - | - | 30.4 | 8.6 | |
| 365/22 | 0.1 | 82.8 | 4.221 | 72.6 | - | 3.1 | - | 17.2 | 7.1 | |
| 2 | 365/40 | 0.5 | 90.0 | 8.790 | 87.9 | 2.1 | - | - | 10.0 | - |
| Retention Time (min) | Wavelength (nm)/Temp. °C | Flow Rate (mL/min) | Conversion (%) | Selectivity (cis/trans Ratio) | Yields of Photoproducts (%) | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| cis-1 | 6 | 7 | 8 | 1 | Undefined | |||||
| 2 | 450/22 | 0.5 | 27.3 | 0.292 | 21.2 | - | 6.1 | - | 72.7 | - |
| 420/22 | 0.5 | 74.1 | 0.923 | 23.9 | - | 7.7 | 3.2 | 25.9 | 39.3 | |
| 405/22 | 0.5 | 83.8 | 2.068 | 33.5 | - | 8.0 | - | 16.2 | 42.3 | |
| 365/22 | The catalysts do not absorb on this wavelength | |||||||||
| 10 | 450/22 | 0.1 | 92.4 | 0.553 | 4.2 | - | 64.8 | 8.2 | 7.6 | 15.2 |
| 420/22 | 0.1 | 100.0 | - | - | - | 76.9 | 11.1 | - | 12.0 | |
| 405/22 | 0.1 | 100.0 | - | - | - | 78.0 | 13.0 | - | 9.9 | |
| 365/22 | The catalysts do not absorb on this wavelength | |||||||||
| 2 | 405/40 | 0.5 | 64.2 | 0.958 | 34.3 | - | 16.4 | - | 35.8 | 18.4 |
| Retention Time (min) | Wavelength (nm)/Temp. °C | Flow Rate (mL/min) | Conversion (%) | Selectivity (cis/trans Ratio) | Yields of Photoproducts (%) | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| cis-1 | 6 | 7 | 8 | 1 | Undefined | |||||
| 2 | 450/22 | 0.5 | 22.7 | 0.107 | 8.3 | - | - | - | 77.3 | 14.4 |
| 420/22 | 0.5 | 88.8 | 0.205 | 2.3 | 32.5 | 44.8 | - | 11.2 | 9.2 | |
| 405/22 | 0.5 | 92.5 | 0.707 | 5.3 | 41.0 | 46.2 | - | 7.5 | - | |
| 365/22 | The catalysts does not absorb on this wavelength | |||||||||
| 10 | 450/22 | 0.1 | 100 | - | - | 34.9 | 32.0 | - | - | 33.1 |
| 420/22 | 0.1 | 100 | - | - | 58.1 | 19.3 | - | - | 22.6 | |
| 405/22 | 0.1 | 100 | - | - | 39.3 | 39.6 | - | - | 21.1 | |
| 365/22 | The catalysts does not absorb on this wavelength | |||||||||
| 10 | 420/40 | 0.1 | 100 | - | - | 4 | 45.1 | - | - | 50.9 |
| Comp. | Catalyst | Retention Time (min) | Wavelength (nm)/Temp. °C | Flow Rate (mL/min) | Conversion (%) | Productivity (μmol mL−1 min−1) | Yields of Photoproducts (%) | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| cis- | Dimer | Cycl. ** | trans- | Undefined | |||||||
| 1 | - | 2 | 365/22 | 0.5 | 94 | 0.470 | 94.0 | - | - | 6.0 | - |
| 2 | - | 2 | 365/22 | 0.5 | 61.6 | 0.308 | 61.6 | - | - | 38.4 | - |
| 3 | - | 2 | 365/22 | 0.5 | 91.7 | 0.458 | 71.0 | - | - | 8.3 | 20.7 |
| 4 | - | 2 | 365/22 | 0.5 | 92.2 | 0.461 | 81.9 | - | - | 7.8 | 10.3 |
| 5 | - | 2 | 365/22 | 0.5 | 91.0 | 0.455 | 22.5 | 4.8 | 29.8 | 9.0 | 30.1 |
| 1 | FbA | 10 | 420/22 | 0.1 | 100 | 0.500 | - | 58.1 | 19.3 | - | 22.6 |
| 2 | FbA | 10 | 420/22 | 0.1 | 40.2 | 0.201 | 8.1 | - | - | 59.8 | 32.1 |
| 3 | FbA | 10 | 420/22 | 0.1 | 100 | 0.500 | - | 6.4 | 93.6 | - | - |
| 4 | FbA | 10 | 420/22 | 0.1 | 100 | 0.500 | - | 58.0 | 13.8 | - | 28.1 |
| 5 | FbA | 10 | 420/22 | 0.1 | 100 | 0.500 | - | - | 90.1 | - | 9.9 |
 | |||
| trans-1 | cis-1 | cis-1-Isom | |
| S0 | 0 | 2.91 | 3.27 |
| S1 | 95.82 | 112.62 | 110.16 |
| S2 | 117.61 | 143.75 | 133.81 |
| S3 | 121.78 | 151.40 | 153.03 |
| FC-cis-isom | S1Mincis-isom | CI-cyc | |
|---|---|---|---|
| S0 | 3.27 | 11.66 | 99.85 |
| S1 | 110.16 | 101.68 | 91.60 |
| S2 | 133.81 | 121.86 | 143.88 |
| S3 | 153.03 | 135.28 | 158.58 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mlakić, M.; Perinić, H.; Vušak, V.; Horváth, O.; Sampedro, D.; Losantos, R.; Odak, I.; Škorić, I. Photochemical Transformations of Diverse Biologically Active Resveratrol Analogs in Batch and Flow Reactors. Molecules 2024, 29, 201. https://doi.org/10.3390/molecules29010201
Mlakić M, Perinić H, Vušak V, Horváth O, Sampedro D, Losantos R, Odak I, Škorić I. Photochemical Transformations of Diverse Biologically Active Resveratrol Analogs in Batch and Flow Reactors. Molecules. 2024; 29(1):201. https://doi.org/10.3390/molecules29010201
Chicago/Turabian StyleMlakić, Milena, Hana Perinić, Vitomir Vušak, Ottó Horváth, Diego Sampedro, Raúl Losantos, Ilijana Odak, and Irena Škorić. 2024. "Photochemical Transformations of Diverse Biologically Active Resveratrol Analogs in Batch and Flow Reactors" Molecules 29, no. 1: 201. https://doi.org/10.3390/molecules29010201