


Dihydropyrimidone Derivatives as Thymidine Phosphorylase Inhibitors: Inhibition Kinetics, Cytotoxicity, and Molecular Docking
 ,
,  , , , , and
, , , , and
Abstract
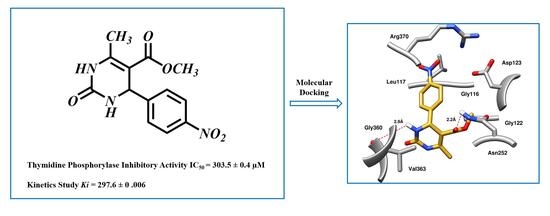
:
1. Introduction
2. Results and Discussion
2.1. In Vitro Study
2.2. In Silico Study
3. Material and Methods
3.1. Thymidine Phosphorylase Inhibition Assay
3.2. Mechanistic Studies
3.3. Cytotoxicity
3.4. Statistical Analysis
3.5. Molecular Docking Studies
3.6. ADME Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Panigrahy, D.; Singer, S.; Shen, L.Q.; Butterfield, C.E.; Freedman, D.A.; Chen, E.J.; Moses, M.A.; Kilroy, S.; Duensing, S.; Fletcher, C.; et al. PPARγ Ligands Inhibit Primary Tumor Growth and Metastasis by Inhibiting Angiogenesis. J. Clin. Invest. 2002, 110, 923–932. [Google Scholar] [CrossRef] [PubMed]
- Vives, M.; Ginestà, M.M.; Gracova, K.; Graupera, M.; Casanovas, O.; Capellà, G.; Serrano, T.; Laquente, B.; Viñals, F. Metronomic Chemotherapy Following the Maximum Tolerated Dose Is an Effective Anti-Tumour Therapy Affecting Angiogenesis, Tumour Dissemination and Cancer Stem Cells. Int. J. Cancer 2013, 133, 2464–2472. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, P.A.; Vetticatt, M.J.; Schramm, V.L. Transition State Analysis of the Arsenolytic Depyrimidination of Thymidine by Human Thymidine Phosphorylase. Biochemistry 2011, 50, 1412–1420. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, S.; Nitanda, T.; Furukawa, T.; Sumizawa, T.; Tani, A.; Nishimoto, K.; Akiba, S.; Miyadera, K.; Fukushima, M.; Yamada, Y.; et al. The Effect of a Thymidine Phosphorylase Inhibitor on Angiogenesis and Apoptosis in Tumors. Cancer Res. 1999, 59, 1911–1916. [Google Scholar]
- Bronckaers, A.; Gago, F.; Balzarini, J.; Liekens, S. The Dual Role of Thymidine Phosphorylase in Cancer Development and Chemotherapy. Med. Res. Rev. 2009, 29, 903–953. [Google Scholar] [CrossRef]
- Mitsiki, E.; Papageorgiou, A.C.; Iyer, S.; Thiyagarajan, N.; Prior, S.H.; Sleep, D.; Finnis, C.; Acharya, K.R. Structures of Native Human Thymidine Phosphorylase and in Complex with 5-Iodouracil. Biochem. Biophys. Res. Commun. 2009, 386, 666–670. [Google Scholar] [CrossRef]
- Focher, F.; Spadari, S. Thymidine Phosphorylase: A Two-Face Janus in Anticancer Chemotherapy. Curr. Cancer Drug. Targets 2001, 1, 141–153. [Google Scholar] [CrossRef]
- Almasmoum, H. Characterization of Mucin 2 Expression in Colorectal Cancer with and without Chemotherapies, in Vivo and in Vitro Study. JUQUMS 2021, 7, 18–22. [Google Scholar] [CrossRef]
- Mayer, R.J.; Van Cutsem, E.; Falcone, A.; Yoshino, T.; Garcia-Carbonero, R.; Mizunuma, N.; Yamazaki, K.; Shimada, Y.; Tabernero, J.; Komatsu, Y.; et al. Randomized Trial of TAS-102 for Refractory Metastatic Colorectal Cancer. N. Engl. J. Med. 2015, 372, 1909–1919. [Google Scholar] [CrossRef]
- Yano, S.; Kazuno, H.; Sato, T.; Suzuki, N.; Emura, T.; Wierzba, K.; Yamashita, J.; Tada, Y.; Yamada, Y.; Fukushima, M.; et al. Synthesis and Evaluation of 6-Methylene-Bridged Uracil Derivatives. Part 2: Optimization of Inhibitors of Human Thymidine Phosphorylase and Their Selectivity with Uridine Phosphorylase. Bioorg Med. Chem. 2004, 12, 3443–3450. [Google Scholar] [CrossRef]
- Balzarini, J.; Gamboa, A.E.; Esnouf, R.; Liekens, S.; Neyts, J.; De Clercq, E.; Camarasa, M.J.; Pérez-Pérez, M.J. 7-Deazaxanthine, a Novel Prototype Inhibitor of Thymidine Phosphorylase. FEBS Lett. 1998, 438, 91–95. [Google Scholar] [CrossRef]
- Sun, L.; Li, J.; Bera, H.; Dolzhenko, A.V.; Chiu, G.N.C.; Chui, W.K. Fragment-Based Approach to the Design of 5-Chlorouracil-Linked-Pyrazolo [1,5-a][1,3,5]Triazines as Thymidine Phosphorylase Inhibitors. Eur. J. Med. Chem. 2013, 70, 400–410. [Google Scholar] [CrossRef]
- Bera, H.; Tan, B.J.; Sun, L.; Dolzhenko, A.V.; Chui, W.-K.; Chiu, G.N.C. A Structure-Activity Relationship Study of 1,2,4-Triazolo [1,5-a][1,3,5]Triazin-5,7-Dione and Its 5-Thioxo Analogues on Anti-Thymidine Phosphorylase and Associated Anti-Angiogenic Activities. Eur. J. Med. Chem. 2013, 67, 325–334. [Google Scholar] [CrossRef]
- Bera, H.; Chui, W.-K.; Gupta, S.D.; Dolzhenko, A.V.; Sun, L. Synthesis, in Vitro Evaluation of Thymidine Phosphorylase Inhibitory Activity, and in Silico Study of 1,3,5-Triazin-2,4-Dione and Its Fused Analogues. Med. Chem. Res. 2013, 22, 6010–6021. [Google Scholar] [CrossRef]
- Dorababu, A. Evolution of Uracil Based Thymidine Phosphorylase Inhibitors, SAR and Electronic Correlation: Revisit. Drug. Dev. Res. 2019, 80, 893–920. [Google Scholar] [CrossRef]
- Ali, F.; Khan, K.M.; Salar, U.; Iqbal, S.; Taha, M.; Ismail, N.H.; Perveen, S.; Wadood, A.; Ghufran, M.; Ali, B. Dihydropyrimidones: As Novel Class of β-Glucuronidase Inhibitors. Bioorganic Med. Chem. 2016, 24, 3624–3635. [Google Scholar] [CrossRef]
- Dimas, K.; Demetzos, C.; Marsellos, M.; Sotiriadou, R.; Malamas, M.; Kokkinopoulos, D. Cytotoxic Activity of Labdane Type Diterpenes against Human Leukemic Cell Lines in Vitro. Planta Med. 1998, 64, 208–211. [Google Scholar] [CrossRef]
- Abd Alhameed, R.; Almarhoon, Z.; Sholkamy, E.N.; Ali Khan, S.; Ul-Haq, Z.; Sharma, A.; de la Torre, B.G.; Albericio, F.; El-Faham, A. Novel 4,6-Disubstituted s-Triazin-2-Yl Amino Acid Derivatives as Promising Antifungal Agents. J. Fungi 2020, 6, 237. [Google Scholar] [CrossRef]
- Aldarhami, A.; Bazaid, A.S.; Alhamed, A.S.; Alghaith, A.F.; Ahamad, S.R.; Alassmrry, Y.A.; Alharazi, T.; Snoussi, M.; Qanash, H.; Alamri, A.; et al. Antimicrobial Potential of Pithecellobium dulce Seed Extract against Pathogenic Bacteria: In Silico and In Vitro Evaluation. BioMed. Res. Int. 2023, 2023, e2848198. [Google Scholar] [CrossRef]
- Alfaifi, G.H.; Farghaly, T.A.; Abdellattif, M.H. Indenyl-Thiazole and Indenyl-Formazan Derivatives: Synthesis, Anticancer Screening Studies, Molecular-Docking, and Pharmacokinetic/ Molin-Spiration Properties. PLoS ONE 2023, 18, e0274459. [Google Scholar] [CrossRef]
- Bronckaers, A.; Aguado, L.; Negri, A.; Camarasa, M.-J.; Balzarini, J.; Pérez-Pérez, M.-J.; Gago, F.; Liekens, S. Identification of Aspartic Acid-203 in Human Thymidine Phosphorylase as an Important Residue for Both Catalysis and Non-Competitive Inhibition by the Small Molecule “Crystallization Chaperone” 5′-O-Tritylinosine (KIN59). Biochem. Pharmacol. 2009, 78, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Hosea, N.A.; Jones, H.M. Predicting Pharmacokinetic Profiles Using in Silico Derived Parameters. Mol. Pharm. 2013, 10, 1207–1215. [Google Scholar] [CrossRef] [PubMed]
- Bakhdar, F. The Role of CYP 450 Isozymes in Drug-Drug Interaction. JUQUMS 2020, 6, 36–40. [Google Scholar] [CrossRef]
- Bera, H.; Dolzhenko, A.V.; Sun, L.; Dutta Gupta, S.; Chui, W.-K. Synthesis and in Vitro Evaluation of 1,2,4-Triazolo [1,5-a][1,3,5]Triazine Derivatives as Thymidine Phosphorylase Inhibitors. Chem. Biol. Drug. Des. 2013, 82, 351–360. [Google Scholar] [CrossRef]
- Iqubal, S.M.S. Review on Kinetic Studies of α-Hydroxy Acids (Glycolic, Mandelic, Citric, Tartaric and Malic) and Some Other Organic Compounds with Water Soluble Nano Particles of Colloidal MnO2 in Absence and Presence of Non-Ionic Surfactant (TX-100). J. Umm. Al-Qura. Univ. Appll. Sci. 2022, 8, 79–84. [Google Scholar] [CrossRef]
- Whiteley, C.G. Mechanistic and Kinetic Studies of Inhibition of Enzymes. Cell Biochem. Biophys. 2000, 33, 217–225. [Google Scholar] [CrossRef]
- Vega-Avila, E.; Pugsley, M.K. An Overview of Colorimetric Assay Methods Used to Assess Survival or Proliferation of Mammalian Cells. Proc. West. Pharmacol. Soc. 2011, 54, 10–14. [Google Scholar]
- Timofeev, V.I.; Abramchik, Y.A.; Fateev, I.V.; Zhukhlistova, N.E.; Murav’eva, T.I.; Kuranova, I.P.; Esipov, R.S. Three-Dimensional Structure of Thymidine Phosphorylase from E. Coli in Complex with 3′-Azido-2′-Fluoro-2′,3′-Dideoxyuridine. Crystallogr. Rep. 2013, 58, 842–853. [Google Scholar] [CrossRef]
- Uddin, I.; Taha, M.; Rahim, F.; Wadood, A. Synthesis and Molecular Docking Study of Piperazine Derivatives as Potent Inhibitor of Thymidine Phosphorylase. Bioorganic Chem. 2018, 78, 324–331. [Google Scholar] [CrossRef]
- Goddard, T.D.; Huang, C.C.; Ferrin, T.E. Software Extensions to UCSF Chimera for Interactive Visualization of Large Molecular Assemblies. Structure 2005, 13, 473–482. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Shahzad, S.A.; Yar, M.; Bajda, M.; Jadoon, B.; Khan, Z.A.; Naqvi, S.A.R.; Shaikh, A.J.; Hayat, K.; Mahmmod, A.; Mahmood, N.; et al. Synthesis and Biological Evaluation of Novel Oxadiazole Derivatives: A New Class of Thymidine Phosphorylase Inhibitors as Potential Anti-Tumor Agents. Bioorg. Med. Chem. 2014, 22, 1008–1015. [Google Scholar] [CrossRef]
- Almandil, N.B.; Taha, M.; Farooq, R.K.; Alhibshi, A.; Ibrahim, M.; Anouar, E.H.; Gollapalli, M.; Rahim, F.; Nawaz, M.; Shah, S.A.A.; et al. Synthesis of Thymidine Phosphorylase Inhibitor Based on Quinoxaline Derivatives and Their Molecular Docking Study. Molecules 2019, 24, 1002. [Google Scholar] [CrossRef]
- Liekens, S.; Hernández, A.-I.; Ribatti, D.; De Clercq, E.; Camarasa, M.-J.; Pérez-Pérez, M.-J.; Balzarini, J. The Nucleoside Derivative 5′-O-Trityl-Inosine (KIN59) Suppresses Thymidine Phosphorylase-Triggered Angiogenesis via a Noncompetitive Mechanism of Action*. J. Biol. Chem. 2004, 279, 29598–29605. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||
|---|---|---|---|---|---|
| Compound Number | R | Thymidine Phosphorylase Inhibitory Activity | Cytotoxicity Studies | ||
| % Inhibition | IC50 ± SEM (µM) | Cell Viability (%) | IC50 ± SD (µM) | ||
| 1 |  Methyl 6-methyl-2-oxo-4-phenyl-1,2,3,4-tetrahydropyrimidine-5-carboxylate | 84.0 | 314.3 ± 0.9 | 57 | NC |
| 2 |  Methyl 4-(4-ethoxyphenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate | 78.2 | 389.2 ± 6.2 | 67 | NC |
| 3 |  Methyl 6-methyl-4-(naphthalen-2-yl)-2-oxo- 1,2,3,4-tetrahydropyrimidine-5-carboxylate | 38.1 | N/A | NC | NC |
| 4 |  Methyl 4-ethyl-6-methyl-2-oxo-1,2,3,4- tetrahydropyrimidine-5-carboxylate | 46.8 | N/A | NC | NC |
| 5 |  Methyl 4-(4-methoxyphenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate | 52.9 | 387.4 ± 2.0 | 72 | NC |
| 6 |  Methyl 4-(2-chlorophenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate | −65.3 | N/A | NC | NC |
| 7 |  Methyl 4-(2-methoxyphenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate | −19.3 | N/A | NC | NC |
| 8 |  Methyl 6-methyl-4-(naphthalen-2-yl)-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate | −10.3 | N/A | NC | NC |
| 9 |  Methyl 4-(furan-2-yl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate | 78.9 | 373.6 ± 2.2 | 72 | NC |
| 10 |  Methyl 4-butyl-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate | 92.7 | 389.0 ± 0.6 | 66 | NC |
| 11 |  Methyl 6-methyl-4-(5-methylfuran-2-yl)-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate | 76.9 | 386.0 ± 0.6 | 77 | NC |
| 12 |  Methyl 6-methyl-4-(4-nitrophenyl)-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate | 91.5 | 303.5 ± 0.4 | 57 | NC |
| 13 |  Methyl 4-(4-hydroxyphenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate | 56.2 | 448.9 ± 4.1 | 72 | NC |
| 14 |  Methyl 4-(2-fluoro-4-methoxyphenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate | 68.4 | 397.1 ± 0.1 | 71 | NC |
| 15 |  Methyl 4-(3-hydroxyphenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate | 38.9 | NA | NC | NC |
| 16 |  Methyl 4-(4-fluoro-3-methoxyphenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate | 47.8 | NA | NC | NC |
| 17 |  Methyl 4-(2,5-dimethoxyphenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate | 46.5 | NA | NC | NC |
| 18 |  Methyl 4-(2,6-dichlorophenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate | −15.4 | NA | NC | NC |
| 19 |  Methyl 4-(4-isopropylphenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate | −18.5 | NA | NC | NC |
| 20 |  Methyl 4-(2,3-dimethoxyphenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate | 67.8 | 414.7 ± 1.6 | 62 | NC |
| 21 |  Methyl 4-isobutyl-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate | 90.7 | 345.4 ± 0.5 | 55 | NC |
| 22 |  Methyl 4-(3-bromo-4,5-dimethoxyphenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate | 61.9 | 394.3 ± 4.3 | 76 | NC |
| 23 |  Methyl 4-(3-(benzyloxy)-4-methoxyphenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate | 7.5 | NA | NC | NC |
| 24 |  Methyl 6-methyl-2-oxo-4-(thiophen-3-yl)-1,2,3,4-tetrahydropyrimidine-5-carboxylate | 79.6 | 350.6 ± 0.6 | 82 | NC |
| 25 |  Methyl 4-(3,4-dimethoxyphenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate | 59.0 | 424.1 ± 0.9 | 99 | NC |
| 26 |  Methyl 4-(4-(dimethylamino) phenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate | 92.3 | 349.9 ± 3.7 | 57 | NC |
| 27 |  Methyl 6-methyl-2-oxo-4-(thiophen-2-yl)-1,2,3,4-tetrahydropyrimidine-5-carboxylate | 47.1 | NA | NC | NC |
| 28 |  Methyl 4-(3-chloro-4-hydroxyphenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate | 67.5 | 404.6 ± 1.0 | 95 | NC |
| 29 |  Methyl 4-(4-acetoxy-3-methoxyphenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate | 76.0 | 400.5 ± 0.6 | 80 | NC |
| 30 |  Methyl 4-(4-hydroxy-3,5-dimethoxyphenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate | 69.6 | 396.7 ± 1.5 | 75 | NC |
| 31 |  Methyl 4-(3-(benzyloxy) phenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate | −41.9 | NA | NC | NC |
| 32 |  Methyl 6-methyl-4-(5-methylthiophen-2-yl)-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate | 58.3 | 485.7 ± 1.5 | 85 | NC |
| 33 |  Methyl 6-methyl-2-oxo-4-(4-(trifluoromethyl) phenyl)-1,2,3,4-tetrahydropyrimidine-5-carboxylate | 73.6 | 322.6 ± 1.6 | 61 | NC |
| 34 |  Methyl 4-(4-chlorophenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate | 66.3 | 443.9 ± 0.9 | 72 | NC |
| 35 |  Methyl 4-(2-fluorophenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate | 9.6 | NA | NC | NC |
| 36 |  Methyl 4-(2-(benzyloxy) phenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate | 5.3 | NA | NC | NC |
| 37 |  Methyl 4-(4-bromophenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate | 15.7 | NA | NC | NC |
| 38 |  Methyl 6-methyl-4-(3-nitrophenyl)-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate | −23.1 | NA | NC | NC |
| 39 |  Methyl 4-(2,4-dichlorophenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate | −1.8 | NA | NC | NC |
| 40 |  Methyl 4-(4-(benzyloxy) phenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate | −273.6 | NA | NC | NC |
| Standard* |  7-Deazaxanthine | 82.0 | 41.0 ± 1.6 | NC | NC |
| Standard* |  Tipiracil-HCl | 92.0 | 0.014 ± 0.04 | NC | NC |
| Standard** |  Cycloheximide | ND | ND | 30 | 0.8 ± 0.2 |
| Compound | Ki ± SEM (µM) | Type of Inhibition |
|---|---|---|
| 1 | 326.3 ± 0.002 | Non-Competitive |
| 12 | 297.6 ± 0.006 | Non-Competitive |
| 33 | 311.4 ± 0.004 | Non-Competitive |
| 7-deazaxanthine | 45.0 ± 0.003 | Non-Competitive |
| Compound | Docking Score (kcal/mol) | Type of Interactions | |
|---|---|---|---|
| Hydrophobic Interactions | Hydrogen Bonding | ||
| 1 | −6.19 | Gly116, Gly122, Asp125, Lys126, Asn252, Arg370, Val363, and Val364 | Gly116, Gly122, Asp125, Lys126, and Asn252 |
| 12 | −5.60 | Gly116, Leu117, Gly122, Asp125, Asn252, Gly360, Arg370, and Val363 | Asn252 and Gly360 |
| 33 | −5.11 | Gly116, Gly121, Lys126, Asn252, Val363, and Asp370 | Gly89, Gly91, Asn252, and Gly360 |
| 7-deazaxanthine | −5.23 | Gly116 and Gly122 | Gly116, Gly122, Asp125, Asn252, and Arg370 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cui, T.-M.; Altaf, M.; Aldarhami, A.; Bazaid, A.S.; Saeedi, N.H.; Alkayyal, A.A.; Alshabrmi, F.M.; Ali, F.; Aladhadh, M.; Khan, M.Y.; et al. Dihydropyrimidone Derivatives as Thymidine Phosphorylase Inhibitors: Inhibition Kinetics, Cytotoxicity, and Molecular Docking. Molecules 2023, 28, 3634. https://doi.org/10.3390/molecules28083634
Cui T-M, Altaf M, Aldarhami A, Bazaid AS, Saeedi NH, Alkayyal AA, Alshabrmi FM, Ali F, Aladhadh M, Khan MY, et al. Dihydropyrimidone Derivatives as Thymidine Phosphorylase Inhibitors: Inhibition Kinetics, Cytotoxicity, and Molecular Docking. Molecules. 2023; 28(8):3634. https://doi.org/10.3390/molecules28083634
Chicago/Turabian StyleCui, Tian-Meng, Muhammad Altaf, Abdu Aldarhami, Abdulrahman S. Bazaid, Nizar H. Saeedi, Almohanad A. Alkayyal, Fahad M. Alshabrmi, Farman Ali, Mohammed Aladhadh, Muhammad Yasir Khan, and et al. 2023. "Dihydropyrimidone Derivatives as Thymidine Phosphorylase Inhibitors: Inhibition Kinetics, Cytotoxicity, and Molecular Docking" Molecules 28, no. 8: 3634. https://doi.org/10.3390/molecules28083634