
Elucidating the Role of Surface Ce4+ and Oxygen Vacancies of CeO2 in the Direct Synthesis of Dimethyl Carbonate from CO2 and Methanol

 and
and
Abstract
:
1. Introduction
2. Results
2.1. Textural Properties of Catalysts
2.2. XRD Analysis of Catalysts
2.3. TEM of Catalysts
2.4. Surface Composition Analysis of Catalysts
2.5. Surface Acidity and Basicity Analysis of Catalysts
2.6. Measurement of Catalytic Activity
3. Discussion
3.1. The Relationship between Catalytic Activity and Surface Acidity/Basicity
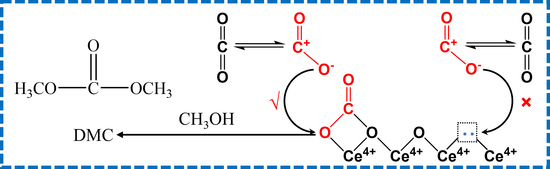
3.2. Deep Insight into the Role of Ce4+ and OV
4. Materials and Methods
4.1. Preparation of CeO2
4.2. Reduction Treatment of CeO2
4.3. Reoxidation Treatment of CeO2–H550 and CeO2–H750
4.4. Characterization of Supports and Catalysts
4.5. Catalytic Evaluation
5. Conclusions
- (1)
- The concentrations of surface Ce4+ and oxygen vacancies of CeO2 were regulated with reduction and reoxidation treatments under different conditions. The reduction treatment promoted the conversion of Ce4+ to Ce3+ and improved the surface concentration of oxygen vacancies, while the reoxidation treatment favored the conversion of Ce3+ to Ce4+ and decreased the concentration of oxygen vacancies.
- (2)
- Catalytic activity was positively correlated with the number of moderate surface acidic/basic sites of catalysts. Moreover, the number of moderate basic sites and catalytic activity were positively correlated with the surface concentration of Ce4+ but negatively correlated with that of oxygen vacancies.
- (3)
- The surface Ce4+ rather than oxygen vacancies served as active Lewis acid sites, and lattice oxygen served as active Lewis base sites to adsorb and activate CO2, promoting the formation of active bidentate carbonates species and DMC.
- (4)
- The cationic oxygen vacancy was beneficial but the anionic oxygen vacancy was detrimental to the formation of active bidentate carbonates species and DMC, which sheds light upon the active sites for CeO2 and metal-doped CeO2 catalysts and provides evidence for the design of efficient catalysts.
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jiang, X.; Nie, X.; Guo, X.; Song, C.; Chen, J.G. Recent Advances in Carbon Dioxide Hydrogenation to Methanol via Heterogeneous Catalysis. Chem. Rev. 2020, 120, 7984–8034. [Google Scholar] [CrossRef] [PubMed]
- Rogelj, J.; Huppmann, D.; Krey, V.; Riahi, K.; Clarke, L.; Gidden, M.; Nicholls, Z.; Meinshausen, M. A new scenario logic for the Paris Agreement long-term temperature goal. Nature 2019, 573, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Yan, B.; Wang, S.; Ma, X. Recent advances in dialkyl carbonates synthesis and applications. Chem. Soc. Rev. 2015, 44, 3079–3116. [Google Scholar] [CrossRef]
- Zhang, M.; Xu, Y.; Williams, B.L.; Xiao, M.; Wang, S.; Han, D.; Sun, L.; Meng, Y. Catalytic materials for direct synthesis of dimethyl carbonate (DMC) from CO2. J. Clean. Prod. 2021, 279, 123344. [Google Scholar] [CrossRef]
- Tamboli, A.H.; Chaugule, A.A.; Kim, H. Catalytic developments in the direct dimethyl carbonate synthesis from carbon dioxide and methanol. Chem. Eng. J. 2017, 323, 530–544. [Google Scholar] [CrossRef]
- Tomishige, K.; Sakaihori, T.; Ikeda, Y.; Fujimoto, K. A novel method of direct synthesis of dimethyl carbonate from methanol and carbon dioxide catalyzed by zirconia. Catal. Lett. 1999, 58, 225–229. [Google Scholar] [CrossRef]
- Jung, K.T.; Bell, A.T. An in Situ Infrared Study of Dimethyl Carbonate Synthesis from Carbon Dioxide and Methanol over Zirconia. J. Catal. 2001, 204, 339–347. [Google Scholar] [CrossRef]
- Li, A.; Pu, Y.; Li, F.; Luo, J.; Zhao, N.; Xiao, F. Synthesis of dimethyl carbonate from methanol and CO2 over Fe-Zr mixed oxides. J. CO2 Util. 2017, 19, 33–39. [Google Scholar] [CrossRef]
- Marin, C.M.; Li, L.; Bhalkikar, A.; Doyle, J.E.; Zeng, X.C.; Cheung, C.L. Kinetic and mechanistic investigations of the direct synthesis of dimethyl carbonate from carbon dioxide over ceria nanorod catalysts. J. Catal. 2016, 340, 295–301. [Google Scholar] [CrossRef]
- Wang, S.-P.; Zhou, J.-J.; Zhao, S.-Y.; Zhao, Y.-J.; Ma, X.-B. Enhancements of dimethyl carbonate synthesis from methanol and carbon dioxide: The in situ hydrolysis of 2-cyanopyridine and crystal face effect of ceria. Chin. Chem. Lett. 2015, 26, 1096–1100. [Google Scholar] [CrossRef]
- Zhao, S.-Y.; Wang, S.-P.; Zhao, Y.-J.; Ma, X.-B. An in situ infrared study of dimethyl carbonate synthesis from carbon dioxide and methanol over well-shaped CeO2. Chin. Chem. Lett. 2017, 28, 65–69. [Google Scholar] [CrossRef]
- Liu, B.; Li, C.M.; Zhang, G.Q.; Yan, L.F.; Li, Z. Direct synthesis of dimethyl carbonate from CO2 and methanol over CaO-CeO2 catalysts: The role of acid-base properties and surface oxygen vacancies. New J. Chem. 2017, 41, 12231–12240. [Google Scholar] [CrossRef]
- Chen, Y.; Tang, Q.; Ye, Z.; Li, Y.; Yang, Y.; Pu, H.; Li, G. Monolithic ZnxCe1-xO2 catalysts for catalytic synthesis of dimethyl carbonate from CO2 and methanol. New J. Chem. 2020, 44, 12522–12530. [Google Scholar] [CrossRef]
- Liu, N.; Xue, Y.; Yu, Z.R.; Li, Y.Y.; Xu, Y.; Xu, J.; Xue, B.; Luo, J.; Wang, F. Zn-Doped CeO2 Nanorods: A Highly Efficient Heterogeneous Catalyst for the Direct Synthesis of Dimethyl Carbonate from CO2 and Methanol. Chemistryselect 2023, 8, e202203472. [Google Scholar] [CrossRef]
- Zhang, G.; Sun, Y.; Shi, Y.; Zheng, H.; Li, Z.; Ju, S.; Liu, S.; Shi, P. Surface Properties of Ce1-xMnxO2 Catalyst on the Catalytic Activities for Direct Synthesis of DMC from CO2 and Methanol. Chem. J. Chin. Univ. 2020, 41, 2061–2069. [Google Scholar]
- Chen, Y.; Wang, H.; Qin, Z.; Tian, S.; Ye, Z.; Ye, L.; Abroshan, H.; Li, G. TixCe1-xO2 nanocomposites: A monolithic catalyst for the direct conversion of carbon dioxide and methanol to dimethyl carbonate. Green Chem. 2019, 21, 4642–4649. [Google Scholar] [CrossRef]
- Luo, M.S.; Qin, T.; Liu, Q.L.; Yang, Z.; Wang, F.L.; Li, H. Novel Fe-modified CeO2 Nanorod Catalyst for the Dimethyl Carbonate Formation from CO2 and Methanol. ChemCatChem 2022, 14, e202200253. [Google Scholar] [CrossRef]
- Kulal, N.; Bhat, S.S.; Hugar, V.; Mallannavar, C.N.; Lee, S.C.; Bhattacharjee, S.; Vetrivel, R.; Shanbhag, G.V. Integrated DFT and experimental study on Co3O4/CeO2 catalyst for direct synthesis of dimethyl carbonate from CO2. J. CO2 Util. 2023, 67, 102323. [Google Scholar] [CrossRef]
- Liu, B.; Li, C.M.; Zhang, G.Q.; Yao, X.S.; Chuang, S.S.C.; Li, Z. Oxygen Vacancy Promoting Dimethyl Carbonate Synthesis from CO2 and Methanol over Zr-Doped CeO2 Nanorods. ACS Catal. 2018, 8, 10446–10456. [Google Scholar] [CrossRef]
- Xu, S.Y.; Cao, Y.X.; Liu, Z.M. Dimethyl carbonate synthesis from CO2 and methanol over CeO2-ZrO2 catalyst. Catal. Commun. 2022, 162, 106397. [Google Scholar] [CrossRef]
- Giordano, F.; Trovarelli, A.; de Leitenburg, C.; Giona, M. A Model for the Temperature-Programmed Reduction of Low and High Surface Area Ceria. J. Catal. 2000, 193, 273–282. [Google Scholar] [CrossRef]
- Hezam, A.; Namratha, K.; Drmosh, Q.A.; Ponnamma, D.; Wang, J.W.; Prasad, S.; Ahamed, M.; Cheng, C.; Byrappa, K. CeO2 Nanostructures Enriched with Oxygen Vacancies for Photocatalytic CO2 Reduction. ACS Appl. Nano Mater. 2020, 3, 138–148. [Google Scholar] [CrossRef]
- Yoshida, Y.; Arai, Y.; Kado, S.; Kunimori, K.; Tomishige, K. Direct synthesis of organic carbonates from the reaction of CO2 with methanol and ethanol over CeO2 catalysts. Catal. Today 2006, 115, 95–101. [Google Scholar] [CrossRef]
- Aneggi, E.; Wiater, D.; de Leitenburg, C.; Llorca, J.; Trovarelli, A. Shape-Dependent Activity of Ceria in Soot Combustion. ACS Catal. 2014, 4, 172–181. [Google Scholar] [CrossRef]
- Wang, X.; Jiang, Z.Y.; Zheng, B.J.; Xie, Z.X.; Zheng, L.S. Synthesis and shape-dependent catalytic properties of CeO2 nanocubes and truncated octahedra. Crystengcomm 2012, 14, 7579–7582. [Google Scholar] [CrossRef]
- Swiatowska, J.; Lair, V.; Pereira-Nabais, C.; Cote, G.; Marcus, P.; Chagnes, A. XPS, XRD and SEM characterization of a thin ceria layer deposited onto graphite electrode for application in lithium-ion batteries. Appl. Surf. Sci. 2011, 257, 9110–9119. [Google Scholar] [CrossRef]
- Cui, Z.; Fan, J.; Duan, H.; Zhang, J.; Xue, Y.; Tan, Y. Effect of calcination atmospheres on the catalytic performance of nano-CeO2 in direct synthesis of DMC from methanol and CO2. Korean J. Chem. Eng. 2017, 34, 29–36. [Google Scholar] [CrossRef]
- Xing, Y.M.; Wu, Y.; Li, L.Y.; Shi, Q.; Shi, J.; Yun, S.N.; Akbar, M.; Wang, B.Y.; Kim, J.S.; Zhu, B. Proton Shuttles in CeO2/CeO2-delta Core-Shell Structure. ACS Energy Lett. 2019, 4, 2601–2607. [Google Scholar] [CrossRef]
- Singhal, R.K.; Kumar, S.; Samariya, A.; Dhawan, M.; Sharma, S.C.; Xing, Y.T. Investigating the mechanism of ferromagnetic exchange interaction in non-doped CeO2 with regard to defects and electronic structure. Mater. Chem. Phys. 2012, 132, 534–539. [Google Scholar] [CrossRef]
- Liu, H.; Zou, W.; Xu, X.; Zhang, X.; Yang, Y.; Yue, H.; Yu, Y.; Tian, G.; Feng, S. The proportion of Ce4+ in surface of CexZr1-xO2 catalysts: The key parameter for direct carboxylation of methanol to dimethyl carbonate. J. CO2 Util. 2017, 17, 43–49. [Google Scholar] [CrossRef]
- Kumar, P.; Srivastava, V.C.; Glaeser, R.; With, P.; Mishra, I.M. Active ceria-calcium oxide catalysts for dimethyl carbonate synthesis by conversion of CO2. Powder Technol. 2017, 309, 13–21. [Google Scholar] [CrossRef]
- Leino, E.; Kumar, N.; Mäki-Arvela, P.; Rautio, A.-R.; Dahl, J.; Roine, J.; Mikkola, J.-P. Synthesis and characterization of ceria-supported catalysts for carbon dioxide transformation to diethyl carbonate. Catal. Today 2018, 306, 128–137. [Google Scholar] [CrossRef]
- Kumar, P.; With, P.; Srivastava, V.C.; Shukla, K.; Glaser, R.; Mishra, I.M. Dimethyl carbonate synthesis from carbon dioxide using ceria-zirconia catalysts prepared using a templating method: Characterization, parametric optimization and chemical equilibrium modeling. RSC Adv. 2016, 6, 110235–110246. [Google Scholar] [CrossRef]
- Akune, T.; Morita, Y.; Shirakawa, S.; Katagiri, K.; Inumaru, K. ZrO2 Nanocrystals As Catalyst for Synthesis of Dimethylcarbonate from Methanol and Carbon Dioxide: Catalytic Activity and Elucidation of Active Sites. Langmuir 2018, 34, 23–29. [Google Scholar] [CrossRef]
- Tomishige, K.; Ikeda, Y.; Sakaihori, T.; Fujimoto, K. Catalytic properties and structure of zirconia catalysts for direct synthesis of dimethyl carbonate from methanol and carbon dioxide. J. Catal. 2000, 192, 355–362. [Google Scholar] [CrossRef]
- Fan, H.-X.; Feng, J.; Li, W.-Y.; Li, X.-H.; Wiltowski, T.; Ge, Q.-F. Role of CO2 in the oxy-dehydrogenation of ethylbenzene to styrene on the CeO2(111) surface. Appl. Surf. Sci. 2018, 427, 973–980. [Google Scholar] [CrossRef]
- Lu, X.Q.; Wang, W.L.; Wei, S.X.; Guo, C.; Shao, Y.; Zhang, M.M.; Deng, Z.G.; Zhu, H.Y.; Guo, W.Y. Initial reduction of CO2 on perfect and O-defective CeO2 (111) surfaces: Towards CO or COOH? RSC Adv. 2015, 5, 97528–97535. [Google Scholar] [CrossRef]
- Bell, A.T. The Impact of Nanoscience on Heterogeneous Catalysis. Science 2003, 299, 1688–1691. [Google Scholar] [CrossRef]
- Schmale, K.; Daniels, M.; Buchheit, A.; Grunebaum, M.; Haase, L.; Koops, S.; Wiemhofer, H.D. Influence of Zinc Oxide on the Conductivity of Ceria. J. Electrochem. Soc. 2013, 160, F1081–F1087. [Google Scholar] [CrossRef]
- Buchheit, A.; Tessmer, B.; Ran, K.; Mayer, J.; Wiemofer, H.D.; Neuhaus, K. The Impact of Fe Addition on the Electronic Conductivity of Gadolinium Doped Ceria. ECS J. Solid. State Sci. Technol. 2019, 8, P41–P50. [Google Scholar] [CrossRef]
- Meng, F.H.; Li, X.J.; Zhang, P.; Yang, L.L.; Liu, S.S.; Li, Z. A facile approach for fabricating highly active ZrCeZnOx in combination with SAPO-34 for the conversion of syngas into light olefins. Appl. Surf. Sci. 2021, 542, 148713. [Google Scholar] [CrossRef]
- Meng, F.H.; Li, X.J.; Zhang, P.; Yang, L.L.; Yang, G.N.; Ma, P.C.; Li, Z. Highly active ternary oxide ZrCeZnOx combined with SAPO-34 zeolite for direct conversion of syngas into light olefins. Catal. Today 2021, 368, 118–125. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalysts | SBET (m2·g−1) | Vp (cm3/g) | Pore Size (nm) |
|---|---|---|---|
| CeO2 | 130.3 | 0.65 | 13.8 |
| CeO2–H450 | 120.9 | 0.59 | 14.6 |
| CeO2–H550 | 106.2 | 0.57 | 14.6 |
| CeO2–H650 | 72.9 | 0.47 | 16.9 |
| CeO2–H750 | 28.2 | 0.27 | 23.1 |
| CeO2–H800 | 18.6 | 0.24 | 30.7 |
| CeO2–H550–O350 | 107.3 | 0.59 | 14.8 |
| CeO2–H550–O450 | 109.2 | 0.64 | 14.9 |
| CeO2–H550–O550 | 107.8 | 0.52 | 12.3 |
| CeO2–H750–O350 | 23.1 | 0.23 | 25.1 |
| CeO2–H750–O450 | 26.2 | 0.27 | 25.6 |
| CeO2–H750–O550 | 24.5 | 0.24 | 23.9 |
| Catalysts | dCeO2 (nm) a | Catalysts | dCeO2 (nm) a |
|---|---|---|---|
| CeO2 | 7.1 | CeO2–H550–O350 | 7.6 |
| CeO2–H450 | 7.4 | CeO2–H550–O450 | 7.3 |
| CeO2–H550 | 7.4 | CeO2–H550–O550 | 7.5 |
| CeO2–H650 | 12.9 | CeO2–H750–O350 | 32.1 |
| CeO2–H750 | 25.5 | CeO2–H750–O450 | 29.7 |
| CeO2–H800 | 46.6 | CeO2–H750–O550 | 31.6 |
| Catalysts | Ce Atom Ratio (%) | O Atom Ratio (%) | |
|---|---|---|---|
| Ce3+ | Ce4+ | OV | |
| CeO2 | 13.5 | 86.5 | 5.3 |
| CeO2–H450 | 15.7 | 84.3 | 6.9 |
| CeO2–H550 | 16.2 | 83.8 | 7.6 |
| CeO2–H650 | 16.7 | 83.3 | 7.8 |
| CeO2–H750 | 16.8 | 83.2 | 8.1 |
| CeO2–H800 | 17.7 | 82.3 | 8.5 |
| CeO2–H550–O350 | 15.7 | 84.3 | 7.1 |
| CeO2–H550–O450 | 15.3 | 84.7 | 6.6 |
| CeO2–H550–O550 | 15.2 | 84.8 | 6.7 |
| CeO2–H750–O350 | 15.8 | 84.2 | 7.2 |
| CeO2–H750–O450 | 15.2 | 84.8 | 6.5 |
| CeO2–H750–O550 | 14.9 | 85.1 | 6.1 |
| Catalysts | CO2 Adsorption (mmol/g) | NH3 Adsorption (mmol/g) | ||||
|---|---|---|---|---|---|---|
| Weak (<200 °C) | Moderate (200~400 °C) | Strong (>400 °C) | Weak (<200 °C) | Moderate (200~400 °C) | Strong (>400 °C) | |
| CeO2 | 0.607 | 0.206 | 0.039 | 0.592 | 0.158 | 0.040 |
| CeO2–H450 | 0.533 | 0.147 | 0.025 | 0.472 | 0.124 | 0.046 |
| CeO2–H550 | 0.525 | 0.136 | 0.020 | 0.586 | 0.134 | 0.043 |
| CeO2–H650 | 0.361 | 0.074 | 0.015 | 0.384 | 0.084 | - |
| CeO2–H750 | 0.142 | 0.023 | 0.002 | 0.199 | 0.018 | - |
| CeO2–H800 | 0.113 | 0.019 | 0.006 | 0.117 | 0.031 | - |
| CeO2–H550–O350 | 0.525 | 0.130 | 0.024 | 0.586 | 0.143 | 0.040 |
| CeO2–H550–O450 | 0.538 | 0.154 | 0.028 | 0.416 | 0.143 | 0.037 |
| CeO2–H550–O550 | 0.590 | 0.156 | 0.028 | 0.598 | 0.152 | 0.041 |
| CeO2–H750–O350 | 0.183 | 0.023 | 0.004 | 0.227 | 0.022 | - |
| CeO2–H750–O450 | 0.192 | 0.024 | 0.006 | 0.232 | 0.023 | - |
| CeO2–H750–O550 | 0.205 | 0.039 | 0.007 | 0.240 | 0.027 | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, G.; Zhou, Y.; Yang, Y.; Kong, T.; Song, Y.; Zhang, S.; Zheng, H. Elucidating the Role of Surface Ce4+ and Oxygen Vacancies of CeO2 in the Direct Synthesis of Dimethyl Carbonate from CO2 and Methanol. Molecules 2023, 28, 3785. https://doi.org/10.3390/molecules28093785
Zhang G, Zhou Y, Yang Y, Kong T, Song Y, Zhang S, Zheng H. Elucidating the Role of Surface Ce4+ and Oxygen Vacancies of CeO2 in the Direct Synthesis of Dimethyl Carbonate from CO2 and Methanol. Molecules. 2023; 28(9):3785. https://doi.org/10.3390/molecules28093785
Chicago/Turabian StyleZhang, Guoqiang, Yuan Zhou, Yanlin Yang, Tiantian Kong, Ya Song, Song Zhang, and Huayan Zheng. 2023. "Elucidating the Role of Surface Ce4+ and Oxygen Vacancies of CeO2 in the Direct Synthesis of Dimethyl Carbonate from CO2 and Methanol" Molecules 28, no. 9: 3785. https://doi.org/10.3390/molecules28093785