


Effective Synthesis of 4-Quinolones by Reductive Cyclization of 2′-Nitrochalcones Using Formic Acid as a CO Surrogate


Abstract
:1. Introduction
2. Results and Discussion
2.1. Optimization of the Reaction Conditions
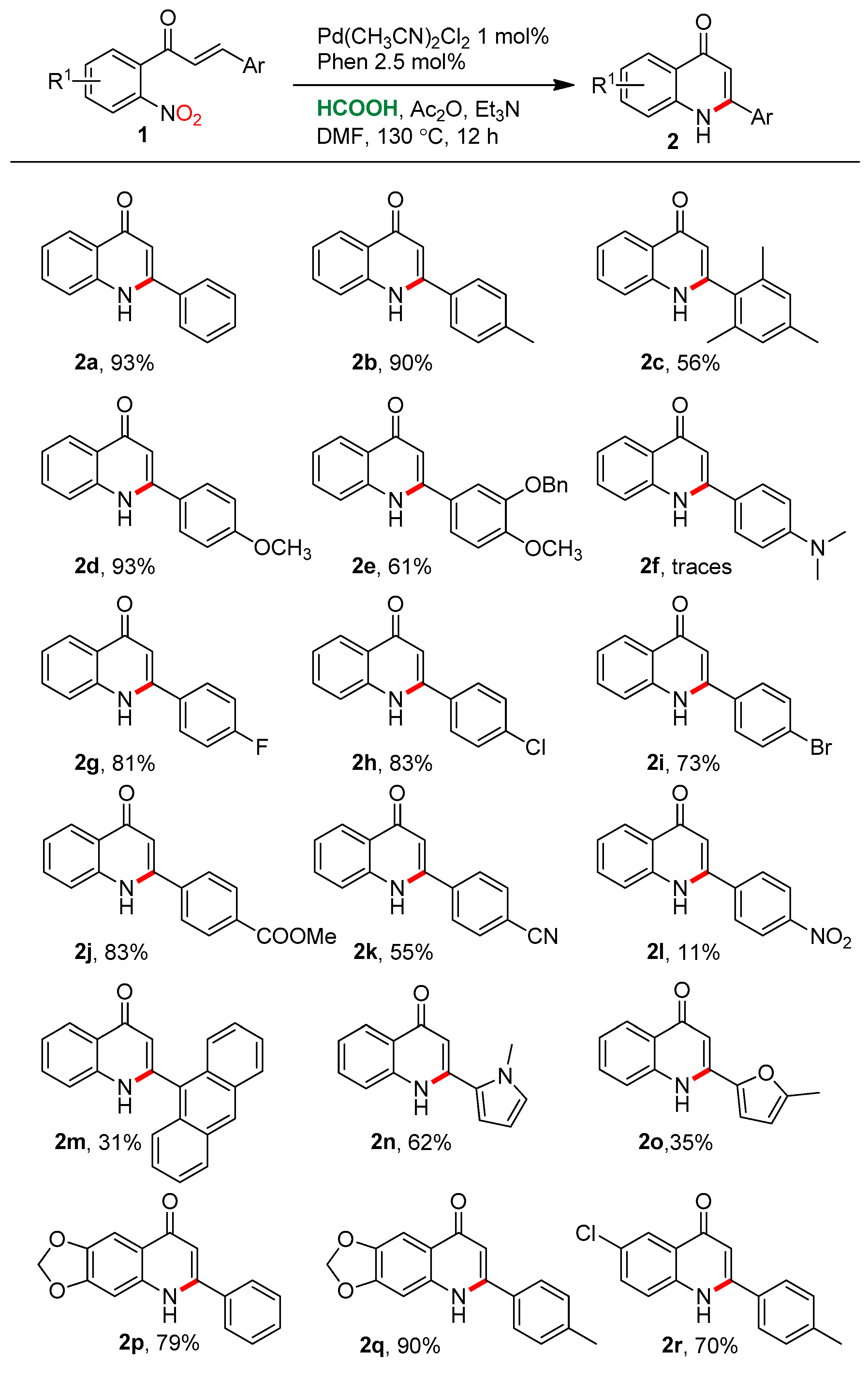
2.2. Substrate Scope
2.3. Synthesis of Graveoline
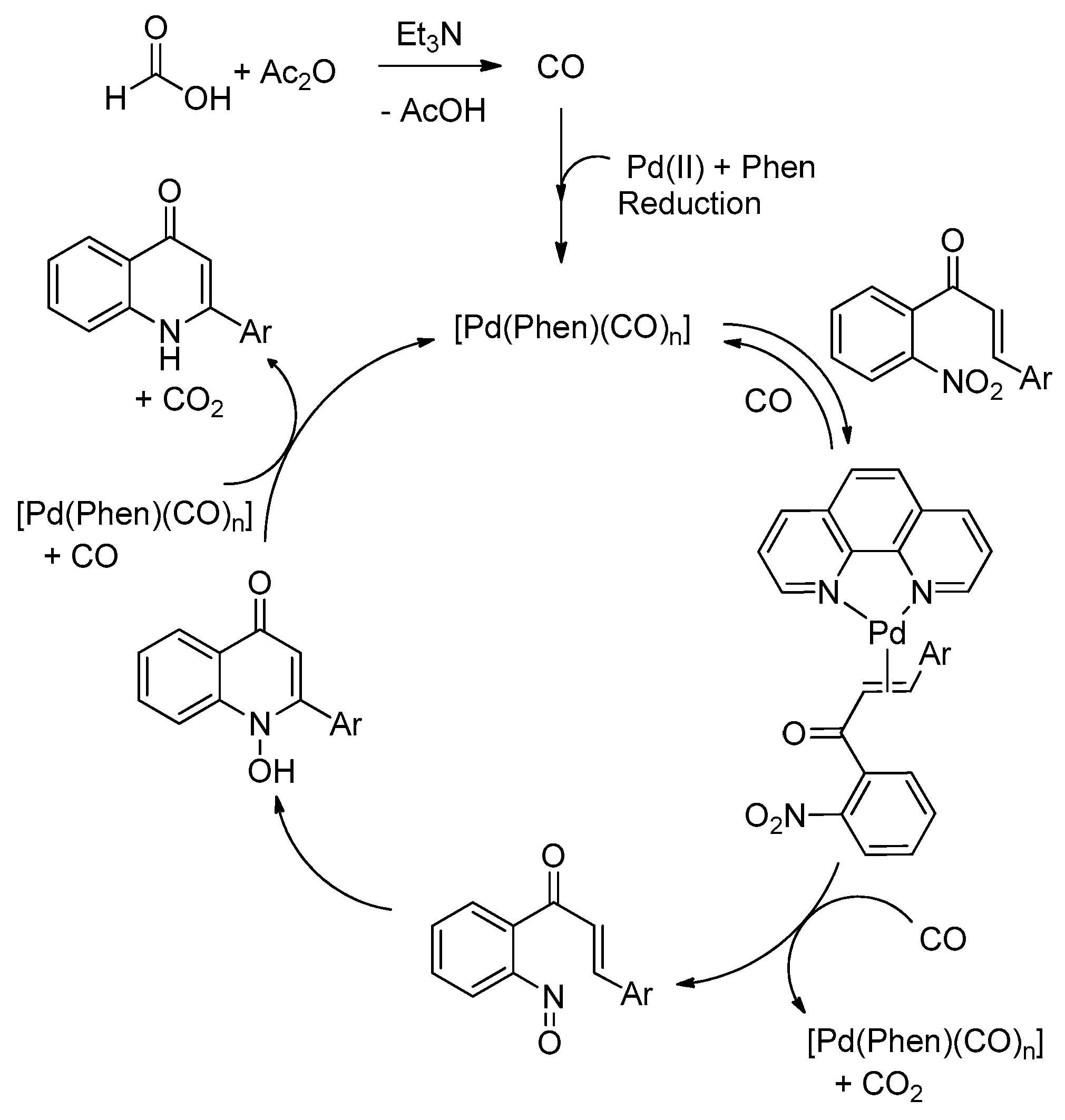
2.4. Reaction Mechanism
3. Materials and Methods
3.1. General Procedures
3.2. Catalytic Reactions
3.3. Procedure for Gram-Scale Reaction
3.4. Characterization Data for Quinolones
- 2-Phenylquinolin-4(1H)-one (2a) [70]. White solid (103 mg, 93% yield). 1H NMR (400 MHz, DMSO-d6) δ 11.70 (br, 1H), 8.11 (d, J = 8.0 Hz, 1H), 7.87–7.80 (m, 2H), 7.77 (d, J = 8.3 Hz, 1H), 7.67 (t, J = 7.6 Hz, 1H), 7.61–7.57 (m, 3H), 7.34 (t, J = 7.5 Hz, 1H), 6.33 (s, 1H) ppm. 13C NMR (100 MHz, DMSO-d6) δ 176.9, 150.0, 140.5, 134.2, 131.8, 130.4, 129.0, 127.4, 124.9, 124.7, 123.2, 118.7, 107.3 ppm.
- 2-(4-Methylphenyl)quinolin-4(1H)-one (2b) [71]. Pale orange solid (106 mg, 90% yield). 1H NMR (DMSO-d6, 400 MHz): δ 11.63 (br, 1H), 8.10 (dd, J = 8.1, 1.2 Hz, 1H), 7.77 (d, J = 8.4 Hz, 1H), 7.74 (d, J = 8.0 Hz, 2H), 7.66 (ddd, J = 8.4, 7.0, 1.5 Hz, 1H), 7.39 (d, J = 8.0 Hz, 2H), 7.33 (t, J = 7.4 Hz, 1H), 6.32 (s, 1H), 2.40 (s, 3H) ppm. 13C NMR (DMSO-d6, 100 MHz): δ 176.9, 149.9, 140.5, 140.3, 131.7, 131.3, 129.5, 127.2, 124.8, 124.7, 123.1, 118.7, 106.9, 20.9 ppm.
- 2-(2,4,6-trimethylphenyl)quinolin-4(1H)-one (2c) White solid (74 mg, 56%yield). 1H NMR (DMSO-d6, 400 MHz): δ 11.74 (br, 1H), 8.23–8.04 (m, 1H), 7.64 (ddd, J = 8.4, 7.0, 1.5 Hz, 1H), 7.56 (d, J = 8.0 Hz, 1H), 7.38–7.29 (m, 1H), 7.02 (s, 2H), 2.30 (s, 3H), 2.13 (s, 6H) ppm. 13C NMR (DMSO-d6, 100 MHz): δ 176.7, 149.8, 140.4, 138.4, 135.6, 132.1, 131.6, 128.1, 124.85, 124.81, 123.1, 118.3, 109.6, 20.6, 19.3 ppm. Elemental analysis for C18H17NO Calcd.: C, 81.10; H, 6.51; N, 5.32%. Found: C, 80.75; H, 6.73; N, 5.10%.
- 2-(4-Methoxyphenyl)quinolin-4(1H)-one (2d) [71] Off-white solid (117 mg, 93% yield). 1H NMR (DMSO-d6, 400 MHz): δ 11.62 (br, 1H), 8.10 (d, J = 7.7 Hz, 1H), 7.87–7.75 (m, 3H), 7.65 (t, J = 7.2 Hz, 1H), 7.32 (t, J = 7.0 Hz, 1H), 7.12 (d, J = 8.1 Hz, 2H), 6.32 (s, 1H), 3.84 (s, 3H) ppm. 13C NMR (DMSO-d6, 100 MHz): δ 176.8, 161.0, 149.7, 140.5, 131.6, 128.8, 126.2, 124.8, 124.7, 123.1, 118.6, 114.4, 106.5, 55.4 ppm.
- 2-(3-Benzyloxy-4-methoxy-phenyl)quinolin-4(1H)-one (2e) Pale pink solid (109 mg, 61% yield). 1H NMR (DMSO-d6, 400 MHz): δ 11.59 (br, 1H), 8.10 (dd, J = 8.0, 1.1 Hz, 1H), 7.78 (d, J = 8.2 Hz, 1H), 7.69–7.64 (m, 1H), 7.55–7.39 (m, 6H), 7.37–7.29 (m, 2H), 7.18 (d, J = 8.5 Hz, 1H), 6.38 (s, 1H), 5.23 (s, 2H), 3.86 (s, 3H) ppm. 13C NMR (DMSO-d6, 100 MHz): δ 176.8, 151.1, 149.8, 147.9, 140.5, 136.9, 131.7, 128.5, 127.94, 127.88, 126.4, 124.73, 124.67, 123.2, 120.5, 118.7, 112.6, 112.1, 106.6, 70.2, 55.8 ppm. Elemental analysis for C23H19NO3 Calcd.: C, 77.29; H, 5.36; N, 3.36 %. Found: C, 77.02; H, 5.08; N, 2.97%.
- 2-(4-Fluorophenyl)quinolin-4(1H)-one (2g) [71] Golden yellow solid (97 mg, 81% yield). 1H NMR (DMSO-d6, 400 MHz): δ 11.71 (br, 1H), 8.10 (ddd, J = 8.1, 1.5, 0.5 Hz, 1H), 7.91 (dd, J = 8.7, 5.6 Hz, 2H), 7.75 (d, J = 8.4 Hz, 1H), 7.67 (ddd, J = 8.4, 6.9, 1.5 Hz, 1H), 7.47–7.40 (m, 2H), 7.34 (ddd, J = 8.1, 7.0, 1.3 Hz, 1H), 6.33 (br, 1H)ppm. 13C NMR (DMSO-d6, 100 MHz): δ 176.9, 163.4 (d 1JC-F, 248.0 Hz), 149.0, 140.5, 131.8, 129.9 (d 3JC-F, 8.7 Hz), 124.7, 123.3, 118.7, 116.0 (d 2JC-F, 21.8 Hz), 107.4 ppm.
- 2-(4-Chlorophenyl)quinolin-4(1H)-one (2h) [72]. Off-white solid (106 mg, 83% yield). 1H NMR (DMSO-d6, 400 MHz): δ 11.72 (br, 1H), 8.10 (d, J = 7.9 Hz, 1H), 7.87 (d, J = 8.2 Hz, 2H), 7.80–7.62 (m, 4H), 7.34 (t, J = 7.3 Hz, 1H), 6.35 (br, 1H) ppm. 13C NMR (DMSO-d6, 100 MHz): 176.9, 148.8, 140.5, 135.2, 131.9, 129.3, 129.0, 124.7, 123.4, 118.8, 107.5 ppm.
- 2-(4-Bromophenyl)quinolin-4(1H)-one (2i) [73]. Pink solid (109 mg, 73% yield). 1H NMR (DMSO-d6, 400 MHz): δ 11.72 (s, 1H), 8.10 (d, J = 7.5 Hz, 1H), 7.80 (s, 4H), 7.75 (d, J = 8.3 Hz, 1H), 7.69 (dd, J = 11.0, 4.1 Hz, 1H), 7.35 (t, J = 7.4 Hz, 1H) ppm. 13C NMR (DMSO-d6, 100 MHz): 177.0, 148.9, 140.5, 133.3, 131.9, 129.5, 124.9, 124.7, 124.0, 123.4, 118.7, 107.5 ppm.
- 2-(4-Carbomethoxyphenyl)quinolin-4(1H)-one (2j) [74]. Pink solid (116 mg, 83% yield). 1H NMR (DMSO-d6, 400 MHz): δ 11.85 (br, 1H), 8.18–8.08 (m, 2H), 8.00 (d, J = 7.8 Hz, 2H), 7.79 (d, J = 8.3 Hz, 1H), 7.70 (t, J = 7.1 Hz, 1H), 7.36 (t, J = 7.4 Hz, 1H), 6.41 (br, 1H), 3.92 (s, 3H) ppm. 13C NMR (DMSO-d6, 100 MHz): 176.9, 165.7, 148.7, 140.5, 138.5, 132.0, 131.1, 129.6, 127.9, 124.9, 124.7, 123.4, 118.8, 108.0, 52.4 ppm.
- 2-(4-Cyanophenyl)quinolin-4(1H)-one (2k) [75]. Light orange solid (68 mg, 55% yield). 1H NMR (DMSO-d6, 400 MHz): δ 11.84 (br, 1H), 8.12 (d, J = 7.9 Hz, 1H), 8.09–8.02 (m, 4H), 7.77 (d, J = 8.3 Hz, 1H), 7.70 (t, J = 7.2 Hz, 1H), 7.37 (t, J = 7.4 Hz, 1H), 6.45 (br, 1H) ppm. 13C NMR (DMSO-d6, 100 MHz): δ 132.8, 132.0, 128.4, 124.6, 123.6, 118.4, 112.8 ppm. Due to the low solubility of the compound in DMSO-d6, no quaternary carbon was detected.
- 2-(4-Nitrophenyl)quinolin-4(1H)-one (2l) [76]. Purified by column chromatography using hexane: EtOAc (7:3). Red solid (14 mg, 11% yield). 1H NMR (DMSO-d6, 400 MHz): δ 10.17 (br, 1H), 8.27 (d, J = 8.7 Hz, 2H), 8.01 (dd, J = 49.0, 8.6 Hz, 2H), 7.62 (d, J = 7.6 Hz, 1H), 7.54 (dt, J = 14.2, 6.8 Hz, 1H), 7.16 (d, J = 8.0 Hz, 1H), 6.98 (t, J = 7.4 Hz, 1H), 6.67 (s, 1H) ppm. 13C NMR (DMSO-d6, 100 MHz): δ 186.6, 154.3, 146.0, 141.3, 137.0, 136.3, 130.4, 124.5, 123.9, 120.5, 119.7, 112.7, 106.1 ppm.
- 2-(Anthracen-9-yl)quinolin-4(1H)-one (2m) [75]. Tan solid (50 mg, 31%yield). 1H NMR (DMSO-d6, 400 MHz): δ 12.22 (br, 1H), 8.84 (s, 1H), 8.28 (dd, J = 8.1, 1.2 Hz, 1H), 8.24–8.19 (m, 2H), 7.80 (d, J = 8.3 Hz, 2H), 7.72 (ddd, J = 8.4, 7.1, 1.5 Hz, 1H), 7.64–7.52 (m, 5H), 7.47–7.41 (m, 1H), 6.20 (s, 1H) ppm. 13C NMR (DMSO-d6, 100 MHz):176.7, 147.9, 140.7, 132.0, 130.6, 129.1, 128.73, 128.74 128.64, 128.57, 127.2, 125.7, 125.3, 125.0, 123.5, 118.5, 112.1 ppm.
- 2-(1-Methyl-1H-pyrrol-2-yl)quinolin-4(1H)-one (2n) Purified by column chromatography using hexane: EtOAc (6:4). Dark solid (70 mg, 62% yield). 1H NMR (DMSO-d6, 400 MHz): δ 9.31 (br, 1H), 7.55 (d, J = 7.6 Hz, 1H), 7.47 (ddd, J = 8.3, 7.3, 1.3 Hz, 1H), 7.15 (d, J = 8.2 Hz, 1H), 7.06 (dd, J = 2.3, 1.6 Hz, 1H), 6.96 (ddd, J = 3.9, 1.6, 0.6 Hz, 1H), 6.88 (ddd, J = 7.6, 7.1, 0.7 Hz, 1H), 6.66 (br, 1H), 6.26 (ddd, J = 3.9, 2.6, 0.6 Hz, 1H), 3.74 (s, 3H) ppm. 13C NMR (DMSO-d6, 100 MHz): δ 184.8, 152.7, 135.3, 130.9, 127.6, 127.3, 123.6, 120.6, 119.2, 113.7, 112.5, 109.5, 99.8, 33.7 ppm. Elemental analysis for C14H12N2O Calcd.: C, 74.98; H, 5.39; N, 12.49%. Found: C, 75.34; H, 5.06; N, 12.67%
- 2-(5-Methylfuran-2-yl)quinolin-4(1H)-one (2o) Purified by column chromatography using hexane: EtOAc (8:2). Orange solid (40 mg, 35% yield). 1H NMR (CDCl3, 400 MHz): δ 7.70 (d, J = 7.7 Hz, 1H), 7.62 (br, 1H), 7.47–7.33 (m, 1H), 6.99 (d, J = 8.1 Hz, 1H), 6.94–6.84 (m, 1H), 6.60 (s, 1H), 6.53 (d, J = 3.3 Hz, 1H), 6.16–6.10 (m, 1H), 2.43 (s, 3H) ppm. 13C NMR (CDCl3, 100 MHz): δ 185.8, 155.1, 152.0, 150.6, 135.8, 132.4, 124.8, 121.5, 120.0, 116.5, 111.5, 109.6, 99.0, 14.4 ppm. Elemental analysis for C14H11NO2 Calcd.: C, 74.65; H, 4.92; N, 6.22%. Found: C, 74.27; H, 5.11; N, 5.97%.
- 6-Phenyl-[1,3]dioxolo[4,5-g]quinolin-8(5H)-one (2p) [77]. Pink solid (105 mg, 79% yield). 1H NMR (DMSO-d6, 400 MHz): δ 11.58 (br, 1H), 7.80 (br, 3H), 7.57 (m, 3H), 7.40 (s, 1H), 7.20 (s, 1H), 6.27 (br, 1H), 6.15 (s, 3H) ppm. 13C NMR (DMSO-d6, 100 MHz): δ 175.8, 151.1, 148.5, 145.20, 137.4, 134.2, 130.2, 129.0, 127.2, 120.4, 106.5, 101.9, 101.2, 97.2 ppm.
- 6-(4-Methylphenyl)-[1,3]dioxolo[4,5-g]quinolin-8(5H)-one (2q) [78]. Pink solid (126 mg, 90% yield). 1H NMR (DMSO-d6, 400 MHz): δ 11.51 (s, 1H), 7.70 (d, J = 8.2 Hz, 2H), 7.38 (m, 3H), 7.21 (s, 1H), 6.24 (d, J = 1.8 Hz, 1H), 6.15 (s, 2H), 2.40 (s, 3H) ppm. 13C NMR (DMSO-d6, 100 MHz): δ 175.7, 151.0, 148.5, 145.0, 140.1, 137.4, 131.2, 129.5, 127.0, 120.4, 106.2, 101.9, 101.2, 97.2, 20.9 ppm.
- 6-Chloro-2-(4-methylphenyl)quinolin-4(1H)-one (2r) Pink solid (95 mg, 70% yield). 1H NMR (DMSO-d6, 400 MHz): δ 11.81 (br, 1H), 8.03 (d, J = 2.4 Hz, 1H), 7.81 (d, J = 8.9 Hz, 1H), 7.75 (d, J = 8.0 Hz, 2H), 7.71 (dd, J = 8.9, 2.4 Hz, 1H), 7.40 (d, J = 8.0 Hz, 2H), 6.38 (br, 1H), 2.41 (s, 3H) ppm. 13C NMR (DMSO-d6, 100 MHz): δ 150.3, 140.5, 131.8, 131.0, 129.57, 127.8, 127.25, 123.58, 107.00, 20.89 ppm. Due to the low solubility of the compound in DMSO, four quaternary carbons were not detected. Elemental analysis for C16H12ClNO Calcd.: C, 71.25; H, 4.48; N, 5.19%. Found: C, 71.04; H, 4.66; N, 4.93%
- 2-(Benzo[d][1,3]dioxol-5-yl)quinolin-4(1H)-one (2s) [72]. Colorless solid (98 mg, 74% yield). 1H NMR (DMSO-d6, 400 MHz): δ 11.56 (br, 1H), 8.08 (dd, J = 8.0, 1.0 Hz, 1H), 7.75 (d, J = 8.2 Hz, 1H), 7.70–7.62 (m, 1H), 7.42 (d, J = 1.6 Hz, 1H), 7.37 (dd, J = 8.1, 1.7 Hz, 1H), 7.32 (t, J = 7.3 Hz, 1H), 7.11 (d, J = 8.1 Hz, 1H), 6.30 (s, 1H), 6.14 (s, 2H) ppm. 13C NMR (DMSO-d6, 100 MHz): δ 176.9, 149.5, 149.2, 147.9, 140.4, 131.7, 128.0, 124.8, 124.7, 123.1, 121.8, 118.6, 108.7, 107.6, 106.8, 101.8 ppm.
3.5. Synthesis of Graveoline from 2s
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Gao, F.; Zhang, X.; Wang, T.; Xiao, J. Quinolone hybrids and their anti-cancer activities: An overview. Eur. J. Med. Chem. 2019, 165, 59–79. [Google Scholar] [CrossRef]
- Pham, T.D.M.; Ziora, Z.M.; Blaskovich, M.A.T. Quinolone antibiotics. MedChemComm 2019, 10, 1719–1739. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, S.; Ba, Y.; Xu, Z. 1,2,4-Triazole-quinoline/quinolone hybrids as potential anti-bacterial agents. Eur. J. Med. Chem. 2019, 174, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Wang, A.; Xu, J.; An, Z.; Loh, K.Y.; Zhang, P.; Liu, X. Recent Advances in the Catalytic Synthesis of 4-Quinolones. Chem 2019, 5, 1059–1107. [Google Scholar] [CrossRef]
- Dine, I.; Mulugeta, E.; Melaku, Y.; Belete, M. Recent advances in the synthesis of pharmaceutically active 4-quinolone and its analogues: A review. RSC Adv. 2023, 13, 8657–8682. [Google Scholar] [CrossRef]
- Wang, J.-S.; Li, C.; Ying, J.; Xu, T.; Lu, W.; Li, C.-Y.; Wu, X.-F. Supported palladium-catalyzed carbonylative cyclization of 2-bromonitrobenzenes and alkynes to access quinolin-4(1H)-ones. J. Catal. 2022, 408, 81–87. [Google Scholar] [CrossRef]
- Ragaini, F.; Cenini, S.; Gasperini, M. Reduction of nitrobenzene to aniline by CO/H2O, catalysed by Ru3(CO)12/chelating diimines. J. Mol. Catal. A Chem. 2001, 174, 51–57. [Google Scholar] [CrossRef]
- Tafesh, A.M.; Beller, M.; Hoechst, A.G. First selective reduction of aromatic nitro compounds using water soluble catalysts. Tetrahedron Lett. 1995, 36, 9305–9308. [Google Scholar] [CrossRef]
- Söderberg, B.C.G.; Berkowitz, W.F. Reductive Cyclization of 2-Nitro- and β-Nitrostyrenes, 2-Nitrobiphenyls, and 1-Nitro-1,3-Dienes to Indoles, Carbazoles, and Pyrroles. Org. React. 2022, 111, 417–640. [Google Scholar] [CrossRef]
- Ferretti, F.; Ramadan, D.R.; Ragaini, F. Transition Metal Catalyzed Reductive Cyclization Reactions of Nitroarenes and Nitroalkenes. ChemCatChem 2019, 11, 4450–4488. [Google Scholar] [CrossRef]
- Tsygankov, A.A.; Makarova, M.; Chusov, D. Carbon monoxide as a selective reducing agent in organic chemistry. Mendeleev Commun. 2018, 28, 113–122. [Google Scholar] [CrossRef]
- Ferretti, F.; Formenti, D.; Ragaini, F. The reduction of organic nitro compounds by carbon monoxide as an effective strategy for the synthesis of N-heterocyclic compounds: A personal account. Rend. Fis. Acc. Lincei 2017, 28, 97–115. [Google Scholar] [CrossRef]
- Ragaini, F.; Cenini, S.; Gallo, E.; Caselli, A.; Fantauzzi, S. Fine chemicals by reductive carbonylation of nitroarenes, catalyzed by transition metal complexes. Curr. Org. Chem. 2006, 10, 1479–1510. [Google Scholar] [CrossRef]
- Soderberg, B.C.G. Synthesis of heterocycles via intramolecular annulation of nitrene intermediates. Curr. Org. Chem. 2000, 4, 727–764. [Google Scholar] [CrossRef]
- Cenini, S.; Ragaini, F. Catalytic Reductive Carbonylation of Organic Nitro Compounds; Kluwer Academic Publishers: Dordrecht, The Netherlands, 1996; p. 352. [Google Scholar] [CrossRef]
- Bernauer, J.; Pölker, J.; von Wangelin, A.J. Redox-Active BIAN-Based Diimine Ligands in Metal-Catalyzed Small Molecule Syntheses. ChemCatChem 2022, 14, e202101182. [Google Scholar] [CrossRef] [PubMed]
- Gasperini, M.; Ragaini, F.; Gazzola, E.; Caselli, A.; Macchi, P. Synthesis of mixed Ar,Ar′-BIAN ligands (Ar,Ar′-BIAN = bis(aryl) acenaphthenequinonediimine). Measurement of the coordination strength of hemilabile ligands with respect to their symmetric counterparts. Dalton Trans. 2004, 3376–3382. [Google Scholar] [CrossRef]
- Tollari, S.; Cenini, S.; Ragaini, F.; Cassar, L. Intramolecular Amination of Olefins—Synthesis of 2-Substituted-4-Quinolones from 2-Nitrochalcones Catalyzed by Ruthenium. J. Chem. Soc. Chem. Commun. 1994, 1741–1742. [Google Scholar] [CrossRef]
- Lin, J.-P.; Long, Y.-Q. Transition metal-free one-pot synthesis of 2-substituted 3-carboxy-4-quinolone and chromone derivatives. Chem. Commun. 2013, 49, 5313–5315. [Google Scholar] [CrossRef]
- Annunziata, R.; Cenini, S.; Palmisano, G.; Tollari, S. 4(1H)-quinolinone alkaloids. An efficient synthesis of graveoline by palladium-catalysed reductive N-heterocyclisation. Synth. Commun. 1996, 26, 495–501. [Google Scholar] [CrossRef]
- Tollari, S.; Penoni, A.; Cenini, S. The unprecedented detection of the intermediate formation of N-hydroxy derivatives during the carbonylation of 2′-nitrochalcones and 2-nitrostyrenes catalyzed by palladium. J. Mol. Catal. A Chem. 2000, 152, 47–54. [Google Scholar] [CrossRef]
- Wu, L.; Liu, Q.; Jackstell, R.; Beller, M. Carbonylations of Alkenes with CO Surrogates. Angew. Chem. Int. Ed. 2014, 53, 6310–6320. [Google Scholar] [CrossRef]
- Gautam, P.; Bhanage, B.M. Recent advances in the transition metal catalyzed carbonylation of alkynes, arenes and aryl halides using CO surrogates. Catal. Sci. Technol. 2015, 5, 4663–4702. [Google Scholar] [CrossRef]
- Friis, S.D.; Lindhardt, A.T.; Skrydstrup, T. The Development and Application of Two-Chamber Reactors and Carbon Monoxide Precursors for Safe Carbonylation Reactions. Acc. Chem. Res. 2016, 49, 594–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konishi, H.; Manabe, K. Formic Acid Derivatives as Practical Carbon Monoxide Surrogates for Metal-Catalyzed Carbonylation Reactions. Synlett 2014, 25, 1971–1986. [Google Scholar] [CrossRef]
- Peng, J.B.; Qi, X.X.; Wu, X.F. Recent Achievements in Carbonylation Reactions: A Personal Account. Synlett 2017, 28, 175–194. [Google Scholar] [CrossRef]
- Cao, J.; Zheng, Z.-J.; Xu, Z.; Xu, L.-W. Transition-metal-catalyzed transfer carbonylation with HCOOH or HCHO as non-gaseous C1 source. Coord. Chem. Rev. 2017, 336, 43–53. [Google Scholar] [CrossRef]
- Grushin, V.V.; Alper, H. Novel palladium-catalyzed carbonylation of organic halides by chloroform and alkali. Organometallics 1993, 12, 3846–3850. [Google Scholar] [CrossRef]
- Gockel, S.N.; Hull, K.L. Chloroform as a Carbon Monoxide Precursor: In or Ex Situ Generation of CO for Pd-Catalyzed Aminocarbonylations. Org. Lett. 2015, 17, 3236–3239. [Google Scholar] [CrossRef] [PubMed]
- Fujihara, T.; Hosoki, T.; Katafuchi, Y.; Iwai, T.; Terao, J.; Tsuji, Y. Palladium-catalyzed esterification of aryl halides using aryl formates without the use of external carbon monoxide. Chem. Commun. 2012, 48, 8012–8014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ueda, T.; Konishi, H.; Manabe, K. Palladium-Catalyzed Carbonylation of Aryl, Alkenyl, and Allyl Halides with Phenyl Formate. Org. Lett. 2012, 14, 3100–3103. [Google Scholar] [CrossRef]
- Chen, Z.; Wang, L.-C.; Wu, X.-F. Carbonylative synthesis of heterocycles involving diverse CO surrogates. Chem. Commun. 2020, 56, 6016–6030. [Google Scholar] [CrossRef]
- Panda, B.; Albano, G. DMF as co surrogate in carbonylation reactions: Principles and application to the synthesis of heterocycles. Catalysts 2021, 11, 1531. [Google Scholar] [CrossRef]
- Mondal, K.; Halder, P.; Gopalan, G.; Sasikumar, P.; Radhakrishnan, K.V.; Das, P. Chloroform as a CO surrogate: Applications and recent developments. Org. Biomol. Chem. 2019, 17, 5212–5222. [Google Scholar] [CrossRef] [PubMed]
- Ragaini, F.; Ferretti, F.; Fouad, M.A. Phenyl Formate as a CO Surrogate for the Reductive Cyclization of Organic Nitro Compounds to Yield Different N-Heterocycles: No Need for Autoclaves and Pressurized Carbon Monoxide. Catalysts 2023, 13, 224. [Google Scholar] [CrossRef]
- Ramadan, D.R.; Ferretti, F.; Ragaini, F. Catalytic reductive cyclization of 2-nitrobiphenyls using phenyl formate as CO surrogate: A robust synthesis of 9H-carbazoles. J. Catal. 2022, 409, 41–47. [Google Scholar] [CrossRef]
- Ferretti, F.; Fouad, M.A.; Ragaini, F. Synthesis of Indoles by Palladium-Catalyzed Reductive Cyclization of β−Nitrostyrenes with Phenyl Formate as a CO Surrogate. Catalysts 2022, 12, 106. [Google Scholar] [CrossRef]
- Fouad, M.A.; Ferretti, F.; Formenti, D.; Milani, F.; Ragaini, F. Synthesis of Indoles by Reductive Cyclization of Nitro Compounds Using Formate Esters as CO Surrogates. Eur. J. Org. Chem. 2021, 2021, 4876–4894. [Google Scholar] [CrossRef]
- Formenti, D.; Ferretti, F.; Ragaini, F. Synthesis of N-Heterocycles by Reductive Cyclization of Nitro Compounds using Formate Esters as Carbon Monoxide Surrogates. ChemCatChem 2018, 10, 148–152. [Google Scholar] [CrossRef]
- EL-Atawy, M.A.; Formenti, D.; Ferretti, F.; Ragaini, F. Synthesis of 3,6-Dihydro-2H-[1, 2]-Oxazines from Nitroarenes and Conjugated Dienes, Catalyzed by Palladium/Phenanthroline Complexes and Employing Phenyl Formate as a CO Surrogate. ChemCatChem 2018, 10, 4707–4717. [Google Scholar] [CrossRef] [Green Version]
- Fouad, M.A.; Ferretti, F.; Ragaini, F. Formic Acid as Carbon Monoxide Source in the Palladium-Catalyzed N-Heterocyclization of o-Nitrostyrenes to Indoles. J. Org. Chem. 2023, 88, 5108–5117. [Google Scholar] [CrossRef]
- Tollari, S.; Cenini, S.; Crotti, C.; Gianella, E. Synthesis of heterocycles via palladium-catalyzed carbonylation of ortho-substituted organic nitro compounds in relatively mild conditions. J. Mol. Catal. 1994, 87, 203–214. [Google Scholar] [CrossRef]
- Ragaini, F.; Ventriglia, F.; Hagar, M.; Fantauzzi, S.; Cenini, S. Synthesis of Indoles by Intermolecular Cyclization of Unfunctionalized Nitroarenes and Alkynes: One-Step Synthesis of the Skeleton of Fluvastatin. Eur. J. Org. Chem. 2009, 2009, 2185–2189. [Google Scholar] [CrossRef]
- Smitrovich, J.H.; Davies, I.W. Catalytic C-H functionalization driven by CO as a stoichiometric reductant: Application to carbazole synthesis. Org. Lett. 2004, 6, 533–535. [Google Scholar] [CrossRef] [PubMed]
- Davies, I.W.; Smitrovich, J.H.; Sidler, R.; Qu, C.; Gresham, V.; Bazaral, C. A highly active catalyst for the reductive cyclization of ortho-nitrostyrenes under mild conditions. Tetrahedron 2005, 61, 6425–6437. [Google Scholar] [CrossRef]
- Clawson, R.W.; Dacko, C.A.; Deavers, R.E.; Akhmedov, N.G.; Soderberg, B.C.G. Attempted synthesis of 3-hydroxy-2-octadecylindole. Proposed structural revision of previously prepared 3-hydroxy-2-octadecylindole and a proposed structure of fistulosin. Tetrahedron 2009, 65, 8786–8793. [Google Scholar] [CrossRef]
- Alessio, E.; Mestroni, G. Catalytic synthesis of aromatic urethanes from nitroaromatic compounds and carbon monoxide, using palladium + 1,10-phenanthroline derivatives as catalyst precursors. J. Mol. Catal. 1984, 26, 337–340. [Google Scholar] [CrossRef]
- Bontempi, A.; Alessio, E.; Chanos, G.; Mestroni, G. Reductive carbonylation of nitroaromatic compounds to urethanes catalyzed by (di-1,10-phenanthroline)palladium bis(hexafluorophosphate) and related complexes. J. Mol. Catal. 1987, 42, 67–80. [Google Scholar] [CrossRef]
- Wehman, P.; Kaasjager, V.E.; Delange, W.G.J.; Hartl, F.; Kamer, P.C.J.; van Leeuwen, P.W.N.M.; Fraanje, J.; Goubitz, K. Subtle Balance between Various Phenanthroline Ligands and Anions in the Palladium-Catalyzed Reductive Carbonylation of Nitrobenzene. Organometallics 1995, 14, 3751–3761. [Google Scholar] [CrossRef] [Green Version]
- Gasperini, M.; Ragaini, F.; Remondini, C.; Caselli, A.; Cenini, S. The palladium-phenanthroline catalyzed carbonylation of nitroarenes to diarylureas: Effect of chloride and diphenylphosphinic acid. J. Organomet. Chem. 2005, 690, 4517–4529. [Google Scholar] [CrossRef]
- Ferretti, F.; Ragaini, F.; Lariccia, R.; Gallo, E.; Cenini, S. New Nonsymmetric Phenanthrolines as Very Effective Ligands in the Palladium-Catalyzed Carbonylation of Nitrobenzene. Organometallics 2010, 29, 1465–1471. [Google Scholar] [CrossRef]
- Ferretti, F.; Gallo, E.; Ragaini, F. Mineral Oil/Methanol: A Cheap Biphasic Reaction Medium with Thermomorphic Properties and Its Application to the Palladium-Catalyzed Carbonylation of Nitrobenzene to Methyl Phenylcarbamate. ChemCatChem 2015, 7, 2241–2247. [Google Scholar] [CrossRef]
- El-Atawy, M.A.; Ferretti, F.; Ragaini, F. Palladium-Catalyzed Intramolecular Cyclization of Nitroalkenes: Synthesis of Thienopyrroles. Eur. J. Org. Chem. 2017, 2017, 1902–1910. [Google Scholar] [CrossRef]
- Stashenko, E.E.; Acosta, R.; Martínez, J.R. High-resolution gas-chromatographic analysis of the secondary metabolites obtained by subcritical-fluid extraction from Colombian rue (Ruta graveolens L.). J. Biochem. Biophys. Methods 2000, 43, 379–390. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.-S.; Shi, L.-S.; Wang, J.-J.; Iou, S.-C.; Chang, H.-C.; Chen, Y.-P.; Kuo, Y.-H.; Chang, Y.-L.; Tenge, C.-M. Cytotoxic and Antiplatelet Aggregation Principles of Ruta Graveolens. J. Chin. Chem. Soc. 2003, 50, 171–178. [Google Scholar] [CrossRef]
- An, Z.-Y.; Yan, Y.-Y.; Peng, D.; Ou, T.-M.; Tan, J.-H.; Huang, S.-L.; An, L.-K.; Gu, L.-Q.; Huang, Z.-S. Synthesis and evaluation of graveoline and graveolinine derivatives with potent anti-angiogenesis activities. Eur. J. Med. Chem. 2010, 45, 3895–3903. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Bishayee, K.; Khuda-Bukhsh, A.R. Graveoline Isolated from Ethanolic Extract of Ruta graveolens Triggers Apoptosis and Autophagy in Skin Melanoma Cells: A Novel Apoptosis-Independent Autophagic Signaling Pathway. Phytother. Res. 2014, 28, 1153–1162. [Google Scholar] [CrossRef]
- Zhou, S.; Huang, G. The biological activities of butyrylcholinesterase inhibitors. Biomed. Pharmacother. 2022, 146, 112556. [Google Scholar] [CrossRef]
- Berman, R.S.; Kochi, J.K. Kinetics and mechanism of oxygen atom transfer from nitro compounds mediated by nickel(0) complexes. Inorg. Chem. 1980, 19, 248–254. [Google Scholar] [CrossRef]
- Skoog, S.J.; Gladfelter, W.L. Activation of Nitroarenes in the Homogenous Catalytic Carbonylation of Nitroaromatics via an Oxygen-Atom-Transfer Mechanism Induced by Inner Sphere Electron Transfer. J. Am. Chem. Soc. 1997, 119, 11049–11060. [Google Scholar] [CrossRef]
- Kunin, A.J.; Noirot, M.D.; Gladfelter, W.L. In situ FTIR spectroscopy at elevated carbon monoxide pressure. Evidence for single electron transfer in the catalytic carbonylation of nitroaromatics. J. Am. Chem. Soc. 1989, 111, 2739–2741. [Google Scholar] [CrossRef]
- Belousov, Y.A. Radical chemistry of iron carbonyls. Russ. Chem. Rev. 2007, 76, 41–58. [Google Scholar] [CrossRef]
- Belousov, Y.A.; Kolosova, T.A. Electron-spin resonance study of the reaction of iron carbonyls with nitro and nitrosoparaffines—A mechanism of the reductive carbonylation of nitrocompounds. Polyhedron 1987, 6, 1959–1970. [Google Scholar] [CrossRef]
- Ragaini, F. Mechanistic study of the phase-transfer-catalyzed reduction of nitrobenzene to aniline by iron carbonyl complexes. Role of the radical anion [Fe3(CO)11)](·−). Organometallics 1996, 15, 3572–3578. [Google Scholar] [CrossRef]
- Liu, P.H.; Liao, H.Y.; Cheng, C.H. Electro-transfer from tetracarbonylrhodate(1-) to nitro aromatics—Novel interaction of nitro radical-anions with a rhodium(I) center. J. Chem. Soc. Chem. Commun. 1995, 2441–2442. [Google Scholar] [CrossRef] [Green Version]
- Ragaini, F.; Cenini, S.; Demartin, F. Mechanistic Study of the Carbonylation of Nitrobenzene Catalyzed by the [Rh(CO)4]- Nitrogen Base System—X-ray Structure of [PPN][Rh(CO)2ON(C6H3Cl2)C(O)O]. Organometallics 1994, 13, 1178–1189. [Google Scholar] [CrossRef]
- Ragaini, F.; Sportiello, P.; Cenini, S. Investigation of the possible role of arylamine formation in the ortho-substituted nitroarenes reductive cyclization reactions to afford heterocycles. J. Organomet. Chem. 1999, 577, 283–291. [Google Scholar] [CrossRef]
- Penoni, A.; Volkmann, J.; Nicholas, K.M. Regioselective synthesis of indoles via reductive annulation of nitrosoaromatics with alkynes. Org. Lett. 2002, 4, 699–701. [Google Scholar] [CrossRef]
- Rimoldi, M.; Ragaini, F.; Gallo, E.; Ferretti, F.; Macchi, P.; Casati, N. Unexpected isomerism in “[Pd(2,9-dimethylphenanthroline)X2]” (X = Cl, Br, I) complexes: A neutral and an ionic form exist. Dalton Trans. 2012, 41, 3648–3658. [Google Scholar] [CrossRef]
- Romek, A.; Opatz, T. Microwave-Assisted Synthesis of Polysubstituted 4-Quinolones from Deprotonated α-Aminonitriles. Eur. J. Org. Chem. 2010, 2010, 5841–5849. [Google Scholar] [CrossRef]
- Haddad, N.; Tan, J.; Farina, V. Convergent Synthesis of the Quinolone Substructure of BILN 2061 via Carbonylative Sonogashira Coupling/Cyclization. J. Org. Chem. 2006, 71, 5031–5034. [Google Scholar] [CrossRef]
- Sun, F.; Zhao, X.; Shi, D. An efficient one-step synthesis of 2-arylquinolin-4(1H)-ones with the aid of a low-valent titanium reagent. Tetrahedron Lett. 2011, 52, 5633–5635. [Google Scholar] [CrossRef]
- Åkerbladh, L.; Nordeman, P.; Wejdemar, M.; Odell, L.R.; Larhed, M. Synthesis of 4-Quinolones via a Carbonylative Sonogashira Cross-Coupling Using Molybdenum Hexacarbonyl as a CO Source. J. Org. Chem. 2015, 80, 1464–1471. [Google Scholar] [CrossRef]
- Lohrer, B.; Bracher, F. Novel access to 2-substituted quinolin-4-ones by nickel boride-mediated reductive ring transformation of 5-(2-nitrophenyl)isoxazoles. Tetrahedron Lett. 2019, 60, 151327. [Google Scholar] [CrossRef]
- Singh, P.; Sahoo, S.K.; Goud, N.S.; Swain, B.; Yaddanapudi, V.M.; Arifuddin, M. Microwave-Assisted Copper-Catalyzed One-Pot Synthesis of 2-Aryl/Heteroaryl-4-Quinolones via Sequential Intramolecular Aza-Michael Addition and Oxidation. Asian J. Org. Chem. 2022, 11, e202200181. [Google Scholar] [CrossRef]
- Malbari, K.; Saha, P.; Chawla-Sarkar, M.; Dutta, S.; Rai, S.; Joshi, M.; Kanyalkar, M. In quest of small-molecules as potent non-competitive inhibitors against influenza. Bioorg. Chem. 2021, 114, 105139. [Google Scholar] [CrossRef]
- Hu, W.; Lin, J.-P.; Song, L.-R.; Long, Y.-Q. Direct Synthesis of 2-Aryl-4-quinolones via Transition-Metal-Free Intramolecular Oxidative C(sp3)–H/C(sp3)–H Coupling. Org. Lett. 2015, 17, 1268–1271. [Google Scholar] [CrossRef]
- Li, L.; Wang, H.-K.; Kuo, S.-C.; Wu, T.-S.; Lednicer, D.; Lin, C.M.; Hamel, E.; Lee, K.-H. Antitumor Agents. 150. 2′,3′,4′,5′,5,6,7-Substituted 2-Phenyl-4-quinolones and Related Compounds: Their Synthesis, Cytotoxicity, and Inhibition of Tubulin Polymerization. J. Med. Chem. 1994, 37, 1126–1135. [Google Scholar] [CrossRef]
- Aksenov, N.A.; Aksenov, D.A.; Arutiunov, N.A.; Aksenova, D.S.; Aksenov, A.V.; Rubin, M. Unexpected cyclization of ortho-nitrochalcones into 2-alkylideneindolin-3-ones. RSC Adv. 2020, 10, 18440–18450. [Google Scholar] [CrossRef]
- Climent, M.J.; Corma, A.; Iborra, S.; Martí, L. Process Intensification with Bifunctional Heterogeneous Catalysts: Selective One-Pot Synthesis of 2′-Aminochalcones. ACS Catal. 2015, 5, 157–166. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||||
| Entry | T (°C) | t (h) | Solvent | FA/Ac2O/Et3N to 1a mol Ratio | Ligand | Conv. % 2 | Select. % 3 |
| 1 4 | 110 | 10 | Acetone | 2.5 | Phen | 98 | 51 |
| 2 | 140 | 10 | CH3CN | 4.4 | Phen | >99 | 69 |
| 3 | 140 | 10 | CH3CN | 3 | Phen | 96 | 76 |
| 4 | 140 | 10 | CH3CN | 2.5 | Phen | 84 | 62 |
| 5 5 | 140 | 10 | CH3CN | 3 | Phen | 37 | 24 |
| 6 | 140 | 10 | CH3CN | 3 | TMPhen | >99 | 47 |
| 7 6 | 140 | 10 | CH3CN | 3 | (MeO)2Phen | 98 | 58 |
| 8 | 150 | 10 | CH3CN | 3 | Phen | 100 | 74 |
| 9 | 130 | 10 | CH3CN | 3 | Phen | 84 | 80 |
| 10 | 120 | 16 | CH3CN | 3 | Phen | >99 | 55 |
| 11 6 | 130 | 10 | CH3CN | 3 | Phen | 57 | 85 |
| 12 6 | 130 | 16 | CH3CN | 3 | Phen | 96 | 76 |
| 13 6 | 130 | 16 | CH3CN/DMF 7 | 3 | Phen | >99 | 75 |
| 14 6 | 130 | 16 | MEK | 3 | Phen | 64 | 72 |
| 15 6 | 130 | 16 | DMF | 3 | Phen | >99 | >99 |
| 16 6 | 130 | 10 | DMF | 3 | Phen | 99 | >99 |
| 17 6 | 130 | 6 | DMF | 3 | Phen | 88 | 93 |
| 18 6 | 130 | 4 | DMF | 3 | Phen | 59 | 93 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferretti, F.; Fouad, M.A.; Abbo, C.; Ragaini, F. Effective Synthesis of 4-Quinolones by Reductive Cyclization of 2′-Nitrochalcones Using Formic Acid as a CO Surrogate. Molecules 2023, 28, 5424. https://doi.org/10.3390/molecules28145424
Ferretti F, Fouad MA, Abbo C, Ragaini F. Effective Synthesis of 4-Quinolones by Reductive Cyclization of 2′-Nitrochalcones Using Formic Acid as a CO Surrogate. Molecules. 2023; 28(14):5424. https://doi.org/10.3390/molecules28145424
Chicago/Turabian StyleFerretti, Francesco, Manar Ahmed Fouad, Cecilia Abbo, and Fabio Ragaini. 2023. "Effective Synthesis of 4-Quinolones by Reductive Cyclization of 2′-Nitrochalcones Using Formic Acid as a CO Surrogate" Molecules 28, no. 14: 5424. https://doi.org/10.3390/molecules28145424